

Abstract
BACKGROUND AND PURPOSE
Despite growing evidence that inhibition of α6β2-containing (α6β2*) nicotinic acetylcholine receptors (nAChRs) may be beneficial for the therapy of tobacco addiction, the lack of good sources of α6β2*-nAChRs has delayed the discovery of α6β2-selective antagonists. Our aim was to generate a cell line stably expressing functional nAChRs with α6β2 properties, to enable pharmacological characterization and the identification of novel α6β2-selective antagonists.
EXPERIMENTAL APPROACH
Different combinations of the α6, β2, β3, chimeric α6/3 and mutant β3V273S subunits were transfected in human embryonic kidney cells and tested for activity in a fluorescent imaging plate reader assay. The pharmacology of rat immune-immobilized α6β2*-nAChRs was determined with 125I-epibatidine binding.
KEY RESULTS
Functional channels were detected after co-transfection of α6/3, β2 and β3V273S subunits, while all other subunit combinations failed to produce agonist-induced responses. Stably expressed α6/3β2β3V273S-nAChR pharmacology was unique, and clearly distinct from α4β2-, α3β4-, α7- and α1β1δε-nAChRs. Antagonist potencies in inhibiting α6/3β2β3V273S-nAChRs was similar to their binding affinity for rat native α6β2*-nAChRs. Agonist affinities for α6β2*-nAChRs was higher than their potency in activating α6/3β2β3V273S-nAChRs, but their relative activities were equivalent. Focussed set screening at α6/3β2β3V273S-nAChRs, followed by cross-screening with the other nAChRs, led to the identification of novel α6β2-selective antagonists.
CONCLUSIONS AND IMPLICATIONS
We generated a mammalian cell line stably expressing nAChRs, with pharmacological properties similar to native α6β2*-nAChRs, and used it to identify novel non-peptide, low molecular weight, α6β2-selective antagonists. We also propose a pharmacophore model of α6β2 antagonists, which offers a starting point for the development of new smoking cessation agents.
Keywords: Nicotine acetylcholine receptor, α6, β3, chimeric subunit, point mutation, stable expression, recombinant, nicotine dependence, Parkinson's disease
Introduction
After the first nicotinic acetylcholine receptor (nAChR) was isolated from muscular tissue and fish electric organ (Changeux et al., 1969; 1970;), 11 nAChR subunits have been cloned from the mammalian nervous system (the α2-α7, α9, α10 and β2-β4 subunits), which can assemble to form a variety of ligand-gated pentameric ion channels permeable to Ca2+, Na+ and K+ ions (Itier and Bertrand, 2001). Three major subtypes of neuronal nAChRs have been extensively described: the α4β2*- and α7-nAChRs, expressed predominantly in the central nervous system (CNS), and the α3β4*-nAChRs, abundant in the peripheral nervous system (for nomenclature, see Lukas et al., 1999; Alexander et al., 2009). These receptors have different pharmacological characteristics and are involved in a multiplicity of distinct pathologies (Lena and Changeux, 1998; Lindstrom, 2003; Gotti et al., 2006b).
In the last decade, another subtype of neuronal nAChRs has been attracting the attention of the scientific community: the α6β2*-nAChR nicotinic acetylcholine receptors containing both the α6 and the β2 subunits (α6β2*-nAChRs) are selectively distributed in a few CNS areas, namely the dopaminergic mesostriatal pathway and the retina, the optic nerve and its visual afferents (Champtiaux et al., 2002; Zoli et al., 2002; Moretti et al., 2004; Gotti et al., 2005; 2010; Cox et al., 2008). Much evidence has been collected on the functions of α6β2*-nAChRs on dopaminergic neurons, indicating that they mediate ACh-elicited currents in the somato-dendritic compartment (Klink et al., 2001; Champtiaux et al., 2003) and nicotine-induced dopamine release at the terminal level (Klink et al., 2001; Champtiaux et al., 2003; Salminen et al., 2004; Grady et al., 2007). The dopaminergic system has a ventral component, comprising cell bodies in the ventral tegmental area and terminals in the nucleus accumbens (nAc) and tuberculum olfactorium, and a dorsal component, comprising cell bodies in the substantia nigra and terminals in the caudate putamen. Notably, different studies suggest that α6β2*-nAChRs preferentially modulate the ventral over the dorsal dopaminergic pathway and therefore dominate control of dopaminergic neurotransmission in the nAc (Drenan et al., 2008; Exley et al., 2008), a main neurochemical target of addictive drugs (Di Chiara, 2000; Wise, 2002). Indeed, some recent papers have started to elucidate the functional role of α6β2*-nAChRs in vivo showing their involvement in mediating the effects of systemic nicotine in dopamine release, locomotion and reinforcement (Drenan et al., 2008; Pons et al., 2008; Brunzell et al., 2010; Gotti et al., 2010). This suggests that α6β2*-selective antagonists able to cross the blood–brain barrier would be promising drugs to affect nicotine addictive properties and therefore for the therapy of tobacco dependence, with higher selectivity for the dopaminergic system and potentially fewer side effects than other therapeutic approaches (Gotti et al., 2010).
Immunoprecipitation and immunopurification techniques coupled with cell-specific lesions have revealed that α6β2*-nAChR in the mesostriatal system are heterogeneous in terms of subunit composition, with α6β2β3 receptors predominant in ventral striatum and α6α4β2β3 receptors dominating in the dorsal striatum and in the midbrain (Gotti et al., 2010). It is generally accepted that heteropentameric receptors have two binding sites per receptor molecule, located at the interface between an α and a β subunit, and that functional receptors comprise two α subunits, carrying the principal component of the ACh binding site, two β subunits, carrying the complementary component of the ACh binding site and a fifth subunit, which does not participate in ACh binding. Assuming that the β3 subunit, which is thought to lack the principal and complementary components of the binding site (Groot-Kormelink et al., 1998), is in the fifth position, it may be argued that native α6β2β3 receptors have two α6β2 binding interfaces and pure α6 pharmacological properties, whereas α6α4β2β3 receptors have an α6β2 and an α4β2 binding interface and mixed α6 and α4 pharmacological properties. A source providing a good quantity of an isolated population of functional, pure α6 receptors (that is, α6β2β3 receptors) would be the optimal reagent for primary screening aimed at identifying α6β2 selective ligands.
Despite the cDNA sequence of the human α6 subunit being first described in 1996 (Elliott et al., 1996), there have been no reports, to date, of the functional expression of human α6β2β3- or α6α4β2β3-nAChRs in mammalian expression systems. The first attempts to obtain the expression of functional nAChRs containing the α6 subunit were made in Xenopus laevis oocytes. Although co-expression of the α6 subunit with the β4 subunit formed functional channels, co-expression of the α6β2 and α6β2β3 subunit combinations either failed to form functional receptors, or was inefficient and yielded channels with very poor responses (Gerzanich et al., 1997; Kuryatov et al., 2000; Broadbent et al., 2006). More promising results were obtained with the construction of chimeric subunits having the extracellular domain (where the principal component of ACh binding site is located) of the α6 subunit, and the transmembrane and intracellular domains of the α3 and the α4 subunit (α6/3 and α6/4 chimeric subunits respectively; Kuryatov et al., 2000; Dowell et al., 2003; Papke et al., 2008). Co-expression of chimeric α6/3 and α6/4 subunit with the β2, or the with the β2 and β3 subunit gave functional receptors potently inhibited by α-conotoxin MII, a high affinity antagonist of native α6β2*-nAChRs (Mogg et al., 2002), suggesting that the α6 properties of the principal component of the ACh binding site are conserved in these chimeras. However, the oocyte expression system does not provide a cellular reagent suitable for continuous passage and is not considered a good source for isolated population of receptors for a functional assay, which is required to identify and develop ligands with selective α6β2 properties.
Functional co-expression of avian or rat α6 with β2 subunits in mammalian cell lines has been reported but had a very low efficiency and/or resulted in very low ACh-activated currents (Fucile et al., 1998; Walsh et al., 2008). In addition, co-expression of the human chimeric α6/4 subunit with the β2 subunit in human embryonic kidney (HEK) cells did not result in functional receptors (Evans et al., 2003), whereas co-expression of the rat or human α6/3 subunit with the β2 and β3 subunits in mammalian cell lines has never been reported. Interestingly, the co-expression of a human β3 subunit with a valine-serine mutation in position 9′ of the second, pore-lining transmembrane domain (M2; the β3V273S subunit) resulted in a gain of function when co-expressed with a variety of nAChR subunit combinations in oocytes, including the α6β2β3 subunit combination (Broadbent et al., 2006). However, co-expression of the α6β2β3V273S combination in mammalian cell lines has not previously been reported. It is noteworthy that Tumkosit et al., 2006 generated α6β2- and α6β2β3-HEK cell lines expressing mature receptors in the cell membrane, which bind cholinergic ligands and are sensitive to nicotine-induced up-regulation; at present, however, there is no report describing functional nAChR activity with these cell lines.
Here we show the generation of a HEK cell line stably co-expressing functional human α6/3β2β3V273S yielded receptors with pharmacological properties similar to rat native α6β2*-nAChRs. Primary screening of focussed compound sets, followed by selectivity comparison of activity against other nAChR subtypes, led to the identification of selective, non-peptide, low molecular weight α6β2*-nAchR antagonists. Superimposition of three of these molecules provides the generation of the first pharmacophore model of α6β2 antagonists.
Methods
Cell lines
Wild-type HEK293 and HEK293T cell lines were obtained from the American Type Culture Collection (CRL-1573 and CRL-11268, respectively). Stable cell lines co-expressing the human nAChR subunits α3 and β4 (α3β4-nAChRs) and the human nAChR subunits α1, β1, δ and ε (α1β1δε-nAChRs) were obtained from Millipore Corporation (Catalogue Numbers: CYL3057 and CYL3052, respectively).
Animals
All animal care and experimental procedures were in accordance with Italian national legislation and the European Communities Council Directive of 24 November 1986 (86/609/EEC). Adult male pathogen-free Sprague Dawley rats (8 weeks old, weight 250–300 g; Charles River) were used.
nAChR subunit cDNA, α6/3 subunit chimera and β3V273S subunit mutant
Human nAChR α6, β2 and β3 subunit cDNAs were amplified from human brain marathon Ready cDNA template (Clontech, Mountain View, CA, USA) using pfu turbo DNA polymerase (Agilent Technologies, Cedar Creek, TX, USA). Primers were derived from published sequences of human α6 (NCBI Reference Sequence: NM_004198), β2 (NM_000748) and β3 (NM_000749) subunits. These subunit cDNAs were subcloned into expression vector pcDNA3.2 (Invitrogen, Carlsbad, CA, USA). The human nAChR α3 subunit cDNA used as a template for chimeric receptor generation (NM_000743) was obtained from Millipore Corporation. nAChR β3V273S mutant was constructed by QuikChange Site-Directed Mutagenesis kit (Agilent Technologies). The primer sequences used were:
Forward-ATCCACATCGGTCTTGTCTTCTCTGACAGTTTTCC
Reverse-GGAAAACTGTCAGAGAAGACAAGACCGATGTGGAT.
A chimeric α6/3 cDNA, encoding a protein of 504 amino acids, in which amino acids 1–237 corresponded to amino acids 1–237 of the N-terminus of α6 (NP_004189, where the unprocessed methionine is amino acid 1) and amino acids 238–504 corresponded to amino acids 239 to 505 of the C-terminus of α3 subunit (NP_000734, again where the unprocessed methionine is amino acid 1), was constructed by In-Fusion technology (Clontech). Essentially, PCR primers were designed to amplify each segment and contain 15 bp overlap with the adjacent segment. The two DNA segments were joined to the vector pcDNA3.2, linearized by restriction enzymes Not1 and Asc1, by the In-Fusion enzyme.
All constructs were sequence confirmed using Big Dye Terminator V1.1 Cycle Sequencing kit and ABI 370XL automated sequencer (Applied Biosystems, Foster City, CA, USA). Sequence analysis was performed with Lasergene (DNA Star, Madison, WI, USA) software program.
Transient transfection and stable cell line generation
Cells were grown in DMEM HAMS F12 medium supplemented with 10% foetal bovine serum and 4 mM glutamine (Invitrogen) at 37°C unless otherwise stated. Transfections were carried out with combinations of nicotinic receptor subunits at equimolar ratios using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions.
Following overnight incubation, the transfected HEK293T cells were plated into 384 well assay plates at 30 000 per well in the presence of 10 mM sodium butyrate and incubated for 4 h at 37°C, followed by 24 h at 30°C before being assayed.
For stable cell line generation, expression vectors for human α6/3, β3V273S and β2 nAChR subunits were transfected into HEK293 cells using Lipofectamine 2000 and the cells were split to 35 mm Petri dishes after overnight incubation. 48 h after transfection, medium was replaced to contain 800 µg·mL−1 geneticin, 200 µg·mL−1 hygromycin B and 1 µg·mL−1 of puromycin. Following 14 days antibiotic selection, the resulting stably transfected population was single cell dilution cloned in 96 well plates to obtain individual colonies. Clonal cell lines were expanded from wells containing single colonies in the continued presence of selection antibiotics. A clonal cell line which gave robust Ca2+ influx responses to both nicotine (80 µM) and epibatidine (200 nM) addition in a fluorescent imaging plate reader (FLIPR) Ca2+-imaging assay was identified for further characterization and cryopreserved at 1.4 × 107 cells·mL−1 in 10% DMSO and 90% dialysed foetal bovine serum.
Stable cell lines co-expressing the human nAChR subunits α4 and β2 (NCBI Reference Sequence: α4 NM_000744 and β2 NM_000748) were generated by electroporation of HEK293 cells with pCIN5-α4 plasmid vector and subsequent lipofectamine transfection of pCIH1-β2 plasmid vector, which carry G418 and hygromycin resistance markers, respectively (Rhodes et al., 1998), selection in 400 µg·mL−1 geneticin G418 and 100 µg·mL−1 hygromycin-B followed by single cell dilution cloning. A clonal cell line that gave functional expression of α4β2 nAChRs was selected following characterization in a FLIPR-Ca2+ imaging assay.
Generation of rat pituitary tumour-derived (GH4C1) cells expressing human α7-nAChRs (Capelli et al., 2010) was performed as described for the generation of the GH4C1 cells expressing rat α7-nAChRs (Virginio et al., 2002). Raw data representative of the functional activity of this cell line are shown as supplementary information (Figure S1).
FLIPR assay
α6/3β2β3V273S-nAChRs
Frozen α6/3β2β3V273S-HEK293 cells were thawed 48 h before an experiment, centrifuged, resuspended in DMEM HAMS F12 growth medium supplemented with 10% foetal bovine serum and 1% non-essential amino acid solution (Invitrogen) at a density of 6 × 105 cells·mL−1 and plated in coated clear bottom black 384 wells plates (Greiner) at 30 000 cells per well. Cells were then incubated at 37°C, 5% CO2 for 24 h and at 30°C, 5% CO2 for 24 h. On the day of the experiment, cells were washed twice with Universal Buffer (145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 20 mM HEPES, 5.5 mM glucose, pH = 7.3) containing 0.5 mM probenecid and the intracellular Ca2+ content measured using the Ca2+ chelating dye FLUO-4-AM (Invitrogen) in conjunction with a FLIPR (Molecular Devices, Sunnyvale, CA, USA). Briefly, the cell permeant dye Fluo-4 was prepared to a concentration of 1 mM in 100% DMSO and 10% Pluronic F127. The dye was then diluted with buffer to a final concentration of 2 µM and placed on the cells. After 45–60 min dye loading incubation at 37°C, the un-incorporated dye was removed from the cells by washing (80 µL, 3 times) with buffer, and a final volume of 30 µL per well of buffer was left in each well. The plates were then placed in the FLIPR, and fluorescence (excitation 488 nm, emission 510–580 nm) measured starting from 30 s before drug addition and monitored for 10 min.
In order to discriminate the agonist/partial agonist and antagonist/desensitizing activity of compounds in the same experiment, a FLIPR dual addition protocol was established. In the first addition, 10 µL compound solution was added to the plate, in order to detect compound ability to activate the channel; the second addition occurred after 10 min and consisted in adding, to the same plate, 10 µL of the standard agonist solution, at a final concentration giving 80% of maximal effect (EC80), in order to determine compound capability to prevent agonist activation. In the case of α6/3β2β3V273S-HEK293 cell assay, the standard agonist solution was 200 nM nicotine. Maximum fluorescence values were recorded after the first and second addition and fitted for activation and inhibition concentration response curves respectively.
Focussed sets of compounds were initially tested for their inhibitory/desensitizing activity at α6/3β2β3V273S receptors at a single concentration (single shot screening), in a FLIPR single addition protocol. Briefly, the compound was added to the cells offline, the plate incubated for 10 min and then placed in the FLIPR; 200 nM nicotine was then added to the cells and the fluorescence measured. After prioritization of positive hits, selected compounds were tested as a range of concentrations in the FLIPR dual addition protocol as explained above, that is with fluorescence measured both after the compound addition (first addition) and the following nicotine addition (second addition), to obtain a complete agonist and antagonist/desensitizing concentration response curve respectively.
α4β2-, α3β4-, α1β1δε- and α7-nAChRs
α4β2-HEK293, α3β4-HEK293, α7-GH4C1 and α1β1δε-HEK293 cell line FLIPR assays were performed with approximately the same procedure of the α6/3β2β3V273S-HEK293 cell line FLIPR assay, with very slight modifications. α4β2-HEK293 cell plates, differently from α6/3β2β3V273S-HEK293 cell plates, were kept at 30°C, 5% CO2 for 48 h; incubation at 30°C was not performed for α3β4-HEK293 and α1β1δε-HEK293 cells. α7-GH4C1 cells were grown in Ham's F10 medium (Invitrogen) supplemented with 15% heat inactivated horse serum, 2.5% fetal bovine serum and 1 mM glutamine. Standard agonist solution was 100 nM epibatidine for α4β2-HEK293 and α3β4-HEK293, 10 µM nicotine for α7-GH4C1 and 100 µM succinylcholine for α1β1δε-HEK293 (final concentration).
Binding to immuno-immobilized rat native α6β2*-nAChRs
Antibodies were produced and characterized as described by Moretti et al. (2010). For preparation of immuno-immobilized native receptors, rats were killed by decapitation, the brain quickly removed from the skull, the superior colliculus dissected and 2% Triton X-100 extracts prepared as reported previously (Gotti et al., 2005).
The affinity-purified anti-α6 antibodies were bound to microwells (MaxiShorp; Nalge Nunc International, Naperville, IL, USA) by incubating overnight at 4°C at a concentration of 10 µg·mL−1 in 50 mM phosphate buffer, pH 7.5. On the following day, the wells were washed to remove the excess of unbound antibodies and then incubated overnight at 4°C with 200 µL of 2% Triton X-100 superior colliculus membrane extracts.
The receptors immobilized by the corresponding subunit-specific antibodies were incubated overnight at 4°C with 200 µL of 125I-epibatidine 0.1 nM (specific activity 2200 Ci·mmol−1 Perkin-Elmer) for inhibition experiments and at concentrations ranging from 0.005 to 1 nM for saturation experiments. All of the incubations were performed in a buffer containing 50 mM Tris-HCl, pH 7, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 2 mg·mL−1 bovine serum albumin, and 0.05% Tween 20. Specific ligand binding was defined as total binding minus the binding in the presence of 100 nM cold epibatidine.
After incubation, the wells were washed seven times with ice-cold phosphate buffer solution containing 0.05% Tween 20, and the bound radioactivity recovered by incubation with 200 µL of 2N NaOH for 2 h. The bound radioactivity was then determined by means of liquid scintillation counting in a gamma counter.
Data analysis
Data analysis from FLIPR experiments was performed with ActivityBase software (IDBS, Guildford, UK). Non-linear curve fitting of the agonist/partial agonist concentration response curves (first addition) led to the evaluation of the maximal effect (Emax) and the concentration giving 50% of maximum effect (EC50). The Emax was expressed as percentage of the maximal activation given by nicotine in the same experiment (Emax%). The intrinsic activity (IA) was defined as the ratio between the Emax of the compound of interest and the Emax of nicotine. Non-linear curve fitting of the inhibition concentration response curves (second addition) lead to the estimation of the concentration giving 50% of maximum inhibition (IC50).
The experimental data obtained from the binding experiments performed using antibody-bound receptors were analysed by means of a non-linear least square procedure using the LIGAND program as described by Munson and Rodbard (1980). The radioligand dissociation constant (KD) value and the unlabelled compounds inhibition constant (Ki) values were obtained by simultaneously fitting the results of 3–4 independent experiments. The selection of the best fit (i.e. one-site vs. two-site model) and the evaluation of the statistical significance of the parameters were based on the F-test for the ‘extra sum of square’ principle. A P-value of <0.05 was considered statistically significant.
Data are expressed as mean ± SEM of n experiments, unless otherwise indicated.
Computational work
Nicotinic set generation
Nicotinic ligands were selected from the AurSCOPE Ion Channels databases (Aureus Sciences, Paris, France) using a query in the Biology field (ephys) with target name defined as ‘nicotinic acetylcholine receptor – all subtypes’ and parameter ‘EC50 OR IC50 OR Ki OR Kb OR Ka OR Kapp (any unit, no range)’. Then, all the compounds and their biological data matching the criteria above were exported in SMILES format (Daylight) and pruned using filters developed in GlaxoSmithKline (GSK) for the removal of reactive and undesirable molecules, as well as of those characterized by non appropriate physico-chemical properties for a CNS drug assessed with the use of a statistical model developed within GSK (Aureus_nicotinic_training_set).
Structure and pXC50 data of the compounds tested in house in the nicotinic assay panel were exported from GSK repositories. Potency and efficacy cut-offs were applied as follows: pEC50≥ 6 AND Emax≥ 20% for agonists; pEC50≥ 6 AND Emax≥ 50% for positive modulators; pIC50≥ 6 for antagonists, except for α1 antagonists where 5.5 ≤pIC50≤ 6 was used. All the compounds and their biological data matching the criteria above were exported and then pruned with the same CNS and structural filters used to filter the derivatives extracted from the Aureus repository. The GSK derivatives selected were then clustered with a standard complete-link clustering. Representatives covering all the clusters were chosen (in_house_nicotinic_representative_set).
The in_house_nicotinic_representative_set and the Aureus_nicotinic_training_set derivatives were described with reduced graph (Gillet et al., 2003; Harper et al., 2004), topological pharmacophore (Schneider et al., 1999) and Daylight fingerprints descriptors. Then, their similarity with respect to in house repository compounds described in the same way was calculated with the use of a tool developed in house. Three similarity searching methods were used to identify compounds from the databases with each query molecule and each similarity method. Both the reduced graph and the topological pharmacophore similarity cut-offs were set to 0.8 while structural similarity (Tanimoto index and Daylight fingerprints as descriptors) was set at 0.85 at maximum. Finally, the compounds retained were filtered by applying a molecular weight cut-off of 400 and calculated lipophilicity of 4 (clogP; Daylight Chemical Information System: http://www.daylight.com, accessed February 2009). Finally, the compounds carrying undesirable chemical features were removed with the use of Daylight SMARTS developed ad hoc.
Post-processing of single shot hits
Focussed set single shot hits were prioritized according to their percentage efficiency index (Abad-Zapatero and Metz, 2005) followed by pruning using filters developed for the removal of derivatives characterized by non-appropriate physico-chemical properties for a CNS drug i.e. MW: 250–400, cLogP: 2–3.5, rotatable_bonds: ≤4, tPSA ≤ 90, aromatic_rings ≤ 3. Focussed set concentration response curve hits characterized by pIC50≥ 6.0 were clustered with the use of Ward cluster algorithms (Ward, 1963)
Pharmacophore model generation
Three selective α6β2 antagonists (compounds 1–3) were chosen to build a 3D pharmacophore model. Their 3D structure was generated starting from their Daylight SMILES with the use of smiles_to_mae script (version 22110, Schrodinger), imported into Maestro (Schrodinger) and minimized with the use of 1000 steps of PRCG minimizer, OPLS-2005 force field, in a water-implicit solvent model (BatchMin V9.6, Schrodinger: http://www.schrodinger.com). All pharmacophore modelling work was performed with program Phase (Schrodinger). Ligand conformational searches were carried out using the ConfGen routine available within Phase and default parameters. Standard Phase pharmacophore features (i.e. H-bond acceptor, H-bond donor, aromatic rings, hydrophobic groups) were selected and default parameters were used to generate common alignments. Pharmacophore models were visually inspected within Maestro.
Materials
N1-4-biphenylyl-N2-(1-methylethyl)glycinamide (compound 1; see Figure 4 for structures of compounds 1–6) was purchased from Enamine Ltd (Kiev, Ukraine), 2-(1-pyrrolidinyl)-N-{4-[(trifluoromethyl)oxy]phenyl}acetamide (2) and 2-[5-(4-fluorophenyl)-3-isoxazolyl]hexahydro-1H-azepine (3) from Asinex Ltd (Moscow, Russia), 5-ethyl-N-(5-methyl-1,3-thiazol-2-yl)-3-thiophenecarboxamide (4) and 2-[(2-chloro-4-fluorophenyl)oxy]-N-(2,2,6,6-tetramethyl-4-piperidinyl)acetamide (5) from Princeton Biomolecular Research (Monmouth Junction, NJ, USA), (1R,5S)-8-methyl-8-azabicyclo[3.2.1]oct-3-yl 4-(ethyloxy)benzoate (6) from Chembridge Corporation (Moscow, Russia). (−)-Nicotine di(+)tartrate salt (+/−)-5-Br-nicotine, 3-methyl-5-[(2S)-1-methyl-2-pyrrolidinyl]isoxazole hydrochloride (ABT-418), succinylcholine, 5-hydroxyindole and ivermectin (also known as MK-933) were purchased from Sigma-Aldrich (Milan, Italy). (+/−)-Epibatidine (−)-cytisine, 6-[5-[(2S)-2-azetidinylmethoxy]-3-pyridinyl]-5-hexyn-1-ol dihydrochloride (sazetidine A), 3-[(2S)-2-azetidinylmethoxy]-5-iodopyridine dihydrochloride (5-I-A-85380), 4-(5-ethoxy-3-pyridinyl)-N-methyl-(3E)-3-buten-1-amine difumarate (TC-2559), N,N,N-triethyl-2-[4-(2-phenylethenyl)phenoxy]ethanaminium iodide (MG-624), (E)-N-methyl-4-(3-pyridinyl)-3-buten-1-amine oxalate (RJR-2403, also known as TC-2403 or metanicotine), α-conotoxin MII, α-conotoxin PIA, methyllycaconitine (MLA), dihydro-β-erythroidine (DHβE), mecamylamine, benzoquinonium, N-(5-chloro-2-hydroxyphenyl)-N′-[2-chloro-5-(trifluorom ethyl)phenyl]urea (NS-1738), ondansetron, etomidate, α-bungarotoxin, α-conotoxin AuIB, pancuronium and naltrexone were from Tocris Cookson (Bristol, UK). 3-[(2S)-2-Azetidinylmethoxy]-pyridine (A-85380) was purchased from Peakdale Molecular Ltd (Chapel-en-le-Frith, High Peak, UK).
Figure 4.

Chemical structure of the novel α6β2*-nAChRs antagonists identified with the screening process (compounds 1–6).
5(R)- or 5(S)-7-(3-Pyridinyl)-1,7-diazaspiro[4.4]nonane (single isomer, unknown absolute stereo) (TC-2216) and α-conotoxin MII[H9A;L15A] were custom synthesized by Syngene International Ltd (Bangalore, India) and NeoMPS (Strasbourg, France) respectively. Varenicline, tilorone hydrochloride, N-(1-azabicyclo[2.2.2]oct-3-yl)(5-(2-pyridyl)thiophene-2-carboxamide) (compound A; Dickinson et al., 2007), 3-(2,4)-dimethoxybenzylidene anabaseine dihydrochloride (GTS-21, aka DMBX), N-[(2S,3R)- or N-[2R,3S)-2-(pyridin-3-ylmethyl)-1-azabicyclo[2.2.2]octan-3-yl]-1-benzofuran-2-carboxamide (single isomer, unknown absolute stereochemistry) (TC-5619) and suberyldicholine were synthesized in GlaxoSmithKline (GSK).
Structural confirmation of selected compounds
Structural confirmation of selected compounds (reported in Figure 4) was carried out on both liquid and solid samples. LC/MS measurements were carried out using an ACQUITY™ UPLC- SQD system (Waters Corporation, Milford, MA, USA) operated in positive and negative ES ion mode (100–1000 amu mass range). The chromatographic conditions used were: column, Acquity BEH C18 (50 × 2.1 mm, 1.7 µm particle size) kept at 40°C; mobile phase A-10 mM acq. NH4HCO3 adjusted to pH 10 with ammonia, B – MeCN; flow rate, 1 mL·min−1; gradient from 3 to 99% B in 1.06 min (linear) lasting for 0.39 min – stop time 1.5 min. UV detection 220–350 nm. 1H NMR spectra were recorded on a Varian Inova (600 MHz) spectrometer.
Results
The α6/3 subunit forms functional receptors only when co-expressed with the β2 and β3V273S subunit
The functional activity of different nicotinic receptor subunit combinations was assessed after transient transfection in HEK293T cells. Co-expression of the wild-type α6 subunit with either the β2 subunit alone (data not shown), or the β2 and the β3 subunits, did not result in any functional response.
The valine-serine mutation in the 9′ position of the M2 segment of the β3 subunit (V273S) is believed to facilitate channel gating, and co-expression of the ‘gain of function’β3V273S mutant with wild-type α6 and β2 subunits resulted in functional receptors in Xenopus laevis oocytes (Broadbent et al., 2006). However, when we co-transfected the wild-type α6 and β2 subunit with the mutant β3V273S in HEK293T cells, we did not obtain functional activity (see Figure 1).
Figure 1.
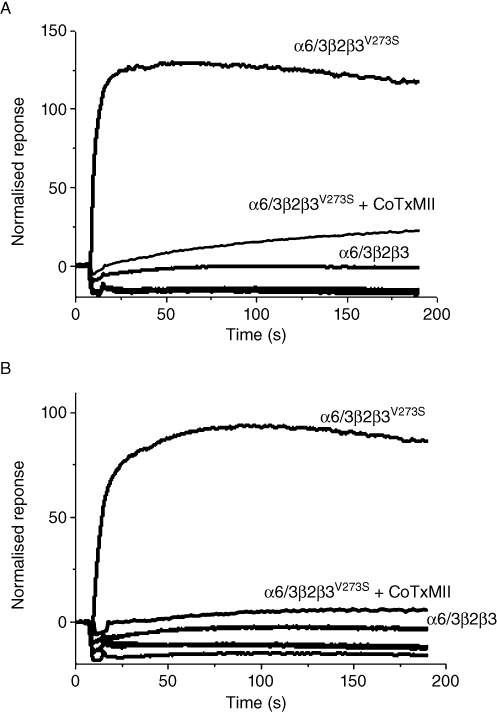
Functional responses obtained with different combinations of wild type (α6, β2 and β3) and modified (α6/3, β3V273S) nAChR subunits following transient transfection into HEK293T cells, as tested in the FLIPR Ca2+ influx assay. Upon addition of 500 µM acetylcholine (A) or 200 nM epibatidine (B) at 10 s, normalized responses (as % of basal fluorescence units) were recorded. Subunit combination labels are above the corresponding curves. Traces from the subunit combinations that were inactive or poorly active, including α6β2β3, α6β2β3V273S, β2β3 and control plasmid transfection, overlie and are un-labelled. CoTxMII, cells were co-incubated with 10 µM α-conotoxin MII.
We therefore considered the chimeric α6/3 subunit strategy, which was known to give functional receptors when co-expressed with the wild-type β2 and β3 subunits in oocytes (Kuryatov et al., 2000). Co-expression of our α6/3 subunit with the wild-type β2 and β3 subunits in HEK293T cells was not functional, whereas co-transfection of the chimeric α6/3, the wild-type β2 and the mutant β3V273S resulted in a robust response in the FLIPR assay, following agonist activation (see Figure 1). The functional calcium activation induced by either ACh (500 µM) or epibatidine (100 nM) was completely blocked by 10 µM α-conotoxin MII. The Ca2+ response was absent in control plasmid transfected cells, or cells co-transfected with the β2 and β3V273S subunits.
Pharmacological profile of α6/3β2β3V273S receptors stably expressed in HEK293 cells
Preliminary experiments, performed following transient co-transfection in HEK293T cells of the α6/3, β2 and β3V273S subunits revealed that α6/3β2β3V273S receptors were activated by different nicotinic receptor agonists and inhibited with high potency by the α6β2-selective antagonists α-conotoxin MII and α-conotoxin PIA (data not shown). We therefore generated a recombinant HEK293 cell line stably expressing α6/3β2β3V273S receptors and characterized their pharmacology with a set of 35 well-known nicotinic ligands (see Table 1).
Table 1.
Potency of representative nicotinic ligands for activation and inhibition of subtypes of nAChRs
| α6/3β2β3V273S | α4β2 | α3β4 | α7 | α1β1δε | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| pEC50 (IA) | pIC50 | pEC50 (IA) | pIC50 | pEC50 (IA) | pIC50 | pEC50 (IA) | pIC50 | pEC50 (IA) | pIC50 | |
| Epibatidine | 10.0 ± 0.07 (0.8) | 9.8 ± 0.1 | 8.0 ± 0.04 (1.6) | 9.3 ± 0.06 | 8.2 ± 0.03 (1.8) | 8.0 ± 0.04 | 7.2 ± 0.01 (1.0) | 7.9 ± 0.09 | 6.4 ± 0.02 (1.2) | 6.9 ± 0.02 |
| A-85380 | 9.5 ± 0.1 (0.8) | 9.8 ± 0.09 | 6.4 ± 0.06 (1.2) | 9.3 ± 0.08 | 6.2 ± 0.04 (1.4) | 5.8 ± 0.06 | 6.8 ± 0.09 (0.8) | 7 ± 0.08 | <6.0 | 5.6 ± 0.1 |
| Sazetidine A | 9.3 ± 0.07 (0.6) | 9.7 ± 0.04 | <6.0 | 9.5 ± 0.03 | 6.2 ± 0.06 (0.9) | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 |
| 5-I-A-85380 | 9.1 ± 0.04 (0.8) | 9.5 ± 0.06 | <6.0 | 9.4 ± 0.08 | 6.2 ± 0.03 (1.1) | 6.1 ± 0.06 | <6.0 | <6.0 | <6.0 | <6.0 |
| Varenicline | 8.0 ± 0.1 (0.3) | 7.9 ± 0.3 | 5.4 ± 0.3 (0.7) | 7.9 ± 0.2 | 5.3 ± 0.09 (1.0) | 5.2 ± 0.04 | 6.6 ± 0.09 (1.1) | 7 ± 0.02 | <4.3 | 4.7 ± 0.09 |
| Cytisine | 7.7 ± 0.1 (0.6) | 7.9 ± 0.09 | 4.3 ± 0.1 (1.2) | 7.1 ± 0.2 | 4.7 ± 0.03 (1.2) | 4.6 ± 0.06 | 5.8 ± 0.02 (1.0) | <5.95 | 5.1 ± 0.09 (0.8) | 5.5 ± 0.06 |
| Nicotine | 7.4 ± 0.09 (1.0) | 7.8 ± 0.2 | 5.6 ± 0.06 (1.0) | 6.9 ± 0.1 | 4.8 ± 0.04 (1.0) | 4.7 ± 0.06 | 5.4 ± 0.02 (1.0) | 5.8 ± 0.1 | 4.6 ± 0.04 (1.0) | 4.8 ± 0.05 |
| Suberyldicholine | 7.1 ± 0.1 (0.5) | 7.2 ± 0.06 | <4.3 | 6.8 ± 0.07 | 4.9 ± 0.03 (1.2) | 4.6 ± 0.1 | 4.5 ± 0.1 (0.9) | <5.0 | 7.6 ± 0.02 (1.6) | 8.1 ± 0.02 |
| TC-2559 | 6.8 ± 0.08 (0.5) | 6.2 ± 0.2 | <4.3 | 7.2 ± 0.07 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 |
| MG-624 | 6.8 ± 0.06 (0.2) | 7.5 ± 0.1 | <4.0 | 6.8 ± 0.05 | <4.0 | 6.6 ± 0.06 | 5.7 ± 0.3 (1.0) | 6.9 ± 0.1 | <4.0 | 7.3 ± 0.05 |
| 5-Br-nicotine | 6.7 ± 0.2 (0.6) | 6.9 ± 0.04 | <5.0 | 6.5 ± 0.3 | <5.0 | <5.0 | 5.2a (0.5) | 5.1a | <5.0 | <5.0 |
| TC-2216 | 6.7 ± 0.07 (0.5) | 6.4 ± 0.07 | <4.3 | 5.9 ± 0.1 | <4.3 | <4.3 | <4.3 | 4.5a | <4.3 | 4.6 ± 0.2 |
| ABT-418 | 6.5 ± 0.06 (0.9) | 6.8 ± 0.1 | 5.8 ± 0.07 (1.2) | 6.3 ± 0.1 | <5.0 | <5.0 | 5.7 ± 0.3 (0.9) | 5.8a | <5.0 | <5.0 |
| RJR-2403 | 5.7 ± 0.07 (0.5) | 5.7 ± 0.07 | 5.0 ± 0.09 (0.9) | 5.9 ± 0.07 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 |
| α-conotoxin MII | <6.0 | 8.3 ± 0.05 | <6.0 | <6.0 | <6.0 | 6.5 ± 0.09 | <6.0 | <6.0 | <6.0 | <6.0 |
| α-conotoxin PIA | <6.0 | 7.6 ± 0.07 | <6.0 | <6.0 | <6.0 | 6.6 ± 0.1 | <6.0 | <6.0 | <6.0 | <6.0 |
| MLA | <6.0 | 7.0 ± 0.07 | <4.0 | 6.2 ± 0.1 | <4.0 | 5.5 ± 0.09 | <4.0 | 8.2 ± 0.03 | <4.0 | 5.4 ± 0.3 |
| α-cntxMII[H9A;L15A] | <6.0 | 6.8 ± 0.09 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 |
| DHβE | <4.7 | 6.7 ± 0.08 | <4.0 | 6.3 ± 0.06 | <4.0 | <4.0 | <4.0 | 4.7 ± 0.3 | <4.0 | 4.2 ± 0.1 |
| GTS-21 | <4.7 | 6.2 ± 0.1 | <4.7 | 5.3 ± 0.1 | <4.7 | 5.4 ± 0.06 | 5.4 ± 0.08 (0.9) | 5.9 ± 0.06 | <4.7 | 5.8 ± 0.09 |
| Mecamylamine | <4.0 | 5.5 ± 0.1 | <4.0 | 6.1 ± 0.05 | <4.0 | 5.6 ± 0.05 | <4.0 | <5.0 | <4.0 | 4.7 ± 0.04 |
| Benzoquinonium | <4.3 | 5.4 ± 0.09 | <4.3 | 4.7 ± 0.06 | <4.3 | <5.0 | <4.3 | <5.0 | <4.3 | 5.6 ± 0.2 |
| TC-5619 | <4.3 | 4.9 ± 0.09 | <4.3 | 4.7 ± 0.3 | <4.3 | <5.0 | 8.4 ± 0.1 (1.0) | >9.1b | <4.3 | 4.3 ± 0.04 |
| Pancuronium | <4.0 | 4.8 ± 0.1 | <4.0 | 4.6 ± 0.5 | <4.0 | <5.0 | <4.0 | <5.0 | <4.0 | 5.9 ± 0.05 |
| NS-1738 | <4.3 | 4.8 ± 0.03 | <4.3 | <5.0 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 |
| Tilorone | <4.3 | 4.7 ± 0.1 | <4.3 | 4.4 ± 0.09 | <4.3 | <5.0 | 7.1 ± 0.1 (0.8) | 6.7 ± 0.2 | <4.3 | 5.3 ± 0.1 |
| Ondansetron | <4.0 | 4.7 ± 0.2 | <4.0 | 4.4 ± 0.07 | <4.0 | 4.5 ± 0.07 | <4.0 | <4.0 | <4.0 | 4.9 ± 0.05 |
| Etomidate | <4.0 | 4.6 ± 0.06 | <4.0 | 4.6 ± 0.08 | <4.0 | 4.5 ± 0.02 | 5.9 ± 0.4 (1.0) | <4.0 | <4.0 | 5.1 ± 0.09 |
| α-bungarotoxin | <5.0 | <5.0 | <5.0 | <5.0 | <5.0 | <5.0 | <5.0 | 7.6a | <5.0 | 7.9 ± 0.06 |
| Succinylcholine | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | 5.8 ± 0.02 (1.3) | 6.4 ± 0.03 |
| Naltrexone | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | 4.6a | <4.0 | 4.5 ± 0.09 |
| Compound A | <4.3 | <4.3 | <4.3 | <6.0 | <4.3 | <4.3 | 7.9 ± 0.01 (1.0) | 8.7 ± 0.1 | <4.3 | 5.1 ± 0.2 |
| α-conotoxin AuIB | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 | <6.0 |
| 5-Hydroxyindole | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 | <4.3 |
| Ivermectin | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 | <4.0 |
α-cntxMII[H9A;L15A],α-conotoxin MII[H9A;L15A]; EC50, concentration giving 50% of maximal activation; IA, intrinsic activity; IC50, concentration giving 50% of maximal inhibition.
Data are from a single experiment.
Compound gave the maximal inhibitory effect at the lowest concentration tested (0.8 nM). Results are the mean ± SEM of at least 2 experiments, unless otherwise indicated.
The ability of 200 nM nicotine (final concentration) to activate the α6/3β2β3V273S receptors in this cell line to yield Ca2+ influx in the FLIPR assay is shown in Figure 2. The addition of nicotine (1st addition) was followed by an increase of Ca2+ influx, as detected by an increase in FLUO-4-AM fluorescence, which reached a maximum in 1–2 min, and then a slow decrease of the response over time, reflecting receptor desensitization and a gradual return to intracellular Ca2+ homeostasis. The addition, after 8 min, of the same nicotine solution (2nd addition), was not able to restore Ca2+ influx to initial peak levels, confirming receptor desensitization. After extensive washing, intracellular Ca2+ returned to homeostatic concentrations and FLUO-4-AM fluorescence returned to background levels. Addition of 200 nM nicotine (3rd addition) was able to restore the Ca2+ influx response to initial levels, revealing the recovery of the receptor from desensitization (see Figure 2).
Figure 2.
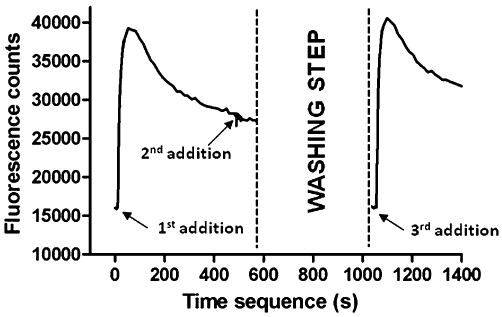
The figure shows the activation, desensitization and recovery from desensitization of human α6/3β2βV273S-nAChRs stably expressed in HEK293 cells, as tested in the FLIPR Ca2+ influx assay. Cells were loaded with the fluorescent Ca2+ indicator dye FLUO-4-AM and transferred to the FLIPR platform for the measurement of increases in intracellular Ca2+. Increases in the relative fluorescence units represent increases in intracellular Ca2+. Following the first addition of 200 nM nicotine, there was a peak of fluorescence change, reflecting channel activation and slow desensitization. Addition of the same quantity of nicotine to the same cells (second addition) did not yield an effect, confirming receptor desensitization. When cells plate was extensively washed with the assay buffer, residual fluorescence disappeared and addition of 200 nM nicotine (third addition) caused a further fluorescence increase, revealing recovery from desensitization. Data are from a representative experiment repeated at least three times with similar results.
In the first addition, nicotine was able to dose-dependently increase Ca2+ influx in α6/3β2β3V273S receptors expressing cells, with a pEC50 value of 7.4 ± 0.09 (mean ± SEM; n = 10). Among the compounds tested, none gave the same level of activation as nicotine. Among the chemicals able to significantly increase Ca2+ influx, epibatidine, A-85380 and I-A-85380 were those with higher IA (0.8) and potency (pEC50= 10.0 ± 0.07, 9.5 ± 0.1 and 9.1 ± 0.04, respectively; n = 10). Also sazetidine A had high potency (pEC50= 9.3 ± 0.07; n = 8), but lower intrinsic activity (IA = 0.6). Varenicline and cytisine are partial agonists, which were slightly more potent than nicotine (pEC50= 8.0 ± 0.1, n = 9 and pEC50= 7.7 ± 0.1, n = 10 for varenicline and cytisine, respectively), with varenicline having a lower intrinsic activity than cytisine (IA = 0.3 and 0.6, respectively). Other compounds, known to have agonist or partial agonist properties at other nAChR subtypes, were partial agonists with lower potency than nicotine. Figure 3A shows representative activation curves for nicotine, A-85380, sazetidine A, varenicline and cytisine.
Figure 3.
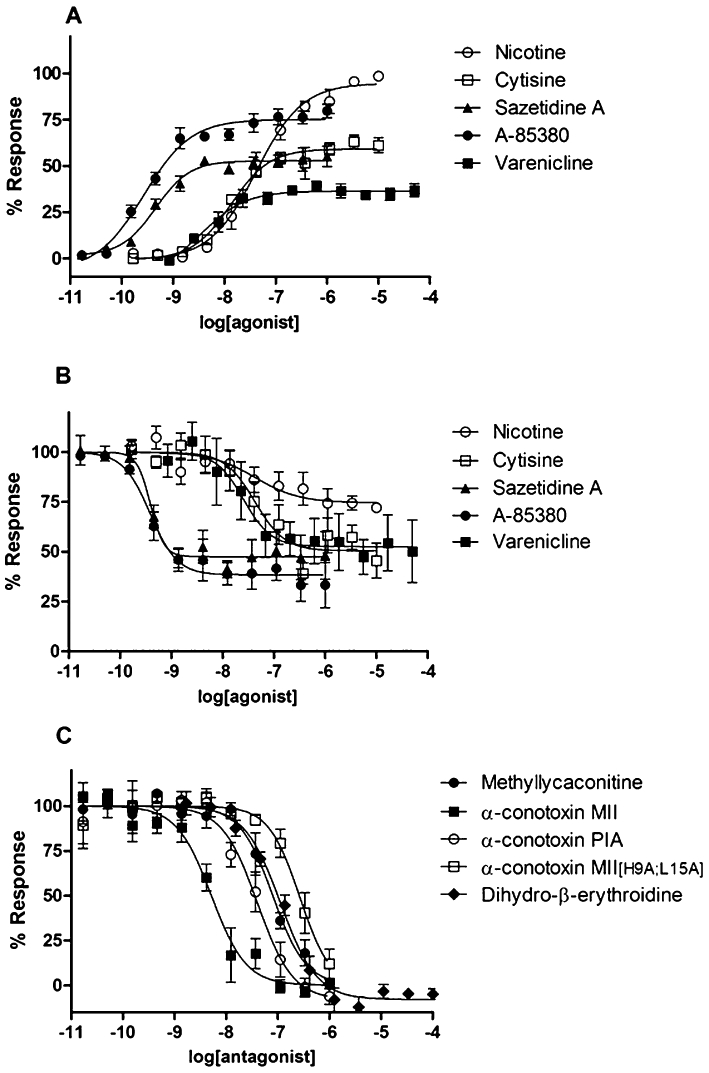
Functional calcium concentration response curves of some nicotinic ligands in a FLIPR Ca2+ influx assay, two-step addition protocol. (A) Representative concentration response curves to the agonists nicotine, varenicline, sazetidine A, cytisine and A-85380, in the first addition. The antagonists α-conotoxin MII, α-conotoxin MII[H9A;L15A], α-conotoxin PIA, methyllycaconitine, and DHβE did not significantly increase calcium response (data not shown). Data were expressed as percentage of the maximum response to nicotine. (B) Representative concentration response curves to the agonists nicotine, varenicline, sazetidine A, cytisine and A-85380 for the inhibition of channel activation induced by the subsequent addition of 200 nM nicotine. Data were expressed as percentage of the response in the absence of inhibitor. (C) Representative concentration response curves to the antagonists α-conotoxin MII, α-conotoxin MII[H9A;L15A], α-conotoxin PIA, methyllycaconitine, and dihydro-β-erythroidine for the inhibition of channel activation induced by the subsequent addition of 200 nM nicotine. Data were expressed as percentage of the response in the absence of inhibitor.
Compounds activating α6/3β2β3V273S receptors (in the first addition), were able to dose-dependently inhibit the Ca2+ influx induced by the addition, after 10 min, of 200 nM nicotine (second addition). Compound inhibitory potency was comparable with their potency in activating receptors in the first addition (see Table 1). Overall, these data confirmed that α6/3β2β3V273S receptors underwent desensitization. Representative desensitization curves for nicotine, varenicline, sazetidine A, cytisine and A-85380 are shown in Figure 3B.
Some nicotinic ligands were not able to significantly increase Ca2+ influx in α6/3β2β3V273S receptors expressing cells, but inhibited the following response to 200 nM nicotine, therefore displaying antagonist properties. The compounds that inhibited nicotine response with higher potency were α-conotoxin MII and α-conotoxin PIA (pIC50 of 8.3 ± 0.05 and 7.6 ± 0.07, respectively; n = 11), followed by MLA, an α7 antagonist that also blocks other nAChR subtypes (pIC50 of 7.0 ± 0.07, n = 7; Gotti et al., 2000; Mogg et al., 2002; Karadsheh et al., 2004; Briggs et al., 2006). Other compounds with submicromolar antagonist potency for α6/3β2β3V273S receptors were the α-conotoxin MII analogue α-conotoxin MII[H9A;L15A], the β2* antagonist DHβE and the α7 agonist/α4β2 antagonist GTS-21 (de Fiebre et al., 1995; Harvey et al., 1996; Briggs et al., 1997; van Haaren et al., 1999; McIntosh et al., 2004). Representative inhibition curves for α-conotoxin MII, α-conotoxin MII[H9A;L15A], α-conotoxin PIA, MLA and DHβE are shown in Figure 3C.
Comparison of α6/3β2β3V273S pharmacology with that of other nAChR subtypes
To better describe the features of α6/3β2β3V273S receptors, we compared their pharmacology with that of the other main nAChR subtypes (see Table 1).
The antagonists α-conotoxin MII, α-conotoxin PIA and α-conotoxin MII[H9A;L15A] were the compounds that best differentiated the α6/3β2β3V273S from the other receptor subtypes. These conotoxins, in fact, while displaying medium to high affinity for α6/3β2β3V273S receptors, were generally inactive (up to 1 µM) at the other nAChR subtypes, with the only exception of α-conotoxin MII and α-conotoxin PIA inhibiting also α3β4-nAChRs, but with 63- and 10-fold lower potency respectively.
In addition to the pharmacology of the conotoxins mentioned above, α6/3β2β3V273S receptors were distinguishable from the α1β1δε, α7 and α3β4 subtypes by their response to other compounds. Unlike α1β1δε-nAChRs, they were completely insensitive to inhibition by the neuromuscular blocking agents, succinylcholine and α-bungarotoxin. The latter compound also differentiated between α6/3β2β3V273S and α7 receptors, being a very potent antagonist at α7 but inactive at α6/3β2β3V273S receptors. Similarly, tilorone, TC-5619 and compound A were potent agonists at α7 receptors and almost inactive at α6/3β2β3V273S. DHβE was a relatively potent antagonist at α6/3β2β3V273S receptors (pIC50= 6.7 ± 0.08, n = 13), but inactive at α3β4-nAChRs. Finally, many ligands activating or inhibiting α6/3β2β3V273S receptors had a much lower potency at the α1β1δε, α7 and α3β4 subtypes: nicotine, for example, was approximately 800-, 100- and 400-fold more potent at α6/3β2β3V273S than α1β1δε, α7 and α3β4 receptors respectively (see Table 1).
Many well-known α4β2 agonists or partial agonists, such as epibatidine, A-85380, sazetidine A, 5-I-A-85380, varenicline, cytisine, nicotine, TC-2559, 5-Br-nicotine, TC-2216, ABT-418 and RJR-2403, were able to activate α6/3β2β3V273S receptors, but failed, or were weak activators of Ca2+ influx in α4β2-HEK293 cells. The same compounds, however, were able to desensitize both α6/3β2β3V273S and α4β2 receptors, with similar potencies (see Table 1). The fact that some agonists or partial agonists were able to desensitize α4β2 receptors without activating them, seems to suggest that the α4β2-HEK293 cell line expresses α4β2-nAChRs with a prevalent (α4)3(β2)2 stoichiometry (Zwart et al., 2008).
Comparison of α6/3β2β3V273S pharmacology with that of rat native α6β2*-nAChRs
The compounds that distinguished α6/3β2β3V273S more clearly from other nicotinic receptor subtypes were tested also in displacement experiments of 125I-epibatidine binding to immuno-immobilized α6β2*-nAChRs, a preparation that contains α6β2(β3) and α6α4β2(β3) subtypes (Gotti et al., 2005). Previous experiments showed that compounds with good selectivity for α6β2*-nAChRs revealed two binding components in similar preparations: a high affinity component, corresponding to the α6β2 interface and a component of low, micromolar affinity, corresponding to the α4β2 interface (Zoli et al., 2002; Gotti et al., 2010).
Agonists and partial agonists at human α6/3β2β3V273S inhibited 125I-epibatidine binding to rat native α6β2*-nAChRs with displacement curves according to a one-site binding site model. A-85380 and sazetidine A were the most potent compounds (pKi= 10.7 ± 0.6 and 10.7 ± 0.3, n = 4, respectively), followed by varenicline, cytisine and nicotine (pKi= 8.9 ± 0.7, 8.7 ± 0.4 and 8.0 ± 0.5, n = 4 respectively). Although the rank order was conserved, the agonists and partial agonists affinity for rat native α6β2*-nAChRs was, on average, 12-fold greater than their potency in activating human α6/3β2β3V273S (see Table 2).
Table 2.
Affinity of representative nicotinic ligands for purified α6β2*-nAChRs from rat superior colliculus. Comparison with their functional potency at human recombinant α6/3β2βV273S receptors and rodent native α6β2*-nAChRs
| Rat α6β2* | Human α6/3β2β3V273S | Rat or mouse α6β2* | |
|---|---|---|---|
| [125I]-epibatidine binding | Ca2+ entrance (FLIPR) | α-conotoxin MII-sensitive [3H] dopamine release | |
| pKi | pEC50(IA) | pEC50(IA) | |
| Agonists | |||
| A-85380 | 10.7 ± 0.6 | 9.5 ± 0.1 (0.8) | 8.9 (0.8)a |
| Sazetidine A | 10.7 ± 0.3 | 9.3 ± 0.1 (0.6) | ND |
| Varenicline | 8.9 ± 0.7 | 8.0 ± 0.1 (0.3) | 7.1 (0.4)b |
| Cytisine | 8.7 ± 0.4 | 7.7 ± 0.1 (0.6) | 7.5 (0.7)a |
| Nicotine | 8.0 ± 0.5 | 7.4 ± 0.1 (1.0) | 6.1 (1.0)a |
| pKi | pIC50 | pIC50 | |
|---|---|---|---|
| Antagonists | |||
| α-Conotoxin MII | 8.3 ± 0.2c | 8.3 ± 0.05 | 8.6a; 9.0d |
| α-Conotoxin PIA | 7.4 ± 0.4c | 7.6 ± 0.1 | 8.8d |
| MLA | 6.7 ± 0.5c | 7.0 ± 0.1 | 6.4a |
| α-Cntx MII[H9A;L15A] | 5.5 ± 0.6 | 6.8 ± 0.1 | ND |
| DHβE | 6.2 ± 0.6 | 6.7 ± 0.1 | 6.0a |
| Mecamylamine | <5.3 | 5.5 ± 0.1 | Full inhibition at 10 µMe |
| α-Bungarotoxin | <5.3 | <5.0 | No inhibition up to 40 nMe |
EC50, concentration giving 50% of maximal activation; IA, intrinsic activity; IC50, concentration giving 50% of maximal inhibition; ND, not determined.
Data from Salminen et al. (2004); pEC50 and pIC50 converted from EC50 and IC50 respectively; IA derived from the maximum release (Rmax) of compound and the Rmax of nicotine; IC50 were determined at a pulse of 3 µM nicotine (submaximal concentration).
Grady et al. (2010); pEC50 converted from EC50; IA derived from efficacy of compound for activation as % nicotine effect for activation.
Data analysis revealed the existence of a second, low affinity binding site in the µM range (not reported).
Azam and McIntosh (2005); pIC50 converted from IC50; IC50 values were determined at a pulse of 3 µM nicotine (submaximal concentration).
Data from Mogg et al., 2002. Results are the mean ± SEM of at least 3 experiments.
The antagonists α-conotoxin MII, α-conotoxin PIA and MLA inhibited 125I-epibatidine binding with displacement curves according to a two-site binding model (P < 0.01), with a high affinity component in the nanomolar range (pKi= 8.3 ± 0.2, n = 6, 7.4 ± 0.4, n = 4 and 6.7 ± 0.5, n = 6, respectively) and a component in the micromolar range (not reported). Conversely, the antagonists DHβE and α-conotoxin MII[H9A;L15A] displaced 125I-epibatidine binding with one-site curves and lower potency (pKi= 6.2 ± 0.6 and 5.5. ± 0.6, n = 6 respectively). In general, there was a good correspondence between the affinity for rat native α6β2*-nAChRs and human α6/3β2β3V273S (see Table 2). Mecamylamine was not able to displace 125I-epibatidine binding, suggesting a non-competitive mechanism of action in the inhibition of human α6/3β2β3V273S receptors. The α7 and α1β1δε antagonist α-bungarotoxin, inactive at human α6/3β2β3V273S receptors, was not able to displace specific binding to native α6β2*-nAChRs.
Generation of the nicotinic focussed set
Three thousand compounds previously tested in the GSK nicotinic receptor assay panel were extracted (in_house_nicotinic_ligand_set) and requested for cherry-picking. Upon cluster analysis, 1000 representatives were selected (in_house_nicotinic_representative_set) and combined with the 116 derivatives selected from the Aureus repository (Aureus_nicotinic_training_set). This combined list of derivatives was used to drive the selection of 1500 in house derivatives according to their reduced graph and pharmacophoric similarities and then requested for cherry-picking. Overall, 1886 compounds were finally plated and tested in single shot mode in the α6/3β2β3V273S-FLIPR Ca2+ assay.
Screening of the focussed sets and novel α6β2 antagonists identification
Three focussed sets were screened in single shot mode in the α6/3β2β3V273S FLIPR assay: the nicotinic set (1886 derivatives), a subset of GSK compounds biased towards the CNS chemical space (CNS set; 92 694 compounds; A. Pozzan, unpubl. data) and a small set of derivatives routinely used for cross-screening (biological fingerprints set; 5237 compounds; A. Feriani, unpubl. data).
As summarized in Table 3, a high proportion of the derivatives exhibiting greater than 50% inhibition were discarded upon percentage efficiency index ranking, biasing the selection towards potent and low molecular weight compounds. The use of stringent CNS-like physico-chemical property cut-offs further narrowed down the number of compounds progressing to determination of concentration response curves (244 derivatives in total). A high proportion (∼30%) of the nicotinic and CNS set hits assessed by concentration response curves showed a pIC50 value greater than 6 and a pEC50 value less than 4.7 (no significant increase of Ca2+ influx was observed up to 20 µM concentration). Several distinct chemotypes were identified as judged by the number of clusters obtained with Ward cluster analysis of each hit list. Furthermore, very limited overlap among the three concentration response curve hit lists was observed as assessed by the use of Tanimoto index and Daylight fingerprints as descriptors.
Table 3.
Summary of the screening performed on human α6/3β2β3V273S-nAChRs
| Number of compounds tested | |||||||
|---|---|---|---|---|---|---|---|
| Total | Positive | ||||||
| SET | Single shota | Progressed to CRCb | pIC50 ≥ 6.0c | Hit rated | Number of chemotypese | Overlapf | |
| Nicotinic | 1 886 | 955 | 40 | 13 | 32.5 | 12 | 2 |
| CNS | 92 694 | 11 997 | 151 | 51 | 33.8 | 45 | 1 |
| BFP | 5 237 | 2 476 | 53 | 3 | 5.7 | 3 | 1 |
IC50, concentration giving 50% of maximal inhibition; BFP, biological fingerprints; CNS, central nervous system.
Compounds giving ≥ 50% inhibition of nicotine-induced Ca2+ entrance at 10 µM (nicotinic and CNS set) or 20 µM (BFP set).
Compounds chosen for determination of activation and inhibition concentration response curves (CRC), according to the filtering criteria described in Materials and Methods.
Compounds inhibiting nicotine response with a pIC50≥ 6.0 and unable to activate the receptors (pEC50≤ 4.7).
Percentage of hits showing a pIC50≥ 6.0, with respect to those progressed to concentration response curve determination.
Number of clusters obtained with Ward cluster analysis of the pIC50≥ 6.0 hits.
Number of the pIC50≥ 6.0 hits structurally similar to those of the other hit lists; similarity was assessed with Daylight fingerprints as descriptors and Tanimoto index greater or equal to 0.85.
Selectivity of the novel α6β2 antagonists
Among the compounds tested, six of them had interesting selectivity profiles (see Table 4).
Table 4.
Potency of compounds for inhibition of subtypes of nAChRs
| α6/3β2β3V273S | α4β2 | α3β4 | α7 | α1β1δε | |
|---|---|---|---|---|---|
| Compound | pIC50 | pIC50 | pIC50 | pIC50 | pIC50 |
| 1 | 6.9 ± 0.3 | 5.5 ± 0.09 | 5.1 ± 0.05 | 5.3 ± 0.1 | 5.3 ± 0.08 |
| 2 | 6.3 ± 0.05 | 4.9 ± 0.07 | <4.7 | <4.7 | 5.1 ± 0.2 |
| 3 | 6.1 ± 0.1 | 4.8 ± 0.03 | 5.1 ± 0.3 | 5.0 ± 0.01 | 5.2 ± 0.1 |
| 4 | 6.4 ± 0.1 | 4.8 ± 0.08 | 5.1 ± 0.09 | <4.7 | <4.8 |
| 5 | 6.4 ± 0.06 | 6.3 ± 0.2 | 5.5 ± 0.07 | <4.8 | 5.4 ± 0.2 |
| 6 | 6.2 ± 0.03 | 5.7 ± 0.05 | 5.2 ± 0.06 | <4.85 | 5.2 ± 0.06 |
Compounds did not activate any nAChR subtype up to 20 µM (pEC50 < 4.7).
EC50, concentration giving 50% of maximal activation; IC50, concentration giving 50% of maximal inhibition. Results are the mean ± SEM of at least 2 experiments.
Compounds 1–4 were α6/3β2β3V273S-selective antagonists: compound 1 displayed the highest potency (pIC50= 6.90 ± 0.3, n = 3) and selectivity (25-, 63-, 40- and 40-fold vs. α4β2, α3β4, α7 and α1β1δε, respectively); compound 2 was less potent (pIC50= 6.30 ± 0.05, n = 3) but similarly selective (25-, >40-, >40- and 16-fold vs. α4β2, α3β4, α7 and α1β1δε, respectively); compound 3 was not as potent (pIC50= 6.10 ± 0.1, n = 3) and less selective (20-, 10-, 13- and 8-fold vs. α4β2, α3β4, α7 and α1β1δε, respectively) and compound 4 had an intermediate potency (pIC50= 6.40 ± 0.1, n = 3) but slightly higher selectivity (40-, 20-, >50- and 40-fold vs. α4β2, α3β4, α7 and α1β1δε respectively). Compounds 5 and 6 were dual α6/3β2β3V273S and α4β2 antagonists (α4β2 : α6/3β2β3V273S IC50 ratio was 1 and 3, respectively), partially selective over the other nAChR subtypes. The chemical structure of compounds 1–6 is reported in Figure 4.
Pharmacophore model generation
Six common-feature pharmacophore models were generated by Phase using compounds 1–3 (Figure 5). All of them consist of three features i.e. a positive ionizable, a hydrogen bond-acceptor and an aromatic ring. Among the top scoring solutions, the most satisfactory pharmacophore model was chosen according to the quality of ligand conformations upon visual inspection and Phase scores associated with each solution. In Figure 5 the best pharmacophore model obtained is shown superimposed with the structures of compounds 1–3. The positive ionizable feature is mapped by the secondary/tertiary amines of the ligands (light blue sphere), a HB-acceptor (purple sphere) mapped by the amide carbonyl group in the side chain of compounds 1–2 and by the isoxazole nitrogen of compound 3, and an aromatic ring, mapped by the aromatic groups (brown ring).
Figure 5.
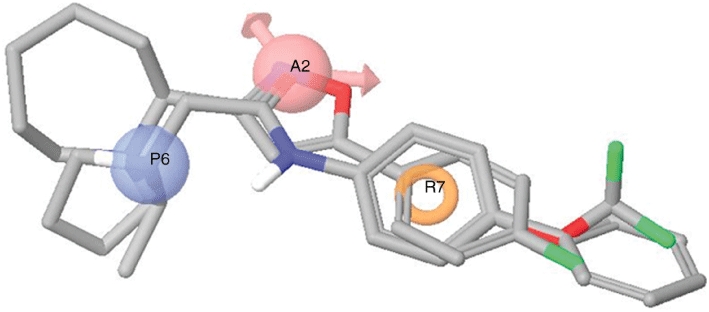
Compounds 1–3 aligned to the top scoring pharmacophore model generated with Phase. The model consists of three features: a positive ionizable (blue), a hydrogen bond-acceptor (pink) and an aromatic ring (orange).
Discussion
The present study reports the stable and functional expression of a human α6β2β3V273S-nAChR with α6(nonα4)β2 properties in a mammalian heterologous expression system. Native receptors with α6(nonα4)β2 properties have an α6β2β3 subunit composition (Gotti et al., 2010), but the functional co-expression of wild-type α6, β2 and β3 subunits was unsuccessful in our hands, consistent with extensive literature reports of failed attempts to form functional receptors with robust responses (Gerzanich et al., 1997; Fucile et al., 1998; Kuryatov et al., 2000; Broadbent et al., 2006; Walsh et al., 2008). In addition, we did not find a functional response after transient transfection of the α6/3β2β3 and α6β2β3V273S subunit combinations in HEK293T cells, even if the same combinations gave functional receptors in oocytes (Kuryatov et al., 2000; Dowell et al., 2003; Broadbent et al., 2006; Papke et al., 2008). These differences remain to be explained; it cannot be excluded, however, that oocytes, differently from mammalian cells, might endogenously express unknown factors that promote efficient assembly of functional α6/3β2β3 and α6β2β3V273S channels, as has been reported for other receptors (Soloviev and Barnard, 1997).
The pharmacology of α6/3β2β3V273S was clearly distinct from that of the other nAChRs in terms of ligand selectivity, with α-conotoxin MII and α-conotoxin PIA showing the greatest selectivity for α6/3β2β3V273S compared with the α4β2, α3β4, α7 and α1β1δε subtypes. Furthermore, the pharmacology of human α6/3β2β3V273S-nAChRs was consistent with that of immuno-immobilized, rat α6β2*-nAChRs. In fact, there was general agreement between the potency of competitive antagonists at α6/3β2β3V273S and their affinity for α6β2*-nAChRs, as detected by displacement of [125I]epibatidine binding (see Table 2). Agonists affinity for α6β2*-nAChRs was generally higher than their potency in activating α6/3β2β3V273S-nAChRs, but the relative activities were similar, suggesting that a conformation with high affinity for agonists, probably reflecting a desensitized state of α6β2*-nAChRs, was predominant at equilibrium conditions with an agonist.
Conversion of the hydrophobic residue in position 9′ of the M2 segment of nicotinic subunits (leucine or valine) to a hydrophilic residue (serine or threonine) has been shown to particularly change the efficacy of gating, increase agonist sensitivity and, in the α7 case, to abolish the inhibitory properties of channel blockers and turn some competitive antagonists into agonists (Revah et al., 1991; Bertrand et al., 1992; Labarca et al., 1995; 2001; Groot-Kormelink et al., 1998; Drenan et al., 2008). The functional properties of α6/3β2β3V273S receptors were similar to those of native α6β2*-nAChRs (as from dopamine release assays in rodent striatal slices or synaptosomes) in terms of compound efficacy and relative activities, but different potencies were observed for some compounds (see Table 2). With the current set of data, however, it is not possible to decide if these discrepancies were due to the V9′S mutation on the β3 subunit or to other factors, such as technical limitations associated with comparing different functional assays (FLIPR vs. dopamine release), the difficulty in separating the α6β2* and α4(nonα6)β2* effects in native tissue, the presence of mixed α6β2 and α4β2 interfaces in the α6β2*-nAChR population, and/or to species differences.
Overall, our results demonstrate that there are no major differences between human, recombinant α6/3β2β3V273S and rodent, native α6β2* receptor in terms of competitive ligand pharmacology; however, because of the V9′S mutation, we cannot exclude the possibility that some agonists may display a higher potency at α6/3β2β3V273S receptors. Moreover, it is important to emphasize that, as only the extracellular N-terminal domain of the α6/3 chimera belongs to the α6 subunit, the α6/3β2β3V273S cell line cannot be used to identify α6β2 channel blockers, or to study α6β2 receptors in terms of pore opening mechanism, desensitization and inactivation, properties that directly depend on residues in the transmembrane segments and the loops between them.
Approximately, 100 000 compounds were screened in the α6/3β2β3V273S FLIPR assay. As summarized in Table 3, approximately 30% of the nicotinic and the CNS sets concentration response curve hits showed a pIC50 in the µM range. Several distinct chemotypes were identified as judged by the high number of Ward clusters of each hit list compounds. At the same time, the hit lists exhibit very limited overlap, suggesting that the focussed screening approach was valuable in providing structurally distinct α6β2 chemotypes.
Among the compounds tested, compounds 1–4 exhibited good selectivity for the α6/3β2β3V273S over all the other nAChR subtypes, including the α4β2. On the contrary, compounds 5 and 6, although showing good selectivity for the α6/3β2β3V273S vs. the α3β4, α7 and α1β1δε subtypes, were also α4β2 antagonists with similar potencies. The chemotypes identified, by virtue of their low molecular weight, low molecular complexity and CNS-like properties, could offer promising starting points for initiating hit-to-lead or lead-optimization processes. Furthermore, these small and relatively rigid molecules could represent an ideal set for exploiting key ligand interactions.
A preliminary pharmacophore model of α6β2 antagonists was generated for the most selective compounds identified in this study (compounds 1–3, Figure 4). It consists of three pharmacophoric features i.e. a positive ionizable group, a hydrogen bond-acceptor and an aromatic ring. As shown in Figure 5, the two most potent compounds 1–2 can also commonly overlap their pendant phenyl ring (compound 1) and the CF3 group (compound 2), which could potentially map an accessory hydrophobic pocket. The presence of an additional hydrophobic feature might explain their higher potency with respect to compound 3. Compound 4 (Figure 4) shares both a hydrophobic feature and a hydrogen bond-acceptor group with compounds 1–3 but it lacks a basic centre. This seems to suggest that an alternative binding mode is also possible and it can be partially common to the binding mode described. Chemical exploration around all these compound series should be carried out to validate this hypothesis.
Compounds 1–3 share some pharmacophoric features with other nicotinic antagonists, like DHβE, MLA and the α7 antagonist chemotypes recently reported by Peng et al. (2010). However, these features (i.e. the presence of a positive ionizable group and a HB-acceptor) have a different spatial orientation in the α6β2 antagonist pharmacophore model that can account for their selectivity towards the other nicotinic subtypes. This observation is consistent with the analysis performed by Horenstein et al. (2008) about the existence of chemotype-dependent motifs of α7 agonists.
It has been known for several years that high levels of α6β2*-nAChRs are expressed in the key areas of the reward circuit, the NAc and the ventral tegmental area, where they mediate dopamine release and neuronal firing, respectively (Le Novere et al., 1996; Mogg et al., 2002; Champtiaux et al., 2003). Nevertheless, because of the lack of appropriate selective antagonists for systemic administration, robust evidence that inhibition of α6β2*-nAChRs interferes with the reinforcing properties of nicotine has been obtained only in recent times. In fact, the first studies showing that local infusion of the non-brain penetrant, α6β2-selective antagonist α-conotoxin MII diminished intravenous nicotine self-administration in rat, date to 2010 (Brunzell et al., 2010; Gotti et al., 2010). These results were in agreement with previous data showing that α6 knock-out mice do not acquire acute, intravenous nicotine self-administration (Pons et al., 2008). Overall, these findings support the use of α6β2 antagonists to reduce the addictive properties of nicotine. Here we report the structure of the first, non-peptide, low molecular weight α6β2-selective antagonists (compounds 1–4), which may represent starting templates for the development of tool compounds with appealing physico-chemical and pharmacokinetic properties for preclinical experiments, as well as for the discovery of novel drugs with an optimum profile for the therapy of tobacco dependence.
Current literature suggests that a strategy involving antagonism at central nAChRs could potentially lead to development of novel antidepressant therapeutics (Shytle et al., 2002; Andreasen et al., 2009). The use of compounds in animal models of depression, concomitant to genetic deletion of specific nAChR subunits, has revealed that the antidepressant-like effects of the non-selective nicotinic antagonist mecamylamine is dependent on the β2 and/or the α7 subunits (Caldarone et al., 2004; Rabenstein et al., 2006). In accord with these findings, the α4β2-selective antagonist TC-2216 has antidepressant activity (Lippiello et al., 2006; 2008;). Here we report the structure of two dual α6β2/α4β2 antagonists (compounds 5 and 6), which may represent starting points for the development of antidepressants with a novel mechanism of action.
The present study has revealed that many nicotinic compounds, commonly known as α4β2 ligands, interact also with α6/3β2β3V273S-nAChRs (see Table 1) and, consequently, α6β2*-nAChRs. Interestingly, we found that the smoking cessation agent varenicline is a partial agonist at α6/3β2β3V273S-nAChRs, with very low intrinsic activity. It cannot be excluded therefore that the efficacy of varenicline, generally attributed to its partial agonist activity at α4β2-nAChRs, may be mediated also by its weak partial agonist/antagonist-like properties at α6β2*-nAChRs. Similarly, TC-2216 is a partial agonist at α6/3β2β3V273S-nAChRs, suggesting that α6β2*-nAChRs may be important for TC-2216 efficacy in animal models of depression.
Recent literature suggests that selective loss of α6β2*-nAChRs is associated with the initial phases of Parkinson's disease and dementia with Lewy bodies (Ray et al., 2004; Quik and McIntosh, 2006; Gotti et al., 2006a). Very recently, the structure of the first α6β2-selective agonist has been reported (compound 20a of Breining et al., 2009) and proposed for the development of therapeutic agents for these diseases. The α6/3β2β3V273S-HEK293 cell line generated in the present study can be used for the identification of additional scaffolds for α6β2-selective agonists, in a way similar to the process that led to the identification of α6β2-selective antagonists.
In conclusion, we have generated a α6/3β2β3V273S-HEK293 cell line that stably expresses receptors with pharmacological properties similar to native α6β2*-nAChRs. In addition, we have identified the first non-peptide, low molecular weight α6β2-selective antagonists. Finally, we describe a pharmacophore model for α6β2-nAChR antagonists, which may help to develop new tools for the investigations of the role of a6β2 receptors in animal models of different pathologies, as well as novel agents for the treatment of tobacco addiction.
Acknowledgments
A special acknowledgement goes to Sergio Senar (GSK, Tres Cantos, Spain) and Caterina Virginio (GSK, Verona, Italy), for their constant commitment to the research programme and frequent, fruitful advice. We thank Margareth Martin, Mark Wigglesworth, Dolores Jimenez-Alfaro (GSK, Harlow, UK and Tres Cantos, Spain) for plate compound dispensing; Stefano Provera, Luca Rovatti and Carla Marchioro (GSK, Verona, Italy) for structural elucidation; Julie Quayle and Irene Areri (GSK, Harlow, UK) for hit quality control and compound purification; Selina Mok, Elena DiDaniel and James Kew (GSK, Harlow, UK), Charlotte Ashby (GSK, Stevenage, UK), George Livi (GSK, Upper Merion, PA, USA), Michela Tessari, Mauro Corsi and Emilio Merlo-Pich (GSK, Verona, Italy), for their important contribution to programme progression. The work was partially supported by the EC Grant Neurocypres and Grant TERDISMENTAL from the Regione Lombardia to Cecilia Gotti.
Glossary
Abbreviations
- 5-I-A-85380
3-[(2S)-2-azetidinylmethoxy]-5-iodopyridine dihydrochloride
- A-85380
3-[(2S)-2-azetidinylmethoxy]-pyridine
- ABT-418
3-methyl-5-[(2S)-1-methyl-2-pyrrolidinyl]isoxazole hydrochloride
- compound A
N-(1-azabicyclo[2.2.2]oct-3-yl)(5-(2-pyridyl)thiophene-2-carboxamide)
- DHβE
dihydro-β-erythroidine
- FLIPR
fluorescent imaging plate reader
- GH4C1
rat pituitary tumour-derived
- GTS-21
3-(2,4)-dimethoxybenzylidene anabaseine dihydrochloride
- HEK
human embryonic kidney
- M2
second transmembrane domain
- MG-624
N,N,N-triethyl-2-[4-(2-phenylethenyl)phenoxy]ethanaminium iodide
- MLA
methyllycaconitine
- nAc
nucleus accumbens
- nAChR
nicotinic acetylcholine receptor
- NS-1738
N-(5-chloro-2-hydroxyphenyl)-N′-[2-chloro-5-(trifluorom ethyl)phenyl]urea
- RJR-2403
(E)-N-methyl-4-(3-pyridinyl)-3-buten-1-amine oxalate
- sazetidine A
6-[5-[(2S)-2-azetidinylmethoxy]-3-pyridinyl]-5-hexyn-1-ol dihydrochloride
- TC-2216
5(R)- or 5(S)-7-(3-pyridinyl)-1,7-diazaspiro[4.4]nonane
- TC-2559
4-(5-ethoxy-3-pyridinyl)-N-methyl-(3E)-3-buten-1-amine difumarate
- TC-5619
N-[(2S,3R)- or N-[2R,3S)-2-(pyridin-3-ylmethyl)-1-azabicyclo[2.2.2]octan-3-yl]-1-benzofuran-2-carboxamide
- α6β2*-nAChRs
nicotinic acetylcholine receptors containing the α6 and β2 subunit
Conflicts of interest
None.
Supporting information
Additional Supporting information may be found in the online version of this article:
Figure S1 Functional responses of human α7-nAChRs stably expressed in rat GH4C1 cells, as tested in a FLIPR Ca2+ influx assay, two-step addition protocol. (A) Representative responses to 11 different concentrations of the agonist nicotine and the antagonist MLA (raw data). Each graph represents the functional activity over time (y axis: relative fluorescence units, representing increases in intracellular Ca2+; x axis: time) in a specific plate well. The time points of the first addition (nicotine or MLA, at varying concentrations) and the second addition (10 µM nicotine) are indicated by a yellow and green arrow respectively. Nicotine and MLA concentration in the first addition is indicated below each graph. (B) Left panel: representative concentration response curve to nicotine, in the first addition; data are expressed as percent of maximum response to nicotine. Right panel: representative concentration response curve to MLA, for the inhibition of channel activation induced by the subsequent addition of 10 µM nicotine; data are expressed as percent of maximum response in the absence of MLA.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abad-Zapatero C, Metz JT. Ligand efficiency indices as guideposts for drug discovery. Drug Discov Today. 2005;10:464–469. doi: 10.1016/S1359-6446(05)03386-6. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen JT, Olsen GM, Wiborg O, Redrobe JP. Antidepressant-like effects of nicotinic acetylcholine receptor antagonists, but not agonists, in the mouse forced swim and mouse tail suspension tests. J Psychopharmacol. 2009;23:797–804. doi: 10.1177/0269881108091587. [DOI] [PubMed] [Google Scholar]
- Azam L, McIntosh JM. Effect of novel alpha-conotoxins on nicotine-stimulated [3H] dopamine release from rat striatal synaptosomes. J Pharmacol Exp Ther. 2005;312:231–237. doi: 10.1124/jpet.104.071456. [DOI] [PubMed] [Google Scholar]
- Bertrand D, Villers-Thiery A, Revah F, Galzi JL, Hussy N, Mulle C, et al. Unconventional pharmacology of a neuronal nicotinic receptor mutated in the channel domain. Proc Natl Acad Sci USA. 1992;89:1261–1265. doi: 10.1073/pnas.89.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breining SR, Bencherif M, Grady SR, Whiteaker P, Marks MJ, Wageman CR, et al. Evaluation of structurally diverse neuronal nicotinic receptor ligands for selectivity at the alpha6(*) subtype. Bioorg Med Chem Lett. 2009;19:4359–4363. doi: 10.1016/j.bmcl.2009.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briggs CA, Anderson DJ, Brioni JD, Buccafusco JJ, Buckley MJ, Campbell JE, et al. Functional characterization of the novel neuronal nicotinic acetylcholine receptor ligand GTS-21 in vitro and in vivo. Pharmacol Biochem Behav. 1997;57:231–241. doi: 10.1016/s0091-3057(96)00354-1. [DOI] [PubMed] [Google Scholar]
- Briggs CA, Gubbins EJ, Marks MJ, Putman CB, Thimmapaya R, Meyer MD, et al. Untranslated region-dependent exclusive expression of high-sensitivity subforms of alpha4beta2 and alpha3beta2 nicotinic acetylcholine receptors. Mol Pharmacol. 2006;70:227–240. doi: 10.1124/mol.105.020198. [DOI] [PubMed] [Google Scholar]
- Broadbent S, Groot-Kormelink PJ, Krashia PA, Harkness PC, Millar NS, Beato M, et al. Incorporation of the beta3 subunit has a dominant-negative effect on the function of recombinant central-type neuronal nicotinic receptors. Mol Pharmacol. 2006;70:1350–1357. doi: 10.1124/mol.106.026682. [DOI] [PubMed] [Google Scholar]
- Brunzell DH, Boschen KE, Hendrick ES, Beardsley PM, McIntosh JM. alpha-Conotoxin MII-sensitive nicotinic acetylcholine receptors in the nucleus accumbens shell regulate progressive ratio responding maintained by nicotine. Neuropsychopharmacology. 2010;35:665–673. doi: 10.1038/npp.2009.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldarone BJ, Harrist A, Cleary MA, Beech RD, King SL, Picciotto MR. High-affinity nicotinic acetylcholine receptors are required for antidepressant effects of amitriptyline on behavior and hippocampal cell proliferation. Biol Psychiatry. 2004;56:657–664. doi: 10.1016/j.biopsych.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Capelli AM, Castelletti L, Salvagno C, Oliosi B, Di Lenarda E, Virginio C, et al. Identification of novel alpha7 nAChR positive allosteric modulators with the use of pharmacophore in silico screening methods. Bioorg Med Chem Lett. 2010;20:4561–4565. doi: 10.1016/j.bmcl.2010.06.014. [DOI] [PubMed] [Google Scholar]
- Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, et al. Distribution and pharmacology of alpha 6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci. 2002;22:1208–1217. doi: 10.1523/JNEUROSCI.22-04-01208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, et al. Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci. 2003;23:7820–7829. doi: 10.1523/JNEUROSCI.23-21-07820.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Changeux JP, Gautron J, Israel M, Podleski T. Separation of excitable membranes from the electric organ of Electrophorus electricus. C R Acad Sci Hebd Seances Acad Sci D. 1969;269:1788–1791. [PubMed] [Google Scholar]
- Changeux JP, Kasai M, Lee C-Y. Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc Natl Acad Sci USA. 1970;67:1241–1247. doi: 10.1073/pnas.67.3.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox BC, Marritt AM, Perry DC, Kellar KJ. Transport of multiple nicotinic acetylcholine receptors in the rat optic nerve: high densities of receptors containing alpha6 and beta3 subunits. J Neurochem. 2008;105:1924–1938. doi: 10.1111/j.1471-4159.2008.05282.x. [DOI] [PubMed] [Google Scholar]
- Di Chiara G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol. 2000;393:295–314. doi: 10.1016/s0014-2999(00)00122-9. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Hanrott KE, Mok MH, Kew JN, Wonnacott S. Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC12 cells. J Neurochem. 2007;100:1089–1096. doi: 10.1111/j.1471-4159.2006.04273.x. [DOI] [PubMed] [Google Scholar]
- Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A, et al. Alpha-conotoxin PIA is selective for alpha6 subunit-containing nicotinic acetylcholine receptors. J Neurosci. 2003;23:8445–8452. doi: 10.1523/JNEUROSCI.23-24-08445.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drenan RM, Grady SR, Whiteaker P, Clure-Begley T, McKinney S, Miwa JM, et al. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha 6 nicotinic acetylcholine receptors. Neuron. 2008;60:123–136. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott KJ, Ellis SB, Berckhan KJ, Urrutia A, Chavez-Noriega LE, Johnson EC, et al. Comparative structure of human neuronal alpha 2-alpha 7 and beta 2-beta 4 nicotinic acetylcholine receptor subunits and functional expression of the alpha 2, alpha 3, alpha 4, alpha 7, beta 2, and beta 4 subunits. J Mol Neurosci. 1996;7:217–228. doi: 10.1007/BF02736842. [DOI] [PubMed] [Google Scholar]
- Evans NM, Bose S, Benedetti G, Zwart R, Pearson KH, McPhie GI, et al. Expression and functional characterisation of a human chimeric nicotinic receptor with alpha6beta4 properties. Eur J Pharmacol. 2003;466:31–39. doi: 10.1016/s0014-2999(03)01540-1. [DOI] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ. Alpha6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology. 2008;33:2158–2166. doi: 10.1038/sj.npp.1301617. [DOI] [PubMed] [Google Scholar]
- de Fiebre CM, Meyer EM, Henry JC, Muraskin SI, Kem WR, Papke RL. Characterization of a series of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative is a selective agonist at neuronal nicotinic alpha 7/125I-alpha-bungarotoxin receptor subtypes. Mol Pharmacol. 1995;47:164–171. [PubMed] [Google Scholar]
- Fucile S, Matter JM, Erkman L, Ragozzino D, Barabino B, Grassi F, et al. The neuronal alpha6 subunit forms functional heteromeric acetylcholine receptors in human transfected cells. Eur J Neurosci. 1998;10:172–178. doi: 10.1046/j.1460-9568.1998.00001.x. [DOI] [PubMed] [Google Scholar]
- Gerzanich V, Kuryatov A, Anand R, Lindstrom J. ‘Orphan’ alpha6 nicotinic AChR subunit can form a functional heteromeric acetylcholine receptor. Mol Pharmacol. 1997;51:320–327. [PubMed] [Google Scholar]
- Gillet VJ, Willett P, Bradshaw J. Similarity searching using reduced graphs. J Chem Inf Comput Sci. 2003;43:338–345. doi: 10.1021/ci025592e. [DOI] [PubMed] [Google Scholar]
- Gotti C, Carbonnelle E, Moretti M, Zwart R, Clementi F. Drugs selective for nicotinic receptor subtypes: a real possibility or a dream? Behav Brain Res. 2000;113:183–192. doi: 10.1016/s0166-4328(00)00212-6. [DOI] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Zanardi A, Gaimarri A, Champtiaux N, Changeux JP, et al. Heterogeneity and selective targeting of neuronal nicotinic acetylcholine receptor (nAChR) subtypes expressed on retinal afferents of the superior colliculus and lateral geniculate nucleus: identification of a new native nAChR subtype alpha3beta2(alpha5 or beta3) enriched in retinocollicular afferents. Mol Pharmacol. 2005;68:1162–1171. doi: 10.1124/mol.105.015925. [DOI] [PubMed] [Google Scholar]
- Gotti C, Moretti M, Bohr I, Ziabreva I, Vailati S, Longhi R, et al. Selective nicotinic acetylcholine receptor subunit deficits identified in Alzheimer's disease, Parkinson's disease and dementia with Lewy bodies by immunoprecipitation. Neurobiol Dis. 2006a;23:481–489. doi: 10.1016/j.nbd.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Gotti C, Riganti L, Vailati S, Clementi F. Brain neuronal nicotinic receptors as new targets for drug discovery. Curr Pharm Des. 2006b;12:407–428. doi: 10.2174/138161206775474486. [DOI] [PubMed] [Google Scholar]
- Gotti C, Guiducci S, Tedesco V, Corbioli S, Zanetti L, Moretti M, et al. Nicotinic acetylcholine receptors in the mesolimbic pathway: primary role of ventral tegmental area alpha6beta2* receptors in mediating systemic nicotine effects on dopamine release, locomotion, and reinforcement. J Neurosci. 2010;30:5311–5325. doi: 10.1523/JNEUROSCI.5095-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, et al. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol. 2007;74:1235–1246. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groot-Kormelink PJ, Luyten WH, Colquhoun D, Sivilotti LG. A reporter mutation approach shows incorporation of the ‘orphan’ subunit beta3 into a functional nicotinic receptor. J Biol Chem. 1998;273:15317–15320. doi: 10.1074/jbc.273.25.15317. [DOI] [PubMed] [Google Scholar]
- van Haaren F, Anderson KG, Haworth SC, Kem WR. GTS-21, a mixed nicotinic receptor agonist/antagonist, does not affect the nicotine cue. Pharmacol Biochem Behav. 1999;64:439–444. doi: 10.1016/s0091-3057(99)00054-4. [DOI] [PubMed] [Google Scholar]
- Harper G, Bravi GS, Pickett SD, Hussain J, Green DV. The reduced graph descriptor in virtual screening and data-driven clustering of high-throughput screening data. J Chem Inf Comput Sci. 2004;44:2145–2156. doi: 10.1021/ci049860f. [DOI] [PubMed] [Google Scholar]
- Harvey SC, Maddox FN, Luetje CW. Multiple determinants of dihydro-beta-erythroidine sensitivity on rat neuronal nicotinic receptor alpha subunits. J Neurochem. 1996;67:1953–1959. doi: 10.1046/j.1471-4159.1996.67051953.x. [DOI] [PubMed] [Google Scholar]
- Horenstein NA, Leonik FM, Papke RL. Multiple pharmacophores for the selective activation of nicotinic alpha7-type acetylcholine receptors. Mol Pharmacol. 2008;74:1496–1511. doi: 10.1124/mol.108.048892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itier V, Bertrand D. Neuronal nicotinic receptors: from protein structure to function. FEBS Lett. 2001;504:118–125. doi: 10.1016/s0014-5793(01)02702-8. [DOI] [PubMed] [Google Scholar]
- Karadsheh MS, Shah MS, Tang X, Macdonald RL, Stitzel JA. Functional characterization of mouse alpha4beta2 nicotinic acetylcholine receptors stably expressed in HEK293T cells. J Neurochem. 2004;91:1138–1150. doi: 10.1111/j.1471-4159.2004.02801.x. [DOI] [PubMed] [Google Scholar]
- Klink R, de Kerchove dA, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–1463. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J. Human alpha6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology. 2000;39:2570–2590. doi: 10.1016/s0028-3908(00)00144-1. [DOI] [PubMed] [Google Scholar]
- Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, et al. Point mutant mice with hypersensitive alpha 4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci USA. 2001;98:2786–2791. doi: 10.1073/pnas.041582598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Novere N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8:2428–2439. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- Lena C, Changeux JP. Allosteric nicotinic receptors, human pathologies. J Physiol Paris. 1998;92:63–74. doi: 10.1016/S0928-4257(98)80140-X. [DOI] [PubMed] [Google Scholar]
- Lindstrom JM. Nicotinic acetylcholine receptors of muscles and nerves: comparison of their structures, functional roles, and vulnerability to pathology. Ann N Y Acad Sci. 2003;998:41–52. doi: 10.1196/annals.1254.007. [DOI] [PubMed] [Google Scholar]
- Lippiello PM, Bencherif M, Gatto GJ, Jordan KG, Traina VM. P.2.d.013 TC-2216: anti-depressant effects through selective modulation of neuronal nicotinic receptors. Eur Neuropsychopharmacol. 2006;16:S339–S340. [Google Scholar]
- Lippiello PM, Beaver JS, Gatto GJ, James JW, Jordan KG, Traina VM, et al. TC-5214 (S-(+)-mecamylamine): a neuronal nicotinic receptor modulator with antidepressant activity. CNS Neurosci Ther. 2008;14:266–277. doi: 10.1111/j.1755-5949.2008.00054.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, et al. Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–952. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S. Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther. 2002;302:197–204. doi: 10.1124/jpet.302.1.197. [DOI] [PubMed] [Google Scholar]
- Moretti M, Vailati S, Zoli M, Lippi G, Riganti L, Longhi R, et al. Nicotinic acetylcholine receptor subtypes expression during rat retina development and their regulation by visual experience. Mol Pharmacol. 2004;66:85–96. doi: 10.1124/mol.66.1.85. [DOI] [PubMed] [Google Scholar]
- Moretti M, Mugnaini M, Tessari M, Zoli M, Gaimarri A, Manfredi I, et al. A comparative study of the effects of the intravenous self-administration or subcutaneous minipump infusion of nicotine on the expression of brain neuronal nicotinic receptor subtypes. Mol Pharmacol. 2010;78:287–296. doi: 10.1124/mol.110.064071. [DOI] [PubMed] [Google Scholar]
- Munson PJ, Rodbard D. Ligand: a versatile computerized approach for characterization of ligand-binding systems. Anal Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- Papke RL, Dwoskin LP, Crooks PA, Zheng G, Zhang Z, McIntosh JM, et al. Extending the analysis of nicotinic receptor antagonists with the study of alpha6 nicotinic receptor subunit chimeras. Neuropharmacology. 2008;54:1189–1200. doi: 10.1016/j.neuropharm.2008.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y, Zhang Q, Snyder GL, Zhu H, Yao W, Tomesch J, et al. Discovery of novel alpha7 nicotinic receptor antagonists. Bioorg Med Chem Lett. 2010;20:4825–4830. doi: 10.1016/j.bmcl.2010.06.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons S, Fattore L, Cossu G, Tolu S, Porcu E, McIntosh JM, et al. Crucial role of alpha4 and alpha6 nicotinic acetylcholine receptor subunits from ventral tegmental area in systemic nicotine self-administration. J Neurosci. 2008;28:12318–12327. doi: 10.1523/JNEUROSCI.3918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quik M, McIntosh JM. Striatal alpha6* nicotinic acetylcholine receptors: potential targets for Parkinson's disease therapy. J Pharmacol Exp Ther. 2006;316:481–489. doi: 10.1124/jpet.105.094375. [DOI] [PubMed] [Google Scholar]
- Rabenstein RL, Caldarone BJ, Picciotto MR. The nicotinic antagonist mecamylamine has antidepressant-like effects in wild-type but not beta2- or alpha7-nicotinic acetylcholine receptor subunit knockout mice. Psychopharmacology (Berl) 2006;189:395–401. doi: 10.1007/s00213-006-0568-z. [DOI] [PubMed] [Google Scholar]
- Ray M, Bohr I, McIntosh JM, Ballard C, McKeith I, Chalon S, et al. Involvement of alpha6/alpha3 neuronal nicotinic acetylcholine receptors in neuropsychiatric features of Dementia with Lewy bodies: [(125)I]-alpha-conotoxin MII binding in the thalamus and striatum. Neurosci Lett. 2004;372:220–225. doi: 10.1016/j.neulet.2004.09.042. [DOI] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Villers-Thiery A, Mulle C, Hussy N, et al. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Rhodes AD, Bevan N, Patel K, Lee M, Rees S. Mammalian expression of transmembrane receptors for pharmaceutical applications. Biochem Soc Trans. 1998;26:699–704. doi: 10.1042/bst0260699. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Schneider G, Neidhart W, Giller T, Schmid G. ‘Scaffold-Hopping’ by topological pharmacophore search: a contribution to virtual screening. Angew Chem Int Ed Engl. 1999;38:2894–2896. [PubMed] [Google Scholar]
- Shytle RD, Silver AA, Lukas RJ, Newman MB, Sheehan DV, Sanberg PR. Nicotinic acetylcholine receptors as targets for antidepressants. Mol Psychiatry. 2002;7:525–535. doi: 10.1038/sj.mp.4001035. [DOI] [PubMed] [Google Scholar]
- Soloviev MM, Barnard EA. Xenopus oocytes express a unitary glutamate receptor endogenously. J Mol Biol. 1997;273:14–18. doi: 10.1006/jmbi.1997.1272. [DOI] [PubMed] [Google Scholar]
- Tumkosit P, Kuryatov A, Luo J, Lindstrom J. Beta3 subunits promote expression and nicotine-induced up-regulation of human nicotinic alpha6* nicotinic acetylcholine receptors expressed in transfected cell lines. Mol Pharmacol. 2006;70:1358–1368. doi: 10.1124/mol.106.027326. [DOI] [PubMed] [Google Scholar]
- Virginio C, Giacometti A, Aldegheri L, Rimland JM, Terstappen GC. Pharmacological properties of rat alpha 7 nicotinic receptors expressed in native and recombinant cell systems. Eur J Pharmacol. 2002;445:153–161. doi: 10.1016/s0014-2999(02)01750-8. [DOI] [PubMed] [Google Scholar]
- Walsh H, Govind AP, Mastro R, Hoda JC, Bertrand D, Vallejo Y, et al. Up-regulation of nicotinic receptors by nicotine varies with receptor subtype. J Biol Chem. 2008;283:6022–6032. doi: 10.1074/jbc.M703432200. [DOI] [PubMed] [Google Scholar]
- Ward JH. Hierarchical grouping to optimize an objective function. J Am Stat Assoc. 1963;58:236–244. [Google Scholar]
- Wise RA. Brain reward circuitry: insights from unsensed incentives. Neuron. 2002;36:229–240. doi: 10.1016/s0896-6273(02)00965-0. [DOI] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002;22:8785–8789. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Carbone AL, Moroni M, Bermudez I, Mogg AJ, Folly EA, et al. Sazetidine-A is a potent and selective agonist at native and recombinant alpha 4 beta 2 nicotinic acetylcholine receptors. Mol Pharmacol. 2008;73:1838–1843. doi: 10.1124/mol.108.045104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.