

Abstract
BACKGROUND AND PURPOSE
Digoxin has been used as an inotropic agent in heart failure for a long time. Troponin I (TnI) phosphorylation is related to cardiac contractility, and the genes are regulated by peroxisome proliferator-activated receptors (PPARs). Our previous studies indicated that cardiac abnormality related to the depressed expression of PPARδ in the hearts of STZ rats is reversed by digoxin. However, the cellular mechanisms for this effect of digoxin have not been elucidated. The aim of the present study was to investigate possible mechanisms for this effect of digoxin using the H9c2 cell line cultured in high glucose (HG) conditions.
METHODS
The effects of digoxin on PPARδ expression, intracellular calcium and TnI phosphorylation were investigated in cultured H9c2 cells, maintained in a HG medium, by using Western blot analysis.
RESULTS
Digoxin increased PPARδ expression in H9c2 cells subjected to HG conditions, and increase the intracellular calcium concentration. This effect of digoxin was blocked by BAPTA-AM at concentrations sufficient to chelate calcium ions. In addition, the calcineurin inhibitor cyclosporine A and KN93, an inhibitor of calcium/calmodulin-dependent protein kinase, inhibited this action. Digoxin also increased TnI phosphorylation and this was inhibited when PPARδ was silenced by the addition of RNAi to the cells. Similar changes were observed on the contraction of H9c2 cells.
CONCLUSION
The results suggest that digoxin appears, through calcium-triggered signals, to reverse the reduced expression of PPARδ in H9c2 cells caused by HG treatment.
Keywords: digoxin, PPARδ, hyperglycaemia, H9c2 cells, troponin
Introduction
Diabetes ranks as one of the main risk factors for the development of congestive heart failure (CHF) (Kannel et al., 1974; Kannel and McGee, 1979). Many patients with CHF and hyperglycaemic symptoms have accompanying abnormalities, including obesity, dyslipidemia and hypertension, which also lead to structural and functional abnormalities of the heart (Herlitz et al., 1988; Malmberg and Ryden, 1988; Frede et al., 2006; Poornima et al., 2006). Digoxin has been used as an effective treatment for CHF for a long time (Besch and Watanabe, 1978). The positive inotropic action of digoxin is caused by the inhibition of sodium-potassium-activated adenosine triphosphatase (Na-K ATPase), leading to an accumulation of intracellular sodium and thereby an increase in intracellular calcium via the Na/Ca exchanger (Pervaiz et al., 2006). The effects of chronic digoxin treatment on cardiac contractility in patients with CHF have been well-established clinically (van Veldhuisen et al., 1993; Pervaiz et al., 2006; Ahmed et al., 2008).
Troponin I (TnI) is an inhibitory unit of the troponin complex associated with thin filaments and inhibits actomyosin interactions at the diastolic level of intracellular Ca2+ (Metzger and Westfall, 2004; Ohtsuki and Morimoto, 2008). Modulation of myofilament properties by alterations in TnI phosphorylation has profound effects on cardiac contractility and pumping (Layland et al., 2005). Phosphorylation of TnI by protein kinase results in a reduction in myofilament Ca2+ sensitivity and an increase in cross-bridge cycling rate, leading to acceleration of relaxation and an increase in power output, but a reduced economy of contraction (Metzger and Westfall, 2004; Layland et al., 2005). Ca2+ is not only involved in muscle contraction, but it is also a ubiquitous intracellular secondary messenger that activates a wide variety of cellular responses including gene transcription (Bootman and Berridge, 1995; Metzger and Westfall, 2004; Layland et al., 2005; Ohtsuki and Morimoto, 2008). Various studies have shown that the activation of calcineurin (Cn) and calcium/calmodulin-dependent protein kinase (CaMK) signalling pathways has a major role in the regulation of gene expression in cardiac muscles (Naya et al., 2000; Passier et al., 2000). These genes are also regulated by peroxisome proliferator-activated receptors (PPARs) (Yang and Li, 2007).
PPARs are ligand-activated transcriptional factors that regulate expression of genes involved in lipid metabolism and inflammation (Yang and Li, 2007). The three subtypes of PPARs, PPARα, PPARγ and PPARδ, modulate the expression of different genes and exert various bioactivities (Yang and Li, 2007). PPARα is relatively abundant in tissues with a high oxidative capacity, such as the liver and the heart. PPARγ expression is confined to a limited number of tissues, primarily adipose tissue (Issemann and Green, 1990; Yang and Li, 2007). The ubiquitously expressed PPARδ enhances lipid catabolism in adipose tissue and muscles (Yang and Li, 2007), and PPARδ-dependent maintenance of inotropic function and metabolic effects is crucial for cardiomyocytes (Cheng et al., 2004a,b; Barish et al., 2006). Deletion of cardiac PPARδ results in an increase in left ventricular end-diastolic pressure, a lower cardiac output, and this leads to decreased contraction and increased incidence of cardiac failure (Cheng et al., 2004a). We have observed a decrease in the expression of PPARδ in the hearts of streptozotocin-induced hyperglycaemic rats (STZ rats) (Su et al., 2008). Previous studies have indicated that impaired relaxation is the main cardiac abnormality observed in STZ rats, and that it is related to depressed troponin function in their hearts (Fein et al., 1980, 1981). In a previous study we showed that this decrease in cardiac PPARδ expression in STZ rats was reversed by digoxin. However, the possible mechanisms for this action of digoxin remain obscure.
In a previous study, it was confirmed that H9c2 cells mimic a wide range of hypertrophic traits displayed by primary cardiomyocytes in response to hypertrophic agents. In addition, other studies have used the H9c2 rat cardiomyoblast cell line, which has the advantage of being an animal-free alternative (Watkins et al., 2010). Hence, in the present study, we used an H9c2 cell line obtained from rats and cultured them under high glucose (HG) conditions to investigate this effect of digoxin.
Methods
Materials
Digoxin and cyclosporine A were purchased from Sigma-Aldrich (St Louis, MO, USA). Caffeine, BAPTA-AM and KN93 were purchased from Calbiochem-Novabiochem Corp (La Jolla, CA, USA). The fluorescent probe, fura-2, was obtained from Molecular Probes (Eugene, OR, USA). The TRIzol RNA extraction reagent, Opti-MEM® I Reduced Serum Medium, Stealth™ Select RNAi [small interfering RNA (siRNA)-PPARδ], scramble siRNA (siRNA-control) and Lipofectamine 2000™ were purchased from Invitrogen (Carlsbad, CA, USA). Antibodies to PPARα, PPARγ, PPARδ and actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies to cardiac TnI and phospho-TnI (Ser 23/24) were purchased from Cell Signaling Technology (Beverly, MA, USA).
Cell cultures
Embryonic rat–heart-derived H9c2 cells (BCRC no. 60096), Madin-Darby canine kidney epithelial cells (MDCK cell line) (BCRC no. 60004) and rat lung L2 cells (BCRC no. 60276) were obtained from the Culture Collection and Research Center of the Food Industry Institute (Hsin-Chiu City, Taiwan). The H9c2 cells were maintained in growth medium composed of Dulbecco's modified Eagle's medium (DMEM) (pH 7.2) supplemented with 10% foetal bovine serum. The H9c2 cells were plated at a density of 6000 cells·cm−2 and allowed to proliferate in growth medium. The medium was changed every 48 h. To induce differentiation of the H9c2 myoblasts into myotubes, the growth medium was replaced with differentiation medium containing DMEM plus 2% horse serum when the cells reached near confluence.
Protocol for treatment
The HG-treated H9c2 cells were generated by treating the cells with 30 mmol·L−1 glucose for 24 h in accordance with our previous study (Yu et al., 2008). Stock solutions of digoxin were prepared with alcohol (0.18%). The HG-treated cells were treated with digoxin (0.01–1 µmol·L−1), or caffeine (1 µmol·L−1) for 30 min, washed twice with phosphate-buffered saline (PBS), and removed by trypsinization. Cells were then collected and subjected to a gene expression assay. Additional pretreatments with, for example, a calcium chelator (25 mmol·L−1 BAPTA-AM), calcineurin inhibitor (1 µmol·L−1 cyclosporine A) (Schaeffer et al., 2004), or CaMK inhibitor (1 µmol·L−1 KN-93) (Schaeffer et al., 2004) were performed for 30 min before the digoxin treatment.
Measurement of intracellular calcium concentration
The changes in intracellular calcium were detected using the fluorescent probe fura-2 (Hallaq and Haupert, 1989). The H9c2 cells were placed in buffered physiological saline solution (PSS) containing 140 mmol·L−1 NaCl, 5.9 mmol·L−1 KCI, 1.2 mmol·L−1 CaCl2, 1.4 mmol·L−1 MgCI2, 11.5 mmol·L−1 glucose, 1.8 mmol·L−1 Na2HPO4 and 10 mmol·L−1 HEPES-Tris, to which 5 µM fura-2 was added, and incubated for 1 h in humidified 5% CO2 and 95% air at 37°C. The cells were washed and incubated for an additional 30 min in PSS. The H9c2 cells were inserted into a thermostatted (37°C) cuvette containing 2 mL of calcium-free PSS. After the recording of baseline value, various doses of digoxin or inhibitor were added to the cuvette for further detection of calcium contents. The fluorescence was continuously recorded using a fluorescence spectrofluorometer (Hitachi F-2000, Tokyo, Japan). Values of [Ca2+]i were calculated from the ratio R = F340/F380 by the formula: [Ca2+]i =KdB (R − Rmin)/(Rmax − R), where Kd is 225 nM, F is fluorescence and B is the ratio of the fluorescence of the free dye to that of the Ca2+-bound dye measured at 380 nm. Rmax and Rmin were determined in separate experiments by using digoxin to equilibrate [Ca2+]j with ambient [Ca2+] (Rmax), and the addition of 0.1 mmol·L−1 MnCl2 and 1 mmol·L−1 EGTA (Rmin). Background autofluorescence was measured in unloaded cells and subtracted from all measurements.
Western blotting analysis
Protein was extracted from tissue homogenates and cell lysates using ice-cold radio-immuno-precipitation assay buffer supplemented with phosphatase and protease inhibitors (50 mmol·L−1 sodium vanadate, 0.5 mmol·L−1 phenylmethylsulphonyl fluoride, 2 mg·mL−1 aprotinin and 0.5 mg·mL−1 leupeptin). Protein concentrations were determined with the Bio-Rad protein assay (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Total proteins (30 µg) were separated by sodium dodecyl sulphate/polyacrylamide gel electrophoresis (10% acrylamide gel) using the Bio-Rad Mini-Protein II system. Protein was transferred to expanded polyvinylidene difluoride membranes (Pierce, Rockford, IL, USA) with a Bio-Rad Trans-Blot system. After transfer, the membranes were washed with PBS and blocked for 1 h at room temperature with 5% (w/v) skimmed milk powder in PBS. The manufacturer's instructions were followed for the primary antibody reactions. Blots were incubated overnight at 4°C with an immunoglobulin-G polyclonal rabbit anti-mouse antibody (Affinity BioReagents, Inc., Golden, CO, USA) (1:500) in 5% (w/v) skimmed milk powder dissolved in PBS/Tween 20 (0.5% by volume) to bind the target protein such as PPARδ. The blots were incubated with goat polyclonal antibody (1:1000) to bind the actin serving as the internal control. After the removal of the primary antibody, the blots were extensively washed with PBS/Tween 20. The blots were then incubated for 2 h at room temperature with the appropriate peroxidase-conjugated secondary antibody diluted in 5% (w/v) skimmed milk powder and dissolved in PBS/Tween 20. The blots were developed by autoradiography using the ECL-Western blotting system (Amersham International, Buckinghamshire, UK). The immune blots of PPARδ (49 kDa), actin (43 kDa), cardiac troponin (28 kDa) and phospho-troponin were quantified with a laser densitometer.
siRNA
Duplexed RNA oligonucleotides for rat PPARδ (Stealth RNAi™) were synthesized by Invitrogen. H9c2 cells were transfected with 40 pmol of PPARδ-specific siRNAs (siRNA-PPARδ) or scramble siRNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols, and treated 48 h post-transfection. In the preliminary experiments, oligonucleotides were observed to be effective at 40 pmol and it was used in the present study. The sequences of the siRNA-PPARδ were UUGCAGAUCCGAUCGCACUUCUCGU (sense strand) and ACGAGAAGUGCGAUCGGAUCUGCAA (antisense strand).
Cell contractility assay
In the contraction study, H9c2 cells were measured by changes in planar surface area as described previously (Chen et al., 2004). H9c2 cells were cultured in 6 mm flat bottom plates about 5 × 105 cells per plate. Culture plates were then mounted on the heated stage of an inverted light microscope for the contractility assay. Changes in the planar surface areas in response to treatment were observed using a video camera, and images of the cells were captured serially at 0, 20, 40 and 60 min. Cells of the same group (n = 6–10) were collected at the same time points. The perimeters of individual cells with clearly defined borders were outlined, and the planar surface area was calculated as percentage change. Control groups were monitored in cultures with vehicle alone. All data were calculated using WIPL∼b software (Foreseen Science and Technology, Tainan, Taiwan).
Statistical analysis
Statistical analysis was carried out using anova and Newman-Keuls post hoc analysis. Statistical significance was set as P < 0.05. Results are expressed as mean ± SEM.
Results
Effects of digoxin on PPARδ expression in H9c2 cells under HG conditions
After incubation with 30 mmol·L−1 glucose for 24 h, as shown in Figure 1A, the H9c2 cells were treated with digoxin at various concentrations (0.01–1 µmol·L−1) for 30 min and the expression of PPARδ in these cells was compared with that in cells treated with 30 mmol·L−1 glucose only (HG-treated cells). The expression of PPARδ protein in H9c2 cells was significantly reduced by HG treatment (Figure 1A). The finding that PPARδ protein levels were unchanged in mannitol-treated H9c2 cells (Figure 1B) suggests that changes in protein levels are not related to hyperosmolarity. Under HG conditions, PPARδ protein expression was markedly increased by digoxin 1 µmol·L−1 in H9c2 cells (Figure 1A). Moreover, digoxin reversed the reduced expression of PPARδ in these cells in a dose-related manner (Figure 1A). We also cultured H9c2 cells in normal medium to investigate the actions of digoxin under normal conditions. Unlike in HG-treated H9c2 cells, digoxin did not affect the expression of PPARδ in H9c2 cells cultured in normal medium (Figure 1B). In addition, we investigated the effects of digoxin on rat lung cells (L2) and MDCK cells. However, digoxin failed to modify PPARδ expression in either the L2 or the MDCK cells incubated in HG medium (Figure 1C). Also, the expressions of PPARα or PPARγ were found to be unaffected by digoxin (Figure 1D). Thus, we continued our studies in H9c2 cells to investigate the possible mechanisms of digoxin-induced changes in PPARδ expression in HG-treated cells. The dose of digoxin used in the flow studies was 1 µmol·L−1.
Figure 1.
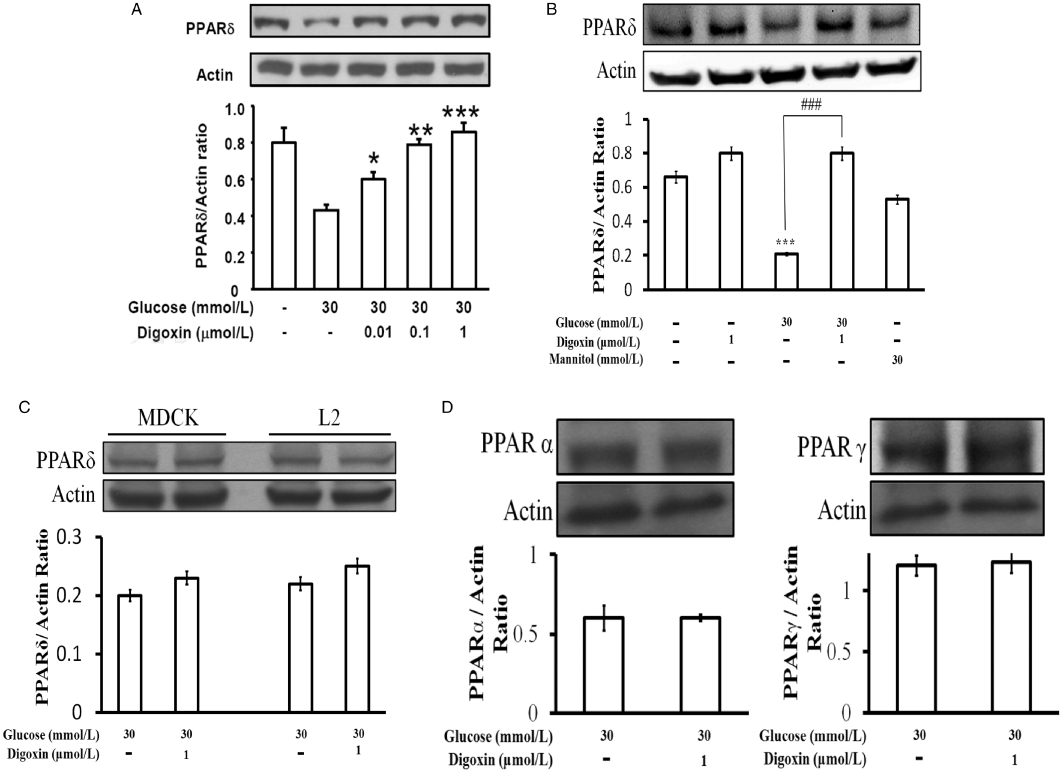
Effects of digoxin on PPARδ expression in H9c2 cells. (A) H9c2 cells were cultured with or without glucose at 30 mmol·L−1 and treated with digoxin, which was also incubated with 5.5 mmol·L−1 glucose. (B) Cells were also exposed to 24.5 mmol·L−1 mannitol to produce the same osmolarity (317 mOsmol·L−1) as that produced with the highest concentration of glucose (30 mmol·L−1). The embryonic rat H9c2 cells (A), rat lung L2 cells and MDCK cells (C) were cultured with 30 mmol·L−1 glucose for 24 h, and then treated with digoxin (0.01–1 µmol·L−1) for 30 min. Also, the possible effects of digoxin (1 µmol·L−1) on the expressions of PPARα and PPARγ in H9c2 cells were also investigated (D). These cells were harvested to determine the protein levels by Western blot analysis. All values are presented as mean ± SEM (n = 4 per group). *P < 0.05, **P < 0.01 and ***P < 0.001 as compared with the HG-treated cells.
Effects of BAPTA-AM on the digoxin-induced increase in intracellular calcium and PPARδ expression in HG -treated H9c2 cells
The fluorescent probe, fura2-AM was used to detect the intracellular calcium concentration in H9c2 cells. We treated H9c2 cell with digoxin at various concentrations and found that digoxin increased the intracellular calcium ion in these cells in a dose-related manner (Figure 2A). Caffeine has been reported to increase the intracellular concentration of calcium (Shkryl and Shirokova, 2006); therefore, it was used as a positive control. Compared with the HG medium, digoxin increased the intracellular calcium concentration from 240.6 ± 11.6 to 450.1 ± 15.6 nmol·L−1 (Figure 2B). We also measured the intracellular calcium concentrations of cells cultured in regular medium, and found that they did not differ significantly from those in the cells of the digoxin-treated HG group. In addition, the increasing effect of digoxin or caffeine on the intracellular calcium concentration was abolished by pretreatment with the calcium chelator BAPTA-AM (BAPTA) at 25 mmol·L−1 (Figure 2B). The digoxin-induced expression of PPARδ protein was also reduced by pretreatment with BAPTA in HG-treated cells (Figure 2C). Similar results were observed in the caffeine-treated group (Figure 2C). However, BAPTA at this concentration (25 mmol·L−1) did not modify the basal calcium level (237.8 ± 12.4 vs. 244.7 ± 10.9 nmol·L−1; n = 6).
Figure 2.
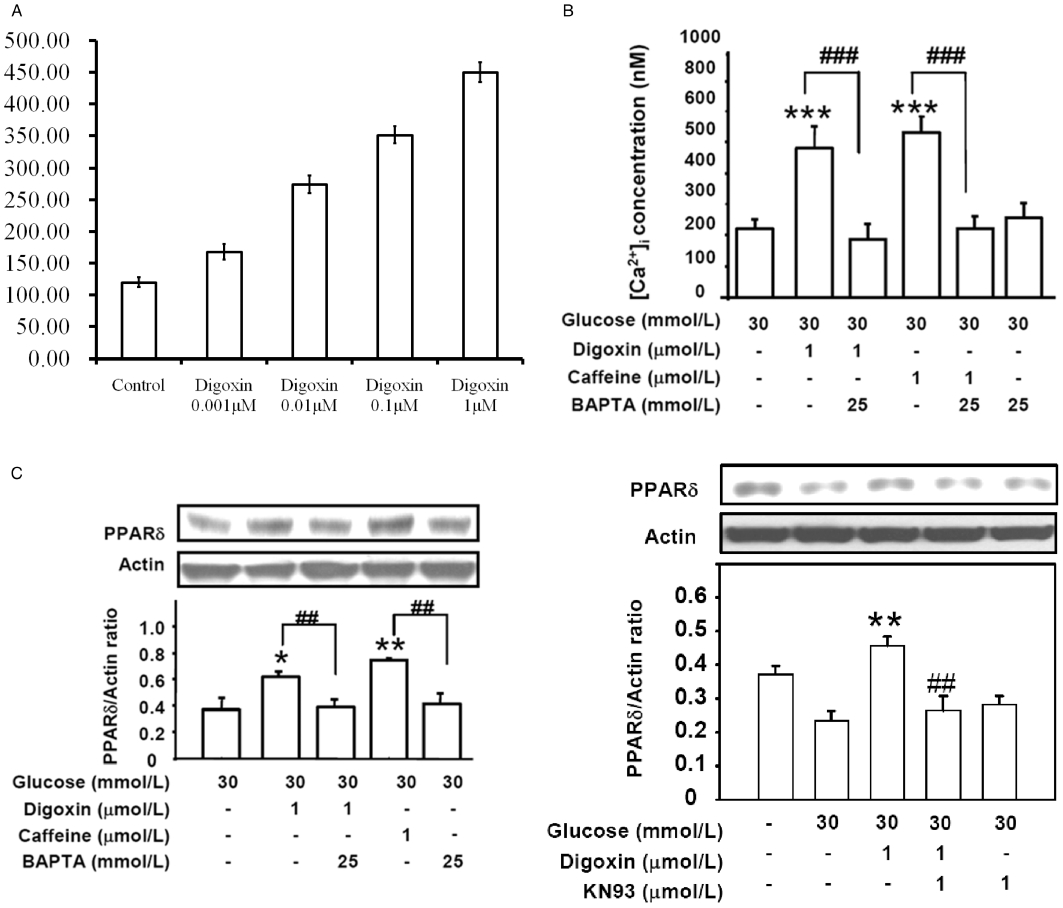
Effects of BAPTA on digoxin- and caffeine-induced calcium release and increased PPARδ expression in HG-treated H9c2 cells. (A) The intracellular calcium ion concentrations in H9c2 cells exposed to digoxin at final concentrations of 0.001, 0.01, 0.1 and 1 µM compared that control cells not treated with digoxin and (B) the effects of the various treatments on the changes in intracellular calcium concentration, as detected by the fura-2 probe using a fluorescence spectrofluorometer. (C) Effect of BAPTA on the digoxin-induced PPARδ expression in the HG-treated cells. Cells were treated with 25 mmol·L−1 BAPTA for 30 min before incubation with 1 µmol·L−1 digoxin or 1 µmol·L−1 caffeine for 30 min, and then harvested to measure PPARδ protein expression by Western blot analysis. The response to caffeine at a concentration sufficient to increase intracellular calcium was used as positive control. All values are expressed as mean ± SEM (n = 4 per group). *P < 0.05, **P < 0.01 and ***P < 0.001 as compared with the HG-treated cells. ##P < 0.01 and ###P < 0.001 as compared with the HG-treated cells incubated with digoxin or caffeine.
The calcium-dependent pathway in the digoxin-induced expression of PPARδ in HG-treated H9c2 cells
Cyclosporine A (CsA, calcineurin inhibitor) and KN93 (calcium/calmodulin kinase inhibitor) were used to investigate whether digoxin-induced PPARδ expression was mediated through the calcium-triggered pathway (Schaeffer et al., 2004). The digoxin-induced increase in PPARδ protein expression was inhibited by 1 µmol·L−1 CsA (Figure 3A) and 1 µmol·L−1 KN93 (Figure 3B). However, treatment with CsA or KN93 at the maximum dose in HG-treated cells had no effect on the levels of PPARδ expression in the absence of digoxin (Figure 3).
Figure 3.
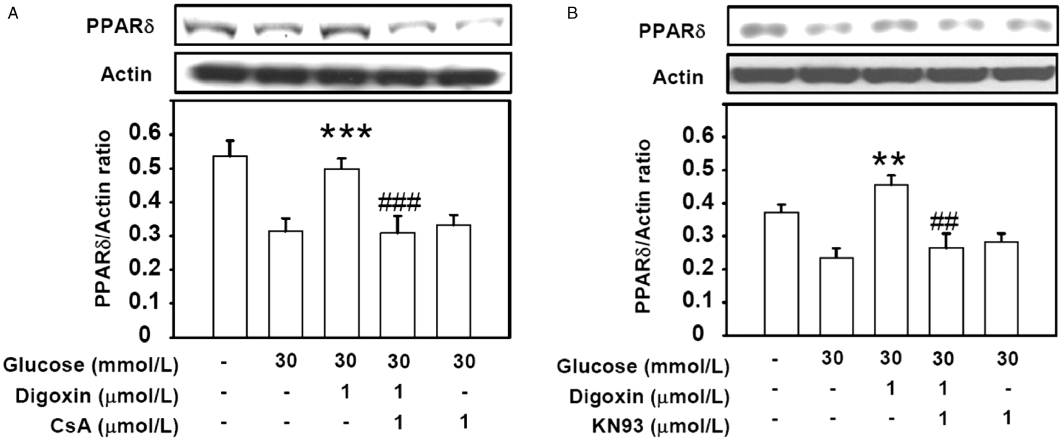
Effects of calcium-mediated signalling inhibitors on digoxin-induced PPARδ expression in HG-treated H9c2 cells. The HG-treated cells were treated with 1 µmol·L−1 cyclosporine A (CsA, calcineurin inhibitor) (A) or 1 µmol·L−1 KN93 (CaMK inhibitor) (B) for 30 min before incubation with 1 µmol·L−1 digoxin for 30 min. The cells were then harvested to measure PPARδ protein expression (A and B) by Western blot analysis. All values are expressed as mean ± SEM (n = 4 per group). **P < 0.01 and ***P < 0.001 as compared with the HG-treated H9c2 cells. ##P < 0.01 and ###P < 0.001 as compared with the HG-treated cells incubated with digoxin.
Effects of transfection with siRNA-PPARδ on the digoxin-induced increased expression of PPARδ and phosphorylation of troponin
The HG-treated H9c2 cells were transfected with siRNA-PPARδ or siRNA-control for 48 h. The level of digoxin-induced PPARδ expression was reduced by transfection with siRNA-PPARδ in HG-treated cells (Figure 4A). Moreover, treatment with the siRNA-control did not influence the digoxin-induced expression of PPARδ in HG-treated cells (Figure 4A). However, the level of digoxin-induced TnI phosphorylation was reduced by siRNA-PPARδ in HG-treated cells (Figure 4B). The levels of digoxin-induced TnI phosphorylation were increased in the siRNA-control in HG conditions (Figure 4).
Figure 4.
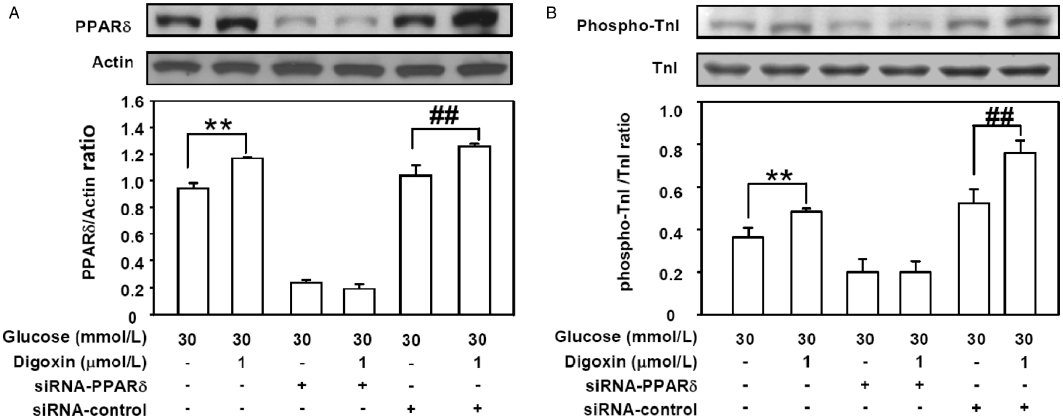
Effects of PPARδ-specific siRNA on digoxin-induced PPARδ expression and cardiac TnI phosphorylation in HG-treated H9c2 cells. The HG-treated H9c2 cells were incubated with siRNA-PPARδ or siRNA-control for 48 h before incubation with 1 µmol·L−1 digoxin for 30 min, and were then harvested to measure PPARδ protein expression (A) and TnI phosphorylation (B) by Western blot analysis. All values are expressed as mean ± SEM (n = 4 per group). **P < 0.01 as compared with the HG-treated cells, and ##P < 0.01 as compared with the HG-treated cells incubated with siRNA-control.
Effects of digoxin on H9c2 cell contraction
The contraction assay was performed in four experimental groups under HG conditions as follows: control (C); digoxin-treated cells (HG + digoxin); siRNA-PPARδ cells treated with digoxin (HG + Si + digoxin); and siRNA-control cells treated with digoxin (HG + Sc + digoxin). Changes in planar surface areas of cells in response to digoxin at 0, 20, 40 and 60 min were recorded (Figure 5). After digoxin stimulation, cell planar areas decreased gradually and were different from their original sizes after 60 min. However, the planar areas of cells in the siRNA-PPARδ plus digoxin group were reversed to a level similar to the control group (P < 0.05). The planar surface areas of H9c2 cells in the siRNA-control treated with digoxin group were the same as those of the digoxin-treated group at 60 min. The perimeter of individual cells with clearly defined borders was outlined, and the planar surface area was calculated. The loss of contraction in the siRNA-PPARδ plus digoxin group was significant compared with the siRNA-control digoxin group (P < 0.05). No marked difference in the planar surface area was observed in the control group under HG conditions compared to the vehicle alone group (Figure 5).
Figure 5.
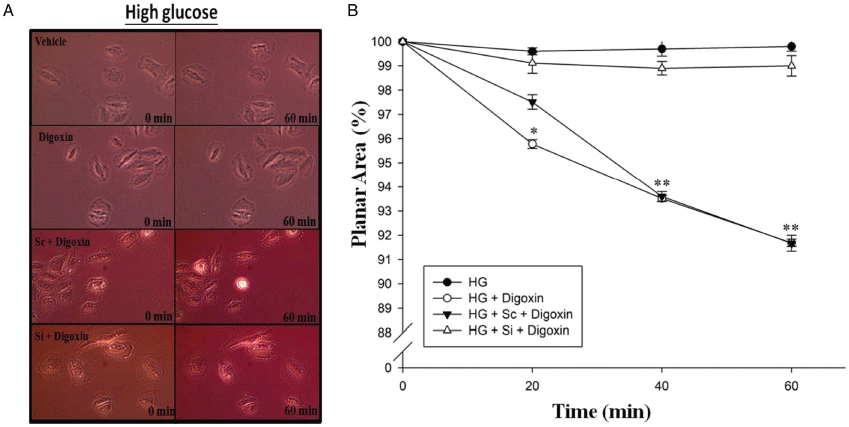
Effects of PPARδ-specific siRNA on digoxin-induced cell contraction in HG-treated H9c2 cells. The HG-treated H9c2 cells were incubated with siRNA-PPARδ (Si) or siRNA-control cells (Sc) for 48 h before incubation with 1 µmol·L−1 digoxin for 30 min and then applied for measurement of the planar surface area. The upper panel (A) shows the representative change of cells in each group. The lower panel (B) expresses the differences between four groups as mean ± SEM (n = 6 per group). Both digoxin-treated group and Sc treated with digoxin group were markedly different compared to the controls (*P < 0.05 and **P < 0.01).
Discussion
Changes in PPARδ expression levels following exposure to glucose (30 mmol·L−1) has been documented previously (Fan et al., 2010) and our results are consistent with this finding. The results obtained with H9c2 cells incubated in mannitol showing the same osmolarity as HG suggest that the changes in PPARδ protein levels induced by digoxin are not related to hyperosmolarity. Also the effect of digoxin on PPARδ expression appears to be specific for HG-treated H9c2 cells as digoxin failed to modify PPARδ expression in either L2 or MDCK cells. In addition, digoxin did not influence the expressions of either PPARα or PPARγ in H9c2 cells.
This digitoxin-induced increase in PPARδ expression in HG-treated cells was blocked by pretreatment with BAPTA, CsA and KN93. Similarly, the digoxin-induced phosphorylation of troponin I in HG-treated cells was markedly reduced in the presence of siRNA-PPARδ. In previous studies, it has been shown that PPARδ is acutely involved in the effects of excitation–transcription coupling (Lunde et al., 2007), and that calcineurin-mediated skeletal muscle reprogramming induces the expression of several transcription regulators, including PPARδ (Long et al., 2007). Hence, we conclude that the increased expression of PPARδ induced by digoxin is dependent on an increase in calcium ions binding with specific proteins and, consequently, the contractility of the cells was also increased (inotropic action).
Contraction of cardiac muscles relies upon interactions between ATP and Ca2+, both of which must be present in adequate amounts (Tate et al., 1991). In addition, impaired cardiac Ca2+ dynamics and reduced ATP production may contribute to the impaired muscle contractility in STZ rats (Fein et al., 1980, 1981). Digoxin increases cardiac contractility by increasing Ca2+ concentration in cardiomyocytes (Besch and Watanabe, 1978). It has recently been found that the maintenance PPARδ levels in cardiomyocytes is crucial for their inotropic function (Cheng et al., 2004a,b; Barish et al., 2006). In a previous study, we showed that PPARδ expression is decreased in the hearts of HG rats (Su et al., 2008). Thus, the ability of digoxin to improve cardiac contractility in the heart appears to be related to the changes in PPARδ.
In the present study, similar to the results in hearts of STZ rats, digoxin increased the PPARδ expression in H9c2 cell line obtained from rats. Moreover, it was observed that this digoxin-induced increase in PPARδ expression was suppressed by BAPTA and reduced by inhibitors of Cn and CaMK. Consistent with our results, PPARδ expression in cardiac muscles was found to be regulated by the Ca2+-triggered signalling pathway (Tate et al., 1991; Schaeffer et al., 2004; Shkryl and Shirokova, 2006). However, this is the first time that Cn and/or CaMK has been shown to be involved in this effect of digoxin.
In the present study, the levels of TnI phosphorylation observed in HG conditions were increased by treatment with digoxin, consistent with the increase in TnI phosphorylation induced by digoxin in the hearts of STZ diabetic rats we observed previously. Also, previous studies have indicated that levels of TnI phosphorylation are increased in rats after induction of diabetes for 8 weeks (Liu et al., 1996). However, in human studies, phosphorylation of TnI was found to be reduced in heart failure (Messer et al., 2007). TnI phosphorylation is thought to act by enhancing the off-rate during Ca2+ exchange with TnC, leading to an acceleration of relaxation and an increase in cardiac output (Tate et al., 1991; Liu et al., 1996; Li et al., 2000; Pi et al., 2002; Messer et al., 2007). Protein kinase C has been shown to be involved in the phosphorylation of TnI induced by high levels of glucose (Liu et al., 1996). Consistent with this, we also found that TnI phosphorylation was elevated in H9c2 cells exposed to high levels of glucose. Moreover, in siRNA-controls of HG-treated cells, the levels of digoxin-induced TnI phosphorylation were increased rather than decreased in the present study. Hence, we hypothesize that intracellular calcium release and increased expression of PPARδ are responsible for the increased TnI phosphorylation induced by digoxin. Any unexpected increase in TnI phosphorylation may have been due to the stress generated in the transfection process and increased intracellular calcium release.
However, the role of PPARδ in the phosphorylation of TnI in cardiomyocytes is still not clear. In the present study, treatment with siRNA-PPARδ suppressed the digoxin-induced increase in PPARδ expression and TnI phosphorylation. Thus, PPARδ is thought to be involved in this digoxin-induced TnI phosphorylation. Cell planar surface area was further characterized in the present study and, as shown in Figure 5, similar changes were observed in the planar surface areas of H9c2 cells, which we believe are linked to contraction, as mentioned earlier.
In conclusion, we found that the mechanism whereby digoxin increases the expression of PPARδ is related to an increase in intracellular calcium through binding with calcineurin and/or CaMK. This finding supports the hypothesis that activation of PPARδ may play an important role in the treatment of heart failure.
Acknowledgments
We appreciate the assistance of Miss Y. J. Wen in immunoblotting analysis. The present study was supported in part by a grant from the National Science Council (NSC 96-2320-B006-010) of the Republic of China.
Glossary
Abbreviations
- BAPTA
BAPTA-AM
- CaMK
calcium/calmodulin-dependent protein kinase
- CHF
congestive heart failure
- Cn
calcineurin
- CsA
Cyclosporine A
- Na-K ATPase
sodium-potassium-activated adenosine triphosphatase
- PPARs
peroxisome proliferator-activated receptors
- PPARδ
peroxisome proliferator-activated receptor δ
- siRNA-PPARδ
PPARδ-specific siRNAs
- STZ
rats, streptozotocin-induced hyperglycaemic rats
- TnI
Troponin I
Conflicts of interest
None.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Ahmed A, Pitt B, Rahimtoola SH, Waagstein F, White M, Love TE, et al. Effects of digoxin at low serum concentrations on mortality and hospitalization in heart failure: a propensity-matched study of the DIG trial. Int J Cardiol. 2008;123:138–146. doi: 10.1016/j.ijcard.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barish GD, Narkar VA, Evans RM. PPAR delta: a dagger in the heart of the metabolic syndrome. J Clin Invest. 2006;116:590–597. doi: 10.1172/JCI27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besch HR, Jr, Watanabe AM. The positive inotropic effect of digitoxin: independence from sodium accumulation. J Pharmacol Exp Ther. 1978;207:958–965. [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Chen JS, Lee HS, Jin JS, Chen A, Lin SH, Ka SM, et al. Attenuation of mouse mesangial cell contractility by high glucose and mannitol: involvement of protein kinase C and focal adhesion kinase. J Biomed Sci. 2004;11:142–151. doi: 10.1007/BF02256557. [DOI] [PubMed] [Google Scholar]
- Cheng L, Ding G, Qin Q, Huang Y, Lewis W, He N, et al. Cardiomyocyte-restricted peroxisome proliferator-activated receptor-delta deletion perturbs myocardial fatty acid oxidation and leads to cardiomyopathy. Nat Med. 2004a;10:1245–1250. doi: 10.1038/nm1116. [DOI] [PubMed] [Google Scholar]
- Cheng L, Ding G, Qin Q, Xiao Y, Woods D, Chen YE, et al. Peroxisome proliferator-activated receptor delta activates fatty acid oxidation in cultured neonatal and adult cardiomyocytes. Biochem Biophys Res Commun. 2004b;313:277–286. doi: 10.1016/j.bbrc.2003.11.127. [DOI] [PubMed] [Google Scholar]
- Fan SC, Yu BC, Chen ZC, Chen LJ, Chung HH, Cheng JT. The decreased expression of peroxisome proliferator-activated receptors delta (PPARdelta) is reversed by digoxin in the heart of diabetic rats. Horm Metab Res. 2010;42:637–642. doi: 10.1055/s-0030-1253373. [DOI] [PubMed] [Google Scholar]
- Fein FS, Kornstein LB, Strobeck JE, Capasso JM, Sonnenblick EH. Altered myocardial mechanics in diabetic rats. Circ Res. 1980;47:922–933. doi: 10.1161/01.res.47.6.922. [DOI] [PubMed] [Google Scholar]
- Fein FS, Strobeck JE, Malhotra A, Scheuer J, Sonnenblick EH. Reversibility of diabetic cardiomyopathy with insulin in rats. Circ Res. 1981;49:1251–1261. doi: 10.1161/01.res.49.6.1251. [DOI] [PubMed] [Google Scholar]
- Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006;396:517–527. doi: 10.1042/BJ20051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallaq HA, Haupert GT., Jr Positive inotropic effects of the endogenous Na+/K(+)-transporting ATPase inhibitor from the hypothalamus. Proc Natl Acad Sci USA. 1989;86:10080–10084. doi: 10.1073/pnas.86.24.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitz J, Malmberg K, Karlson BW, Ryden L, Hjalmarson A. Mortality and morbidity during a five-year follow-up of diabetics with myocardial infarction. Acta Med Scand. 1988;224:31–38. doi: 10.1111/j.0954-6820.1988.tb16735.x. [DOI] [PubMed] [Google Scholar]
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Kannel WB, McGee DL. Diabetes and cardiovascular disease. The Framingham study. JAMA. 1979;241:2035–2038. doi: 10.1001/jama.241.19.2035. [DOI] [PubMed] [Google Scholar]
- Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. 1974;34:29–34. doi: 10.1016/0002-9149(74)90089-7. [DOI] [PubMed] [Google Scholar]
- Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Li L, Desantiago J, Chu G, Kranias EG, Bers DM. Phosphorylation of phospholamban and troponin I in beta-adrenergic-induced acceleration of cardiac relaxation. Am J Physiol Heart Circ Physiol. 2000;278:H769–H779. doi: 10.1152/ajpheart.2000.278.3.H769. [DOI] [PubMed] [Google Scholar]
- Liu X, Takeda N, Dhalla NS. Troponin I phosphorylation in heart homogenate from diabetic rat. Biochim Biophys Acta. 1996;1316:78–84. doi: 10.1016/0925-4439(96)00007-5. [DOI] [PubMed] [Google Scholar]
- Long YC, Glund S, Garcia-Roves PM, Zierath JR. Calcineurin regulates skeletal muscle metabolism via coordinated changes in gene expression. J Biol Chem. 2007;282:1607–1614. doi: 10.1074/jbc.M609208200. [DOI] [PubMed] [Google Scholar]
- Lunde IG, Ekmark M, Rana ZA, Buonanno A, Gundersen K. PPARdelta expression is influenced by muscle activity and induces slow muscle properties in adult rat muscles after somatic gene transfer. J Physiol. 2007;582:1277–1287. doi: 10.1113/jphysiol.2007.133025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmberg K, Ryden L. Myocardial infarction in patients with diabetes mellitus. Eur Heart J. 1988;9:259–264. doi: 10.1093/oxfordjournals.eurheartj.a062494. [DOI] [PubMed] [Google Scholar]
- Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Metzger JM, Westfall MV. Covalent and noncovalent modification of thin filament action: the essential role of troponin in cardiac muscle regulation. Circ Res. 2004;94:146–158. doi: 10.1161/01.RES.0000110083.17024.60. [DOI] [PubMed] [Google Scholar]
- Naya FJ, Mercer B, Shelton J, Richardson JA, Williams RS, Olson EN. Stimulation of slow skeletal muscle fiber gene expression by calcineurin in vivo. J Biol Chem. 2000;275:4545–4548. doi: 10.1074/jbc.275.7.4545. [DOI] [PubMed] [Google Scholar]
- Ohtsuki I, Morimoto S. Troponin: regulatory function and disorders. Biochem Biophys Res Commun. 2008;369:62–73. doi: 10.1016/j.bbrc.2007.11.187. [DOI] [PubMed] [Google Scholar]
- Passier R, Zeng H, Frey N, Naya FJ, Nicol RL, McKinsey TA, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervaiz MH, Dickinson MG, Yamani M. Is digoxin a drug of the past? Cleve Clin J Med. 2006;73:821–824. doi: 10.3949/ccjm.73.9.821. 826, 829–832 passim. [DOI] [PubMed] [Google Scholar]
- Pi Y, Kemnitz KR, Zhang D, Kranias EG, Walker JW. Phosphorylation of troponin I controls cardiac twitch dynamics: evidence from phosphorylation site mutants expressed on a troponin I-null background in mice. Circ Res. 2002;90:649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]
- Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- Schaeffer PJ, Wende AR, Magee CJ, Neilson JR, Leone TC, Chen F, et al. Calcineurin and calcium/calmodulin-dependent protein kinase activate distinct metabolic gene regulatory programs in cardiac muscle. J Biol Chem. 2004;279:39593–39603. doi: 10.1074/jbc.M403649200. [DOI] [PubMed] [Google Scholar]
- Shkryl VM, Shirokova N. Transfer and tunneling of Ca2+ from sarcoplasmic reticulum to mitochondria in skeletal muscle. J Biol Chem. 2006;281:1547–1554. doi: 10.1074/jbc.M505024200. [DOI] [PubMed] [Google Scholar]
- Su JC, Li ZD, Yu BQ, Cao LH, Zhang CC. [Diagnosis and treatment of peripheral nerve injury in Wenchuan earthquake: a report of 14 cases] Zhongguo Gu Shang. 2008;21:739–740. [PubMed] [Google Scholar]
- Tate CA, Hyek MF, Taffet GE. The role of calcium in the energetics of contracting skeletal muscle. Sports Med. 1991;12:208–217. doi: 10.2165/00007256-199112030-00005. [DOI] [PubMed] [Google Scholar]
- van Veldhuisen DJ, Man in ′t Veld AJ, Dunselman PH, Lok DJ, Dohmen HJ, Poortermans JC, et al. Double-blind placebo-controlled study of ibopamine and digoxin in patients with mild to moderate heart failure: results of the Dutch Ibopamine Multicenter Trial (DIMT) J Am Coll Cardiol. 1993;22:1564–1573. doi: 10.1016/0735-1097(93)90579-p. [DOI] [PubMed] [Google Scholar]
- Watkins SJ, Borthwick GM, Arthur HM. The H9C2 cell line and primary neonatal cardiomyocyte cells show similar hypertrophic responses in vitro. In Vitro Cell Dev Biol Anim. 2010 doi: 10.1007/s11626-010-9368-1. [Epub ahead of print]. doi: 10.1007/s11626-010-9368-1. [DOI] [PubMed] [Google Scholar]
- Yang Q, Li Y. Roles of PPARs on regulating myocardial energy and lipid homeostasis. J Mol Med. 2007;85:697–706. doi: 10.1007/s00109-007-0170-9. [DOI] [PubMed] [Google Scholar]
- Yu BC, Chang CK, Ou HY, Cheng KC, Cheng JT. Decrease of peroxisome proliferator-activated receptor delta expression in cardiomyopathy of streptozotocin-induced diabetic rats. Cardiovasc Res. 2008;80:78–87. doi: 10.1093/cvr/cvn172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.