

Abstract
BACKGROUND AND PURPOSE
Exenatide is a 39-amino-acid peptide widely used to manage type 2 diabetes mellitus. However, it has a short plasma half-life and requires a twice daily injection regime. To overcome these drawbacks we used maleimide-polyethylene glycol to induce site-specific PEGylation.
EXPERIMENTAL APPROACH
The analogue PB-105 (ExC39) was produced by replacing cysteine at position 39 of exenatide to provide a free thiol group. PB-105 showed the same glucoregulatory activity as exenatide in mice. Site-specific PEGylation of PB-105 was performed to produce PB-110 (ExC39PEG5kDa), PB-106 (ExC39PEG20kDa), PB-107 (ExC39PEG30kDa) and PB-108 (ExC39PEG40kDa). Their effects on intracellular cAMP, acute glucoregulatory activity and pharmacokinetic profile were compared in mice and rats.
KEY RESULTS
PEGylation shifted the concentration–response curve of PB-105 to the right in a parallel, polyethylene glycol mass-dependent manner but with an inflexion point of at least 20 kDa. The activities of PB-107 and PB-108 but not PB-106 were reduced by 90% and 99%. PEGylation affected in vivo glucoregulatory activity in the same ‘Inflexion-Shift’ fashion at least at 20 kDa, but linearly increased plasma duration and systemic exposure without inflexion. PB-106 had a plasma t1/2 approximately 10-fold that of PB-105, and exhibited superior glucoregulatory activity compared with PB-105 in normal and diabetic mice.
CONCLUSIONS AND IMPLICATIONS
Site-specific PEGylation of exenatide with a permanent amide linkage affects its activity in a new type of ‘Inflexion-Shift’ fashion. PB-106 is a putative new analogue for treating diabetes; it possesses no loss of in vitro activity, prolonged plasma duration and superior, improved in vivo glucoregulatory activity compared with exenatide.
Keywords: exenatide, site-specific PEGylation, cAMP, glucoregulation
Introduction
It is known that gut-derived mammalian incretins, such as glucagon-like peptide-1 (GLP-1), regulate blood glucose levels following their release into the circulation. GLP-1 exhibits glucoregulation via activation of GLP-1 receptors, located in pancreatic periductal cells, α- and β-cells, as well as the kidney, heart, stomach and brain (Holst, 2007), to increase glucose-dependent insulin secretion, which decreases risks of hypoglycaemia (Parkes et al., 2001; Egan et al., 2002; Kolterman et al., 2003; Buse et al., 2004). In addition, GLP also suppresses inappropriately high glucagon secretion in a glucose-dependent manner (Kolterman et al., 2003; 2005; Buse et al., 2004), slows gastric emptying (Kolterman et al., 2005), reduces food intake, which in turn promotes weight loss (Szayna et al., 2000; Edwards et al., 2001) and promotes β-cell proliferation and islet neogenesis in diabetic animals (Farilla et al., 2002). Exenatide, the active ingredient of Byetta®, is a synthetic version of the 39-amino-acid peptide originally isolated from the saliva of the gila monster lizard, with 53% amino acid sequences overlapping with GLP-1 (Eng et al., 1992). Exenatide acts as a potent agonist of GLP-1 receptors (Montrose-Rafizadeh et al., 1997) and shows very similar glucoregulatory activities to GLP-1 (DeFronzo et al., 2005; Holst, 2007). As a first-in-class incretin mimetic agent, exenatide has been widely used to improve glycaemic control in type 2 diabetes mellitus patients featured by less hypoglycaemia and progressive weight loss (DeFronzo et al., 2005; Triplitt and Defronzo, 2006).
However, exenatide exhibits a short plasma half-life of 1.5–4 h and clinical effects lasting for up to 8 h (Parkes et al., 2001; Kolterman et al., 2005) mainly due to its fast kidney excretion (Copley et al., 2006), and requires s.c. injection twice daily for the treatment (Egan et al., 2003; DeFronzo et al., 2005) This has lead to main drawbacks of less than optimal clinical compliance (medication possession radio of 68%) and poorer quality of life (Fabunmi et al., 2009). Reduction of the required frequency of s.c. injections is one way that would significantly enhance compliance. It has been reported that continuous exenatide therapy reached compliance of 100% (Cuddihy et al., 2010). Many drug delivery technologies have been developed to improve exenatide's pharmacokinetic profiles. A once a week long-acting release version of exenatide employing microsphere technology has been demonstrated to be effective for the treatment of type 2 diabetes (Kim et al., 2007). Meanwhile, the exclusive disposition of exenatide via kidney secretion (Copley et al., 2006) offers a unique opportunity for site-specific PEGylation technology to improve exenatide's short plasma duration and poor systemic exposure possibly without reducing its activity.
PEGylation, the covalent attachment of polyethylene glycol (PEG) to targeted drug molecules, has become one of the most mature, valid and widely used drug delivery technologies ever developed. It has generated several blockbuster protein drugs with markedly improved therapeutic properties mainly by extending their plasma lifetimes compared with their corresponding unmodified predecessor molecules (Bailon et al., 2001; Fishburn, 2008; Bailon and Won, 2009). Rational design of a PEGylated exenatide analogue involves several considerations. In order to be able to perform site-specific PEGylation with a maleimide method, exenatide analogues need to be engineered to have a cysteine residue providing a free thiol group, so that a specific C-terminal permanent amide conjugation with maleimide PEGs can be accomplished (Bailon and Won, 2009). It is generally believed that the intact amino acid sequence from 1 through 8 (H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser) of the N-terminal is essential for exenatide to bind GLP-1 receptors and maintain its biological activity, such as stimulation of intracellular cAMP (Lopez de Maturana et al., 2003; Mann et al., 2007). Therefore, the added cysteine should be placed towards the C-terminus site as far as possible from the active site.
Application of random PEGylation usually causes an exponential reduction in binding affinity and bioactivity due to the attachment of PEGs (as little as 4 kDa) near the active site and subsequent steric hindrance of target molecule binding (Bowen et al., 1999; Bailon et al., 2001; Bailon and Won, 2009). In order to minimize the loss of bioactivity, site-specific PEGylation technologies have emerged using a variety of techniques such as the protein mutation method (Yamamoto et al., 2003; Rosendahl et al., 2005), transglutaminase method (Fontana et al., 2008) and maleimide method (Manjula et al., 2003; Bailon and Won, 2009), which also make the manufacturing process simple and products homogeneous (Bailon and Won, 2009; French et al., 2009). Site-specific PEGylation to proteins of large molecular size, particularly antibody fragments, usually results in no loss of activity up to a PEG50kDa (Yamamoto et al., 2003; Bailon and Won, 2009). However, preserving activity through this type of site-specific PEGylation may not be valid for small-to-medium size peptides such as exenatide due to the short distances between the active site and the conjugation site relative to the molecular size of PEGs.
On the other hand, PEGylation always results in higher plasma levels of PEGylated protein drugs, due both to increased actual and apparent molecular size resulting from the linear structure and hydrophilicity of PEGs (Caliceti and Veronese, 2003; Youn et al., 2006; Bailon and Won, 2009). The in vivo efficacy of PEGylated proteins or peptides would be the net effects from PEG-affected activity and prolonged activity produced by extended plasma lifetime (Fishburn, 2008; Bailon and Won, 2009). It is a challenge to optimally select a PEG size based on a balance between the activity and plasma duration by PK/PD modelling. Therefore, the PEG molecular mass-dependency of PEGylation on activities and pharmacokinetics of exenatide analogues needed to be systemically evaluated.
The present study first compared activities of exenatide and PB-105 (ExC39) and PB-102 (ExC35), cysteine-replacement analogues of exenatide at the position 39 and 35, respectively, as well as PB-111 (ExY40), the PB-105 analogue with tyrosine added at the position of 39, on intracellular cAMP stimulation, acute glucoregulatory activity and pharmacokinetic profile. Based on the preservation of its activities, we selected PB-105 as the active intermediate for site-specific PEGylation. The study further investigated PEG mass-dependency of site-specific PEGylation on PB-105's in vitro and in vivo activities as well as its pharmacokinetic profile. Finally, the study selected PB-106 (ExC39PEG20kDa) for further study based on its favourable combination of activity and plasma duration, and showed it to be twofold to fivefold more efficacious than exenatide in acute glucoregulation in normal and diabetic mice. Preliminary results were presented at the 10th Chinese National Pharmacology Conference (Gong et al., 2009).
Methods
Chemicals
Streptozotocin (STZ) was purchased from Sigma (St Louis, MO, USA). Exenatide and its analogues were synthesized by Kaijie Bio-Pharmaceutical Co. (Chengdu, China) using a solid phase method by a multiple peptide synthesizer where an F-moc strategy was employed. As presented in Figure 1, PB-102 (ExC35) and PB-105 (ExC39) are analogues of exenatide formed by replacing alanine and serine with cysteine at positions of 35 and 39, respectively, while PB-111 (ExY40) is a PB-105 analogue formed by addition of tyrosine at the position 39. Peptides were characterized by high-performance liquid chromatography and mass spectrometry, as well as sequence analysis in the case of PB-105, with all purities of more than 98%. PEGylated exenatide analogues were manufactured by PegBio Co. (Suzhou, China) employing site-specific PEGylation technology. PB-110 (ExC39PEG5kDa), PB-106 (ExC39PEG20kDa), PB-107 (ExC39PEG30kDa) and PB-108 (ExC39PEG40kDa) were produced by site-specific conjugation of PB-105 linked with a free thiol group provided by the replaced cysteine at the C-terminal with maleimide-derivatized PEG of different molecular masses, that is, 5, 20, 30 and 40 kDa respectively. The purity of all PEGylated peptides was greater than 95% (UV detection). All test substances were dissolved in normal saline except for STZ, which was freshly dissolved in sodium citrate buffer (pH 4.5).
Figure 1.
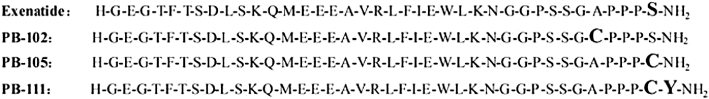
Amino acid sequences of exenatide and its analogues, PB-102 (ExC35), PB-105 (ExC39) and PB-111 (ExY40).
Intracellular cAMP assay
The phaeochromocytoma PC12 cells (obtained from American Type Culture Collection, Rockvill, MD, USA) expressing GLP-1 receptors (Perry et al., 2002) were routinely cultured in RPMI 1640 medium containing 10% fetal bovine serum at 37°C in a humidified 5% CO2 air incubator. PC12 cells were plated 48 h prior to the experiment on 24-cell plates at a density of 105 cells·mL−1. On the day of the experiment when PC12 cells reached 60–70% confluency, the plates were washed twice with PBS buffer and blocked by adding 1 mL of 1% BSA in PBS, then incubated with test compounds and 3-isobutyl-1-methylxanthine (100 µM) for 30 min. The intracellular cAMP was extracted by the addition of 500 µL of 0.1 M hydrochloric acid followed by sonication for 15 s. Intracellular cAMP was determined by using a cAMP elisa kit (R&D Systems Inc, Minneapolis, MN, USA) by a Microplate Reader (Multiskan MK3, Thermo Labsystems, Vantaa, Finland) while the protein content was quantified by using the BCA method.
Animals and surgical procedures
Adult male Swiss mice for blood sugar studies, weighing 20–30 g, and adult male Sprague-Dawley rats for pharmacokinetics studies, weighing 250–300 g, were purchased from Shanghai Laboratory Animal Center (Shanghai, China). Following shipment, mice and rats were housed for at least 3 days allowing acclimatization prior to experimental studies. The animals were housed at 23 ± 1°C, and were given access to food and water ad libitum unless otherwise noted. All procedures were approved by the Laboratory Animal Use Committee of Shanghai Jiao Tong University School of Pharmacy.
For the pharmacokinetic study, rats were anaesthetized with an i.p. injection of chloral hydrate (300 mg·kg−1). Cannulations of the right femoral vein for i.v. injection and of the right femoral artery for sampling were performed with a PE-50 polyethylene cannula (BD Biosciences, San Jose, CA, USA). Cannulas were flushed with 0.2 mL of heparin-treated saline (200 IU·mL−1). Experiments were started 16 h after cannulation while animals were fully conscious in home cages, with free access to food and water.
Blood glucose measurement in normal and diabetics animals
Blood glucose levels were measured by using a glucose oxidase method by OneTouch® Ultra® 2 Blood Glucose Meter (Johnson & Johnson Company, Shanghai, China). Blood samples were diluted in normal saline at a ratio of 3:4 for further measurements in case the blood glucose concentration was higher than 33.3 mmol·L−1 in some diabetic mice. Diabetes was induced by s.c. injection of STZ at a dose of 120 mg·kg−1 in mice deprived of food for 12 h. These mice developed diabetes 3 days after STZ injection and those with blood glucose readings higher than 16.7 mM during the 3 day measurements were used in the study.
Pharmacokinetic and measurement of plasma exenatide analogues by elisa method
Blood samples (200 µL) were taken (the same volume of heparin-treated saline was flushed back) at different time points before and after injections of exenatide analogues, initially via an indwelling cannula up to 24 h and subsequently from gently squeezing of the tail-tip. The blood samples were centrifuged immediately and stored at −20°C until analysis. Plasma concentrations of exenatide and its analogues were determined using a commercially available enzyme-linked immunosorbent assay kits (Phoenix Pharmaceuticals, Inc., Burlingame, CA, USA) that had been validated for determination of exenatide over the concentration range of 0.06–0.68 ng·mL−1. The cross-activity of the assay included exenatide, exendin-3(9–39)NH2 and exendin-4(3–39) (100%), but not human and rat GLP-1(7–36)NH2, GLP-1(7–37) and GLP-2; glucagon; human, rat and mouse oxyntomodulin (0%). The activity signal was measured with a Microplate Reader and concentrations of samples were determined by comparison with a calibration curve run at the same time. The linear standard curve (0.1–1 ng·mL−1) of exenatide was exactly the same as PB-105 (ExC39), indicating that the replacement of alanine with cysteine at the position of 39 did not affect the immunogenicity of exenatide detected by this assay and the exenatide elisa kit could be used for detection of PB-105. Furthermore, a slight, insignificant right-shift of the standard curve of exenatide by PEGylated PB-105 analogues also indicated that site-specific PEGylation had negligible effects on the immunogenicity of exenatide assayed in a PEG mass-dependent manner and this kit was suitable for determination of plasma concentrations of PB-110 (ExC39PEG5kDa), PB-106 (ExC39PEG20kDa), PB-107 (ExC39PEG30kDa) and PB-108 (ExC39PEG40kDa). The intra-assay coefficient of variation for all experiments was 7.2 ± 0.4% and inter-assay coefficient of variation for all experiments was 8.7 ± 0.9%.
Data calculations and statistical analysis
Sigmoidal concentration (dose)–response curve analysis was performed to calculate four-parameter logistic regression, including the median effective concentration (dose) (EC50 or ED50) and maximum effective dose (Emax), by using the v5.0 GraphPad Prism Program (GraphPad Software Inc., San Diego, CA, USA). Biological half recovery time (t1/2) was calculated by performance of linear regression of the time course curves. Pharmacokinetic parameters were also calculated according to the non-compartmental model using Kinetica Version 5.0 (Thermo Scientific, Inc., Two Rivers, WI, USA). Area under time course curve (AUC) or area above lowered blood glucose versus time course curve (AAC) was calculated from the actual data or extrapolated by AAC = 1/2 ×[(2 × biological half-life value + peak time value) × peak effect value] as noted.
All data are expressed as means ± SEM or 95% confidence intervals. Data were analysed for statistical differences by a two-way anova followed by post hoc Student-Newman-Keuls test. A P-value less than 0.05 was taken to indicate a statistically significant difference between data.
Results
Bioactivities of exenatide and its mutant analogues
Effects of exenatide and its analogues on stimulation of intracellular cAMP in PC12 cells
The stimulant effect of PB-105 (ExC39), an exenatide analogue with a cysteine substituted at the C-terminus, on intracellular cAMP production in PC12 cells was compared with that of exenatide. After incubation for 30 min, exenatide at varying concentrations (10−10–10−6M) stimulated cAMP production in a concentration-dependent manner, with an Emax of 129.4 ± 6.8 pmol·mg−1 protein and an EC50 of 2.3 nM (95% confidence intervals: 1.3–3.8 nM). PB-105 produced the same concentration-dependent production in cAMP, with an Emax of 133.2 ± 7.2 pmol·mg−1 protein and an EC50 of 2.9 nM (95% confidence intervals: 1.7–5.0 nM) (Figure 2A).
Figure 2.
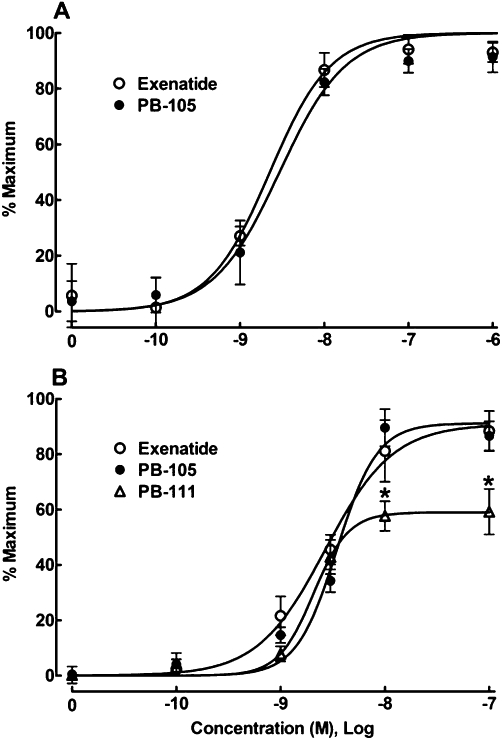
Comparisons of concentration–response curves for PB-105 (ExC39) with exenatide (A) and PB-111 (ExY40) (B) on stimulation of intracellular cAMP in PC12 cells. Concentration–response analysis was best fitted by a non-linear least-squares method. All the readings are means ± SEM of triplicate results, repeated two to three times. *Denotes statistically significant difference compared with either exenatide treatment or PB-105 treatment (P < 0.05 by two-way anova followed by Student-Newman-Keuls test).
PB-111 (ExY40), an analogue of PB-105 with a tyrosine added after position 39 (i.e. position 40), was also compared with PB-105 and exenatide. PB-111 caused a concentration-dependent increase in intracellular cAMP and exhibited the same potency (EC50= 2.1 nM, 95% confidence intervals: 1.5–2.9 nM) as exenatide (2.7 nM, 95% confidence intervals: 1.7–4.2 nM) or PB-105 (3.5 nM, 95% confidence intervals: 2.7–4.5 nM), but its maximal effect was significantly lower than those of either PB-105 or exenatide by 35.0% (P < 0.05 by anova) (Figure 2B).
Effects of exenatide and its analogues on random and fasted blood glucose levels in normal mice
Each of three groups of Swiss mice (n = 6 in each group) received s.c. injections of normal saline (10 mL·kg−1), PB-105 (ExC39, 10 µg·kg−1) or exenatide (10 µg·kg−1). During random blood glucose measurement, mice were allowed free access to food and water. Random blood glucose (representing postprandial sugar) values in saline control mice were in the range of 9.1–11.9 mM and remained stable during the observation period. Both PB-105 and exenatide reduced the random blood glucose levels with a comparable time-dependency, the peak effect occurred after approximately 1 h and the recovery time was approximately 8 h after injection (Figure 3A). The biological half recovery life (t1/2) values of PB-105 and exenatide were 4.4 ± 0.2 h and 4.7 ± 0.2 h, and the decreases were 44.1 ± 8.9% and 45.1 ± 3.4% of initial values. The glucoregulatory activity of exenatide was consistent with previous reports (Young et al., 1999; Hargrove et al., 2007). On the other hand, each of the three groups of mice deprived of food for 16 h but with free access to water (fasted groups; n = 5 for each group) also received s.c. injections of saline, exenatide (10 µg·kg−1) or PB-105 (10 µg·kg−1). As shown in Figure 3B, blood glucose levels in control mice deprived of food dropped to 3.2–7.3 mM and remained stable during the observation period. Neither exenatide nor PB-105 further lowered fasted blood glucose levels at any time point observed.
Figure 3.
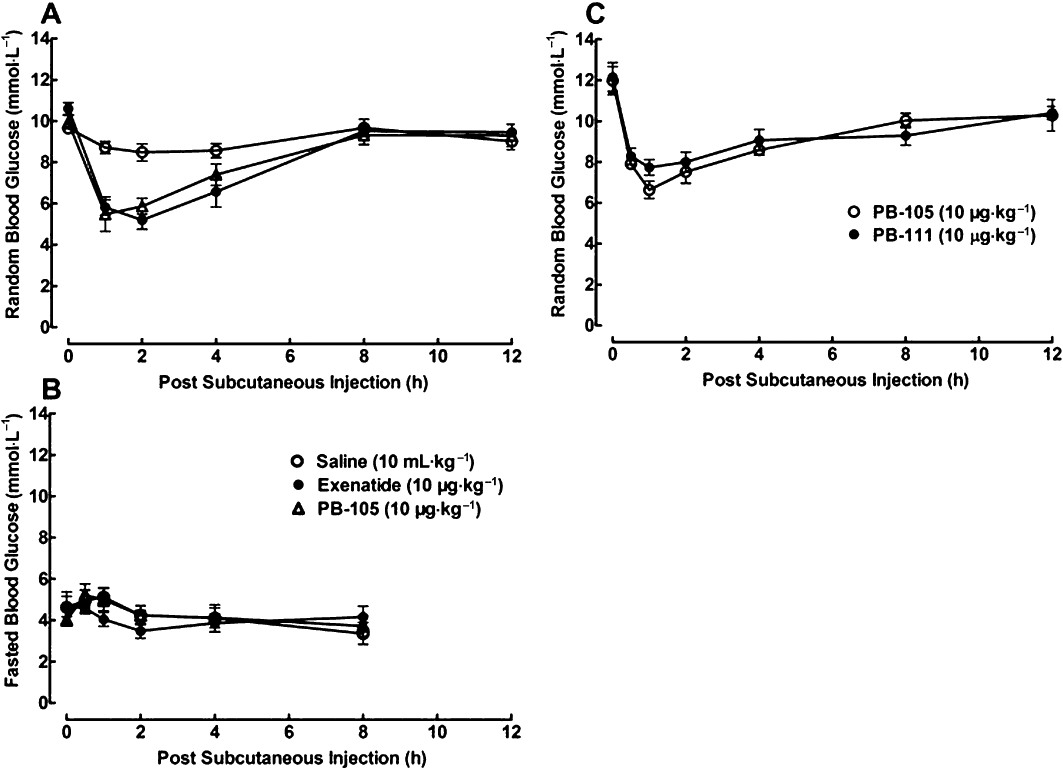
Time courses for the effects of exenatide, PB-105 (ExC39) and PB-111 (ExY40) on random and fasted blood glucose in normal Swiss mice. Data are presented as means ± SEM. (A) Three groups of non-fasted mice received single bolus s.c. injections of normal saline (10 mL·kg−1), PB-105 (10 µg kg−1) or exenatide (10 µg·kg−1); n = 8 in each group. (B) Three groups of fasted (over 16 h) mice received s.c. bolus injections of normal saline (10 mL·kg−1), exenatide (10 µg·kg−1) or PB-105 (10 µg·kg−1); n = 5 in each group. (C) Two groups of non-fasted mice received single bolus s.c. injections of PB-105 (10 µg·kg−1) and PB-111 (10 µg·kg−1) (dosing volume of 10 mL·kg−1); n = 6 in each group.
The time course for the ability of PB-111 (ExY40) to reduce random blood glucose was also compared with PB-105. Each of two groups of non-fasted mice (n = 6 in each group) received s.c. injections of PB-105 (10 µg·kg−1) or PB-111 (10 µg·kg−1). Both PB-105 and PB-111 reduced blood sugar with a comparable time-dependency; peak time at approximately 1 h and recovery time approximately 8 h after injection (Figure 3C). The t1/2 values of PB-105 and PB-111 were 4.6 ± 0.5 h and 6.0 ± 0.8 h, and the peak effect of PB-111 (36.4 ± 3.1% of initial values) was slightly and statistically insignificantly smaller than that of PB-105 (44.7 ± 3.6% of initial value) by 18.6%.
Dose–response curves for the effects of PB-105 (ExC39) and exenatide on random blood glucose were determined for normal non-fasted mice. Sixteen groups of mice (n = 18 in each group) received s.c. injections of saline or PB-105 or exenatide at doses of 0.01–100 µg·kg−1 and the peak blood sugar values were obtained 1 h after injection. Both exenatide and PB-105 caused a dose-dependent reduction in random blood glucose (Figure 4A), with roughly the same maximal reductions of 33.3% and 37.5% of initial values, and ED50 values of 0.6 µg·kg−1 (95% confidence intervals: 0.4–1.2 µg·kg−1) and 1.2 µg·kg−1 (95% confidence intervals: 0.5–3.2 µg·kg−1).
Figure 4.
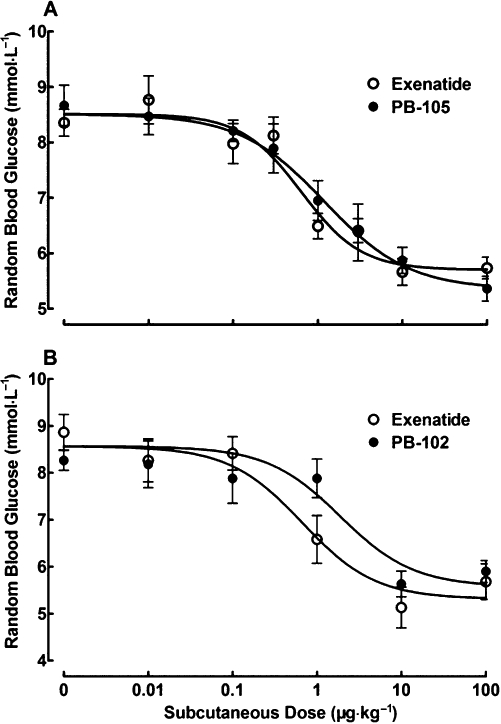
Dose-dependent comparisons of exenatide with PB-105 (ExC39) (A) and PB-102 (ExC35) (B) on random blood glucose in normal Swiss mice. Data are presented as means ± SEM. (A) Sixteen groups of non-fasted mice received a single bolus s.c. injection of normal saline or PB-105 or exenatide at the doses indicated. The peak blood sugar values were obtained 1 h after injection; n = 18 in each group. (B) Twelve groups of non-fasted mice received single bolus s.c. injections of normal saline, exenatide or PB-102 as indicated. The peak blood sugar values were obtained 1 h after injection; n = 6 in each group.
The dose–response curve for PB-102 (ExC35), an exenatide analogue with a cysteine substituted at the position 35, was also compared with that of exenatide. Twelve groups of non-fasted mice (n = 6 in each group) received s.c. injections of saline, exenatide or PB-102 at doses of 0.01–100 µg·kg−1 and the peak blood sugar values were obtained at 1 h after injection. Both exenatide and PB-102 produced a dose-dependent reduction of random blood sugar (Figure 4B), with maximal effects of 37.9% and 34.6% of initial values. PB-102 was less potent (but not significantly) than exenatide, with ED50 values of 0.7 µg·kg−1 (95% confidence intervals: 0.2–2.1 µg·kg−1) and 2.0 µg·kg−1 (95% confidence intervals: 0.6–6.5 µg·kg−1).
Pharmacokinetics of exenatide and PB-105 (ExC39) in rats
Two groups of cannulated rats received i.v. injections of exenatide or PB-105 (ExC39) at a dose of 5 µg·mL−1·kg−1. After the i.v. injection, both plasma exenatide and PB-105 first rapidly decreased in an exponential fashion in the distribution phase followed by a slow-decay elimination phase (Figure 5A). Pharmacokinetic parameters were calculated using the non-compartment model. Exenatide and PB-105 had elimination phase half-life values of 4.8 ± 0.7 and 4.9 ± 1.0 h respectively. Other pharmacokinetic parameters are also listed in Figure 5B.
Figure 5.
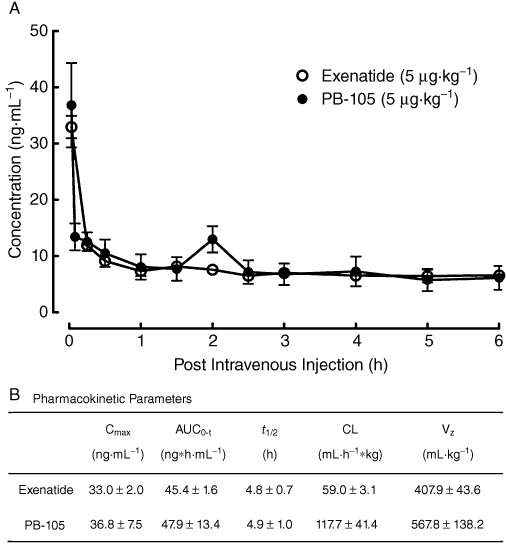
Pharmacokinetic profiles (A) and pharmacokinetic parameters (B) of exenatide and PB-105 (ExC39) in normal Sprague-Dawley rats. Two groups of cannulated rats received i.v. injections of exenatide or PB-105 at a dose of 5 µg·mL−1·kg−1. Blood samples (200 µL) were taken (replaced by the same volume of heparin-treated saline) at different time points as indicated. Data are presented as means ± SEM; n = 3 in each group. Pharmacokinetic parameters were calculated by using a non-compartment model.
Effects of PB-105 (ExC39) and its PEGylated analogues on the stimulation of intracellular cAMP in PC12 cells
Concentration–response curves for the stimulant effects of PB-105 (ExC39) and its PEGylated analogues with different molecular weights on intracellular cAMP were obtained in PC12 cells. As shown in Figure 6A, PB-105 increased intracellular cAMP in a concentration-dependent manner, with an EC50 of 1.4 nM (95% confidence intervals: 1.2–1.6 nM) and Emax of 103.9 ± 1.5 pmol·mg−1 protein, consistent with the above results. PEGylation with PEGs of up to at least 40 kDa did not affect PB-105's maximal production of intracellular cAMP. However, PEGylation shifted the concentration–response curve of PB-105 to the right in a parallel, PEG mass-dependent manner but with an inflexion point of at least PEF20kDa. EC50 values of PB-110 (ExC39PEG5kDa), PB-106 (ExC39PEG20kDa), PB-107 (ExC39PEG30kDa) and PB-108 (ExC39PEG40kDa) were 1.3 nM (95% confidence intervals: 1.0–1.8 nM), 1.2 nM (95% confidence intervals: 0.9–1.5 nM), 14.2 nM (95% confidence intervals: 9.9–20.4 nM) and 114.1 nM (95% confidence intervals: 89.7–145.2 nM). The relationship between their PEG mass and log EC50 values is exhibited in Figure 6B and PEG 20 kDa (at least) was shown to be an inflexion point whereas PB-110 and PB-106 did not affect PB-105's potency, while PB-107 and PB-108 displaced PB-105's potency by approximately 10- and 100-fold respectively.
Figure 6.
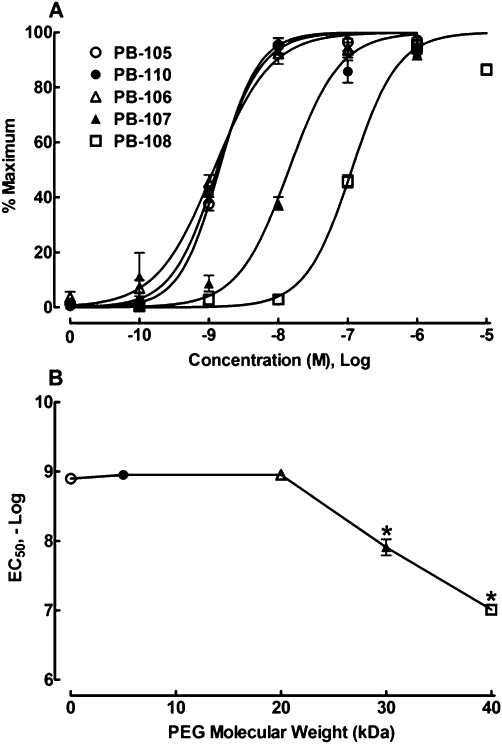
Concentration–response curves for the effect of PB-105 (ExC39) and its PEGylated analogues on intracellular cAMP in PC12 cells (A) and the relationship between PEG mass (kDa) and potencies of PB-105 and its PEGylated analogues (B). PEGylated analogues conjugated with different molecular masses of PEGs: PB-110 (ExC39PEG5kDa) with PEG5kDa, PB-106 (ExC39PEG20kDa) with PEG20kDa, PB-107 (ExC39PEG30kDa) with PEG30kDa and PB-108 (ExC39PEG40kDa) with PEG40kDa. Concentration–response analysis was best fitted by a non-linear least-squares method. All the readings are means ± SEM of triplicate results, repeated two to three times. *Denotes statistically significant difference compared with PB-105 treatment (P < 0.05 by a two-way anova followed by post hoc Student-Newman-Keuls test).
Effects of PB-105 (ExC39) and its PEGylated analogues on random blood glucose in normal and diabetic mice
In order to compare the biological durations of PB-105 (ExC39) and its PEGylated analogues, five groups of non-fasted normal mice (n = 12 in each group except for PB-110 group where n = 6) received s.c. injections of PB-105, PB-110 (ExC39PEG5kDa), PB-106 (ExC39PEG20kDa), PB-107 (ExC39PEG30kDa) or PB-108 (ExC39PEG40kDa) at a dose of 10 µg·kg−1. As shown in Figure 7A, PB-105 produced a reversible reduction in blood glucose the same as shown in Figure 3A, with a peak of 38.0 ± 4.3% of initial values 1 h after injection and biological t1/2 of 4.9 ± 0.1 h. PEGylation reduced the peak response to PB-105, slowed and prolonged biological duration of PB-105 in a PEG mass-dependent fashion but also had an inflexion point of at least 20 kDa of PEG. The peak values of PB-110, PB-106, PB-107 and PB-108 were 36.3 ± 1.2%, 36.5 ± 3.2%, 24.3 ± 2.8% and 22.6 ± 2.9% of initial values with a peak time of approximately 1 h (PB-110) and 4 h (PB-106, PB-107 and PB-108); biological t1/2 values were 7.0 ± 2.0, 13.4 ± 0.5, 12.5 ± 2.0, 10.8 ± 2.0 and 9.2 ± 2.2 h. The relationship between PEG mass and the peak reduction in random blood sugar induced by PEGylated PB-105 analogues as well as biological duration is shown in Figure 7B and C, indicating that at least PEG 20 kDa was an inflexion point where PB-106 prolonged PB-105's biological duration most without reducing its glucoregulatory activity. To evaluate the overall effect on random blood glucose, the AAC was used. The AAC value for PB-105 was 322.3 ± 57.4 mmol*h·L−1, which was significantly increased by PB-106 by 61.1%, but not by PB-110, PB-107 or PB-108 (Figure 7D).
Figure 7.
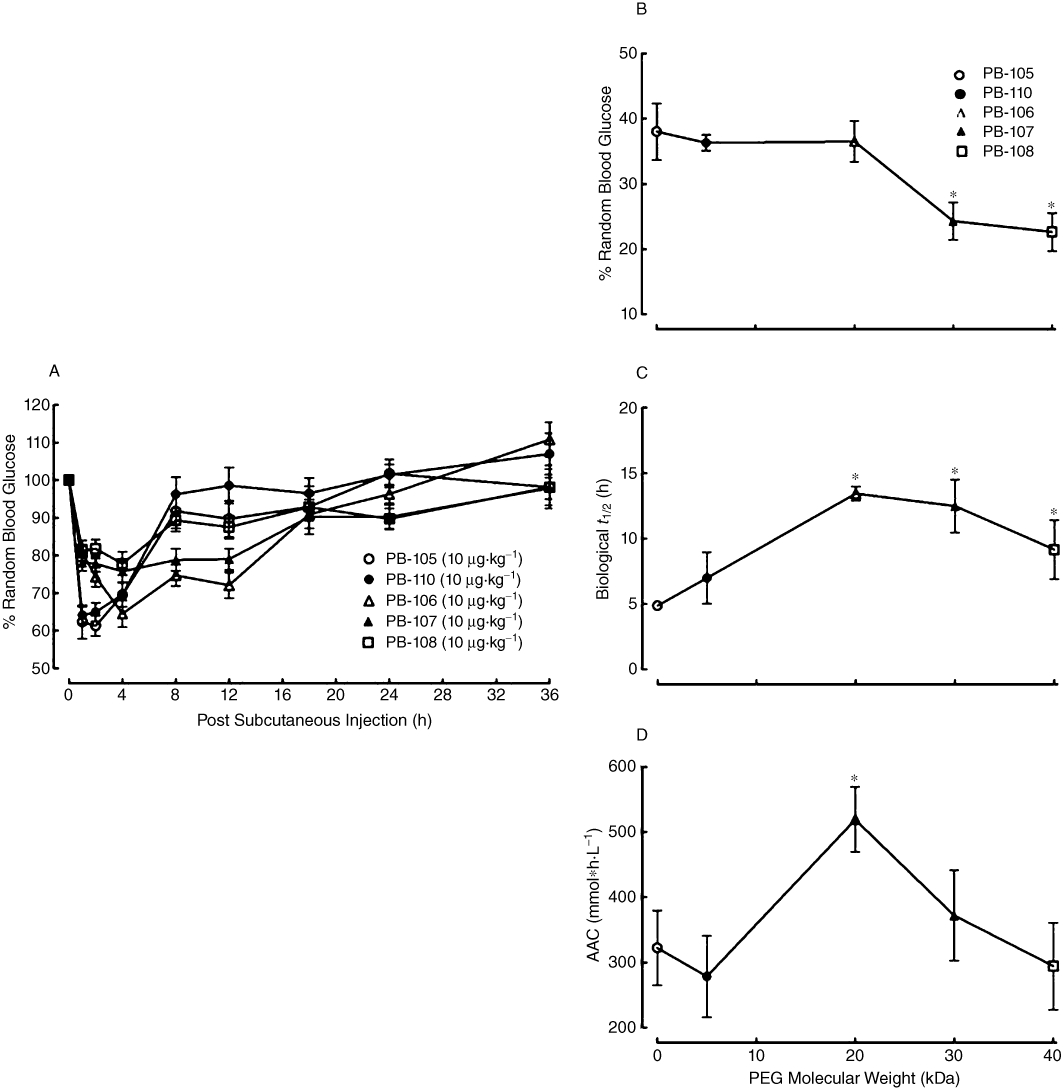
Time courses for the effects of PB-105 (ExC39) and its PEGylated analogues on random blood sugar levels in normal mice (A), and the correlation between PEG molecular weight and the maximal lowering effect (Emax) (B), biological half-life (t1/2) (C) and area above time course curve (AAC) (D). PEGylated analogues conjugated with different molecular masses of PEGs: PB-110 (ExC39PEG5kDa) with PEG5kDa, PB-106 (ExC39PEG20kDa) with PEG20kDa, PB-107 (ExC39PEG30kDa) with PEG30kDa and PB-108 (ExC39PEG40kDa) with PEG40kDa. Six groups of non-fasted normal mice received bolus s.c. injections of PB-105, PB-110, PB-106, PB-107 or PB-108 at a dose of 10 µg·kg−1 (dosing volume: 10 mL·kg−1). Data are presented as means ± SEM; n = 12 in each group except for the PB-110 group where n = 6. *Denotes statistically significant difference compared with PB-105 group (P < 0.05 by a two-way anova followed by post hoc Student-Newman-Keuls test).
Dose–response curves for the effects of PB-105 (ExC39) and PB-106 (ExC39PEG20kDa) on random blood sugar were further compared. Thirteen groups of normal non-fasted mice (n = 6 in each group) each received s.c. injections of normal saline, PB-105 or PB-106 at doses ranging from 0.1 to 30 µg·kg−1. Blood glucose levels were assessed 1 h (saline group and PB-105 groups) or 4 h (PB-106 groups) after administration. Both peptides reduced blood sugar in a dose-dependent manner (Figure 8A). Maximal falls in blood glucose with each peptide approached 33.5% and 38.4% of initial values. ED50 values of PB-105 and PB-106 were 1.1 µg·kg−1 (95% confidence intervals: 0.5–2.6 µg·kg−1) and 3.0 µg·kg−1 (95% confidence intervals: 1.2–9.3 µg·kg−1). AACs were extrapolated by the formula of AAC = 1/2 × [(2 × biological half-life value + peak time value) × peak effect value], where mean biological t1/2 values of 4.9 h for PB-105 and 13.4 h for PB-106, respectively, were derived from Figure 7A, and were selected to integrate biological durations and magnitudes of reduction. As shown in Figure 8B, PB-105 lowered blood sugar in a dose-dependent manner, with an ED50 value of 1.8 µg·kg−1 (95% confidence intervals: 1.1–2.8 µg·kg−1) and an AAC Emax of 11.6 ± 0.9 (mmol*h L−1). PB-106 also dose-dependently lowered blood sugar with an ED50 value of 4.5 µg·kg−1 (95% confidence intervals: 1.9–10.7 µg·kg−1) and an AAC Emax of 37.8 ± 7.1 (mmol*h L−1), which was 3.2-fold that of PB-105's potency (P < 0.05).
Figure 8.
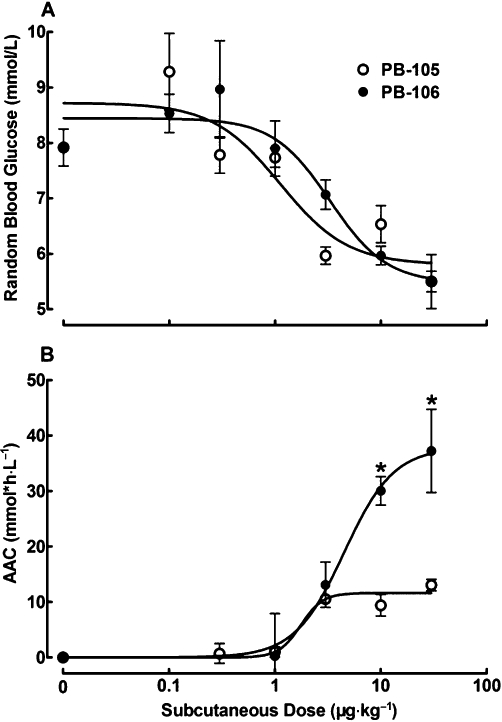
Dose–response curves for the effects of PB-105 (ExC39) and PB-106 (ExC39PEG20kDa) on random blood glucose levels (A) and areas above curves (AACs, B) in normal Swiss mice. Thirteen groups of non-fasted mice received bolus s.c. injections of normal saline, PB-105 or PB-106 at the doses indicated. Blood glucose levels were assessed 1 h (normal saline group and PB-105 groups) or 4 h (PB-106 groups) after administration. (B) AACs were selected to integrate biological durations and drop magnitudes and were extrapolated by the formula AAC = 1/2 ×[(2 × biological half-life value + peak time value) × peak effect value], where the mean biological t1/2 values of 4.9 h for PB-105 and 13.4 h for PB-106, respectively, were derived from (A). Data are presented as means ± SEM; n = 6 in each group. *Denotes statistically significant difference compared with PB-105 group (P < 0.05 by two-way anova followed by Student-Newman-Keuls test).
The lowering of blood sugar by exenatide, PB-105 (ExC39) and PB-106 (ExC39PEG23kDa) were further tested in diabetic mice. Swiss mice developed diabetes with random blood sugar ranging from 16.7 to 44.4 mmol·L−1 3 days after s.c. injection of STZ (120 mg·kg−1). Three groups of STZ-induced non-fasted diabetic mice (n = 7–11 in each group) each received s.c. injections of exenatide, PB-105 or PB-106 at a dose of 10 µg·kg−1. As shown in Figure 9A, exenatide and PB-105 exhibited reversible lowering of blood glucose, with similar peak drops in random blood glucose (37.7 ± 5.7% of initial values for exenatide vs. 40.4 ± 8.8% of initial values for PB-105) and biological duration (t1/2 of 6.1 ± 0.6 h for exenatide vs. 5.6 ± 0.3 h for PB-105), close to the results for normal mice suggesting the diabetic phenotype did not affect the ability of thee peptides to decrease blood glucose. Compared with exenatide or PB-105, PEG-106 produced a larger, but not statistically significant, decrease in random blood sugar of 55.1 ± 6.4% and a longer biological duration (t1/2 of 23.0 ± 1.3 h). To integrate the overall glucoregulatory activity with biological duration and magnitude of reductions in blood sugar, the calculated AACs were selected for a comparison. PB-106 produced profound glucoregulatory effects (P < 0.05 vs. PB-105 or exenatide), which were 5.9-fold and 4.6-fold those of exenatide and PB-105 respectively (Figure 9B).
Figure 9.
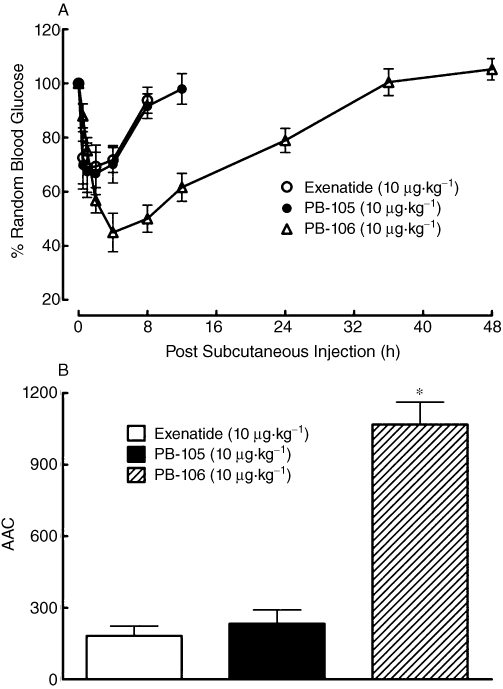
Time courses for the effects of exenatide, PB-105 (ExC39) and PB-106 (ExC39PEG20kDa) on random blood glucose levels expressed as fall in magnitude (A) and areas above curves (AACs, B) in streptozotocin (STZ)-induced diabetic mice. These mice developed diabetes with initial random blood sugar ranging from 16.7 to 44.4 mmol·L−1 3 days after s.c. injection of STZ (120 mg·kg−1). Data are presented as means ± SEM; n = 7–11 in each group. *Denotes statistically significant difference compared with either exenatide group or PB-105 group (P < 0.05 by anova followed by Student-Newman-Keuls test).
Pharmacokinetics of PB-105 (ExC39) and its PEGylated analogues in normal rats
Five groups of normal Sprague-Dawley rats (n = 3 in each group) each received bolus i.v. injections of PB-105 (ExC39) or its PEGylated analogues at a dose of 5 µg·mL−1·kg−1. Figure 10A illustrates plasma concentration profiles of these peptides during scheduled experimental periods. Pharmacokinetic parameters calculated by using a non-compartment model are listed in Figure 10B. Unmodified PB-105 was rapidly removed from the circulation first in an exponential fashion in the distribution phase followed by a slow-decay elimination phase, with an elimination phase t1/2 of 2.9 ± 0.1 h, whereas PEGylated PB-105 analogues were found to have extended plasma lifetimes with increasing PEG molecular weights from 5 to 40 kDa. Specifically, the plasma t1/2 values of PB-110 (ExC39PEG5kDa), PB-106 (ExC39PEG20kDa), PB-107 (ExC39PEG30kDa) and PB-108 (ExC39PEG40kDa) were linearly increased to 6.1 ± 0.8, 49.4 ± 7.7, 70.5 ± 2.6 and 76.4 ± 7.4 h (Figure 10C). PEGylation also linearly increased the AUC of PB-105 with an increasing in PEG mass (Figure 10D), whereas the apparent volume of distribution (Vz) and systemic clearance (CL) of PB-105 were reduced in a PEG size-dependent fashion.
Figure 10.
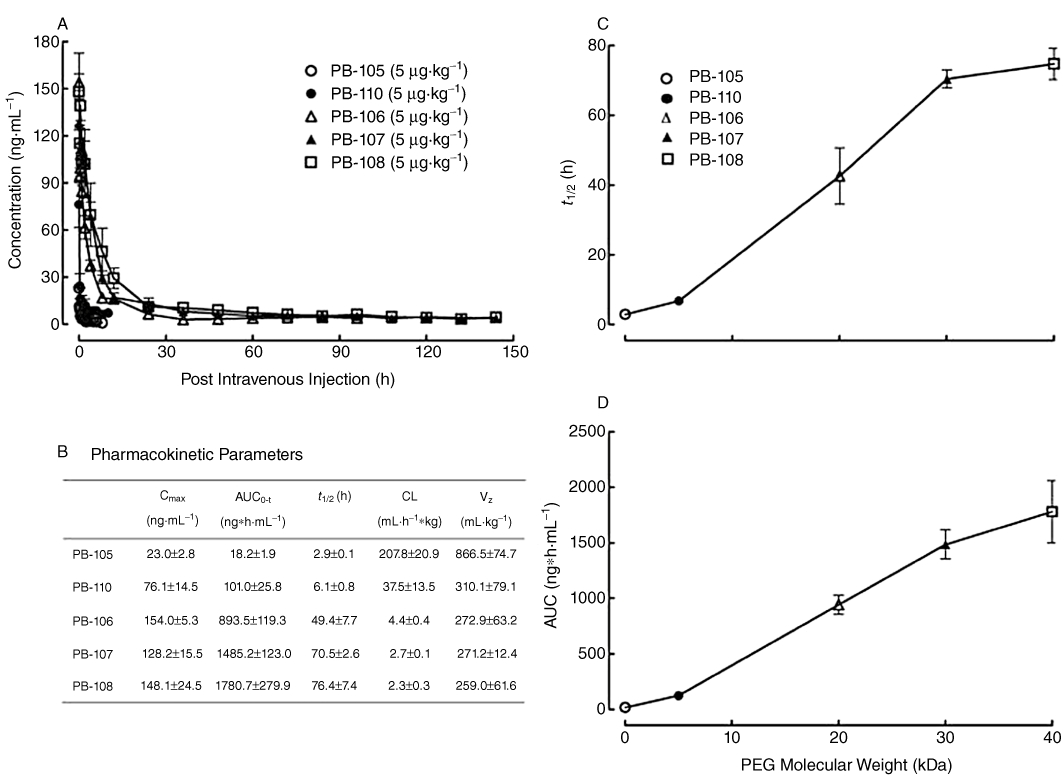
Pharmacokinetic profiles (A) and parameters (B) of PB-105 and its PEGylated analogues in normal Sprague-Dawley rats, and the correlation between PEG mass (kDa) and plasma half-life (t1/2) (C) and systemic exposure (AUC) (D). PEGylated analogues conjugated with different molecular masses of PEGs: PB-110 (ExC39PEG5kDa) with PEG5kDa, PB-106 (ExC39PEG20kDa) with PEG20kDa, PB-107 (ExC39PEG30kDa) with PEG30kDa and PB-108 (ExC39PEG40kDa) with PEG40kDa. Five groups of cannulated rats received bolus i.v. injections of PB-105 or its PEGylated analogues at a dose of 5 µg·mL−1·kg−1. Blood samples (200 µL) were taken (replaced by the same volume of heparin-treated saline) at different time points ranging from 0.03 h to 6 h (PB-105), 10 h (PB-110), 96 h (PB-106), 144 h (PB-107) and 168 h (PB-108) post administration. Data are presented as means ± SEM; n = 3 in each group. Pharmacokinetic parameters were calculated by using a non-compartmental model.
Discussion
In contrast to the intact amino acid sequence from 1 through to 8 (H-His-Gly-Glu-Gly-Thr-Phe-Thr-Ser) of the N-terminal is required for exenatide to bind GLP-1 receptors and maintain its biological activity (Lopez de Maturana et al., 2003; Mann et al., 2007), its extension of nine amino acid residues (Leu 21-Pro 38), known as the ‘Trp cage’ of exendin-4, was reported to be negligibly involved in GLP-1 receptor binding (Neidigh et al., 2001; Runge et al., 2007; 2008;). Our data support this notion and show that replacement of alanine with cysteine at position of 35 of exenatide (PB-102, ExC35) slightly, but not significantly, reduced the potency of exenatide.
The importance of the serine residue at position 39 in the binding of the compound to GLP-1 receptors has not been studied, although it was found that deletion of the last three amino acid residues slightly, but significantly, reduced the GLP-1 receptor binding affinity (Doyle et al., 2003). We extensively studied this and found that (i) both PB-105 (ExC39) and exenatide had the same potency and efficacy at increasing the production of intracellular cAMP; (ii) both PB-105 and exenatide displayed the same acute reduction of random blood sugar but not fasting blood sugar in normal and diabetic mice, induced by comparing their biological durations and dose–response curves; and (iii) both peptides also exhibited the same pharmacokinetic profiles, including plasma duration, systemic exposure, volume of distribution and systemic clearance. Thus, substitution of serine with cysteine at position 39 not only preserves the activity of exenatide, but also provides a free thiol group for conjugation. PB-105 was therefore selected as an active intermediate for further site-specific PEGylation. PB-105 would be expected to keep the same or have better glucoregulatory properties in humans, as it was recently reported that the C-terminal region of exenatide is more important for binding to rat GLP-1 receptors than to human GLP-1 receptors (Mann et al., 2010).
Random PEGylation of interferon (Bailon et al., 2001) and of granulocyte colony-stimulating factor (Bowen et al., 1999) has been shown to impair their in vitro activity in an exponential manner, starting with as little as 4 kDa of PEGs. The typical ‘Exponential-Shift’ of bioactivity by random PEGylation usually means the doses needed to produce a particular effect have to be increased, which possibly results in more adverse effects. On the other hand, site-specific PEGylation with proteins of large molecular size, particularly antibody fragments, usually results in no activity loss up to PEG50kDa in a ‘No-Shift’ manner (Chapman et al., 1999; Chapman, 2002; Bailon and Won, 2009). For example, site-specific PEGylated Fab′ with PEG molecule of 40 kDa was shown to preserve full binding activity, in contrast to random PEGylation resulting in 50% loss of binding activity (Chapman et al., 1999; Chapman, 2002).
However, in the present study a new type of ‘Inflexion-Shift’ by site-specific PEGylation of small peptides was demonstrated. We found that site-specific PEGylation did not reduce either PB-105's intracellular cAMP activity or its acute glucoregulatory activity until PEGs reached at least 20 kDa in molecular mass. The feature of ‘Inflexion-Shift’ by site-specific PEGylation with permanent (not cleavable) linkage to the PEG molecule is particularly important for small peptides of a few kDa in molecular size such as exenatide, as it is generally believed that cleavable linkages should be used for these peptides or small organic compounds (Bailon and Won, 2009).
Our study showed that PEGylation greatly improved the poor pharmacokinetic profile of exenatide by increasing the elimination phase t1/2 and AUC, as well as reducing Vz and CL, in a PEG mass-dependent manner. Of the PEGylated PB-105 analogues studied, PB-106 (ExC39PEG20kDa) was found to have a plasma half-life of 43 h, approximately 10-fold higher than exenatide. Increased effective molecular size has two contrary effects on biological activity and metabolic stability. Thus selection of PEG molecular weight should be based on a balance between both factors, preferably increasing plasma duration and systemic exposure without sacrificing intrinsic biological activity. PB-106, with its increased plasma half-life and preserved in vitro activity and in vivo glucoregulatory activity (expressed as the drop magnitude compared with initial values) would be expected to be effective in stabilizing postprandial glycaemic challenges.
PB-106 was actually 2.2-fold more effective than PB-105 in postprandial random glucoregulation when the increased plasma duration and drop in magnitude of blood glucose were integrated (expressed as the area above the time course curves). The enhancement of glucoregulation by PEGylation was amplified in diabetic mice, where PB-106 was roughly 3.4- and 4.9-fold more superior to exenatide or PB-105. The excellent efficacy of PB-106 along with its improved pharmacokinetic profile suggests that this exenatide analogue could be useful in the long-term management of diabetes. It is known that long-term treatment by twice daily exenatide is used to successfully manage type 2 diabetic animals and humans leading to a reduction of blood HbA1c (a measurement of prior 3 month blood glucose level) as well as body weight and food intake (which are beneficial effects for diabetes patients) (Greig et al., 1999; Szayna et al., 2000). PB-106's increases in acute glucoregulatory effects in both normal mice (twofold to threefold superior) and diabetic mice (fourfold to fivefold superior) relative to exenatide or PB-105 translated to its long-term effects in GK diabetic rats. This was demonstrated by showing that PB-119 (ExC39PEG23kDa), another PEGylated (PEG23kDa) analogue of PB-105 (ExC39) administered once every 2 days was as equally effective as twice-a-day exenatide or PB-105 in reducing blood HbA1c and body weight as well as food intake in GK diabetic rats (N. Gong et al., unpubl. data).
Our results showed that exenatide and PB-105 produced an acute and reversible glucoregulatory effect, consistent with previous reports (Young et al., 1999; Hargrove et al., 2007). The acute glucoregulatory activity of exenatide and PB-105 observed had the following characteristics: (i) although the maximal reduction in random blood glucose (representing postprandial sugar) was greater in STZ diabetic mice, that were initially hyperglycaemic, than in normal mice with normal blood glucose, the magnitude of the reduction in blood glucose was independent of the initial random blood sugar levels and was roughly 35–45% of initial values, as demonstrated by the same peak effect in normal Swiss mice (the present study) and C57/BL/6 mice (Hargrove et al., 2007), STZ-induced diabetic mice (the present study), db/db diabetic mice (Young et al., 1999; Hargrove et al., 2007) and ob/ob diabetic mice (Young et al., 1999), and in diabetic rhesus monkeys (Young et al., 1999). (ii) This effect is species-specific, as acute administration of exenatide or PB-105 were effective in mice (the present study; Young et al., 1999; Hargrove et al., 2007) and monkeys (Young et al., 1999) but did not reduce random blood sugar levels in normal Wistar rats or GK diabetic rats (N. Gong et al., unpubl. data). Acute but not chronic administration of exenatide has been shown to exhibit a paradoxical increase in glucose in rats (but not in mice) probably through activation of the sympathetic nervous system (Malendowicz et al., 2001; Perez-Tilve et al., 2010). (iii) Neither exenatide nor PB-105 produced an acute regulatory effect in the fasted mice, similar to previous studies (Nauck et al., 1993; Qualmann et al., 1995; Baggio et al., 2000). This is due to the strict glucose-dependence of the insulinotropic effect of GLP-1 that limits the amount of insulin secreted when high doses of GLP-1 or similarly acting analogues are administered at fasted plasma glucose concentrations, demonstrated in normal or type 2 diabetic animals (Young et al., 1999) and diabetic mellitus humans (Kolterman et al., 2003; Kendall et al., 2005). (iv) Acute glucoregulatory effects (magnitudes) of exenatide and PB-105 can be integrated with their biological durations and expressed as area above the curve, which more precisely predicts their long-term anti-diabetic properties. Thus the mouse acute blood glucose measurement might provide a simple, reliable and predictable assay for reflecting the glucoregulatory nature of the GLP-1 system in normal and diabetic conditions, and might be useful for screening for GLP-1 receptor agonists.
In summary, site-specific PEGylation shifted the concentration–response curve of PB-105 (ExC39) to the right in a parallel, PEG mass-dependent manner but had an inflexion point of at least PEG20kDa. PEGylation also affected acute in vivo glucoregulatory activity (the magnitude) in an ‘Inflexion-Shift’ fashion at PEG20kDa (at least), but linearly increased plasma duration and overall systemic exposure in a PEG mass-dependent manner without an inflexion point. PB-106 (ExC30PEG20kDa) had a plasma t1/2 of 43 h, approximately 10-fold that of PB-105 or exenatide. PB-106 was also found to exhibit much better glucoregulatory activity compared with exenatide or PB-105, twofold to threefold better in normal mice and fourfold to fivefold in diabetic mice. Our results show that site-specific PEGylation of small peptides in permanent amide linkage affects their activity in a new type of ‘Inflexion-Shift’ fashion, in contrast to the previously demonstrated ‘Exponential-Shift’ pattern in random PEGylation or the ‘No-Shift’ pattern in site-specific PEGylation with protein drugs of large molecular mass. PB-106 was designed to be a putative new molecular entity for treating diabetes, with no loss of in vitro activity but prolonged plasma duration and consequently markedly improved in vivo glucoregulatory activity.
Acknowledgments
This work is financially supported by the Mega New Drug Development Program of China (Grant No: 2009ZX09102-257) and a Shanghai Jiao Tong University School of Pharmacy Predoctoral Fellowship to N.G. We thank Dr Qing-Shan Zheng at Shanghai University of Chinese Medicine (Shanghai, China) for discussion on data calculation and statistics and Dr George Miljanich at Airmid Inc. (Redwood City, CA, USA) for editing the manuscript.
Glossary
Abbreviations
- AAC
area above lowering blood glucose time course curve
- AUC
area under time course curve
- GLP-1
glucagon-like peptide-1
- PEG
polyethylene glycol
- STZ
streptozotocin
- t1/2
half recovery time
Conflict of interest
LJZ, XSL, YHZ and MX are employees of PegBio Co. Ltd. that is developing PEGylated exenatide analogues for the treatment of type 2 diabetes and financially sponsored the study, in part.
Supporting Information
Teaching Materials; Figs 1–10 as PowerPoint slide.
References
- Baggio L, Kieffer TJ, Drucker DJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, regulates fasting glycemia and nonenteral glucose clearance in mice. Endocrinology. 2000;141:3703–3709. doi: 10.1210/endo.141.10.7720. [DOI] [PubMed] [Google Scholar]
- Bailon P, Won CY. PEG-modified biopharmaceuticals. Expert Opin Drug Deliv. 2009;6:1–16. doi: 10.1517/17425240802650568. [DOI] [PubMed] [Google Scholar]
- Bailon P, Palleroni A, Schaffer CA, Spence CL, Fung WJ, Porter JE, et al. Rational design of a potent, long-lasting form of interferon: a 40 kDa branched polyethylene glycol-conjugated interferon alpha-2a for the treatment of hepatitis C. Bioconjug Chem. 2001;12:195–202. doi: 10.1021/bc000082g. [DOI] [PubMed] [Google Scholar]
- Bowen S, Tare N, Inoue T, Yamasaki M, Okabe M, Horii I, et al. Relationship between molecular mass and duration of activity of polyethylene glycol conjugated granulocyte colony-stimulating factor mutein. Exp Hematol. 1999;27:425–433. doi: 10.1016/s0301-472x(98)00051-4. [DOI] [PubMed] [Google Scholar]
- Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylureatreated patients with type 2 diabetes. Diabetes Care. 2004;27:2628–2635. doi: 10.2337/diacare.27.11.2628. [DOI] [PubMed] [Google Scholar]
- Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly(ethylene glycol) protein conjugates. Adv Drug Del Rev. 2003;55:1261–1277. doi: 10.1016/s0169-409x(03)00108-x. [DOI] [PubMed] [Google Scholar]
- Chapman AP. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv Drug Deliv Rev. 2002;54:531–545. doi: 10.1016/s0169-409x(02)00026-1. [DOI] [PubMed] [Google Scholar]
- Chapman AP, Antoniw P, Spitali M, West S, Stephens S, King DJ. Therapeutic antibody fragments with prolonged in vivo half-lives. Nat Biotechnol. 1999;17:780–783. doi: 10.1038/11717. [DOI] [PubMed] [Google Scholar]
- Copley K, McCowen K, Hiles R, Nielsen LL, Young A, Parkes DG. Investigation of exenatide elimination and its in vivo and in vitro degradation. Curr Drug Metab. 2006;7:367–374. doi: 10.2174/138920006776873490. [DOI] [PubMed] [Google Scholar]
- Cuddihy R, Rosenstock J, Henry R, Alessi T, Luskey K. Comparing ITCA 650, continuous subcutaneous delivery of exenatide via DUROS® device, vs. twice daily exenatide injections in metformin-treated type 2 diabetes. 2010. European Association for the Study of Diabetes (EASD) 46th Annual Meeting: Abstract 78.
- DeFronzo R, Ratner R, Han J, Kim D, Fineman M, Baron A. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. 2005;28:1083–1091. doi: 10.2337/diacare.28.5.1092. [DOI] [PubMed] [Google Scholar]
- Doyle ME, Theodorakis MJ, Holloway HW, Bernier M, Greig NH, Egan JM. The importance of the nine-amino acid C-terminal sequence of exendin-4 for binding to the GLP-1 receptor and for biological activity. Regul Pept. 2003;114:153–158. doi: 10.1016/s0167-0115(03)00120-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards CMB, Stanley SA, Davis R, Brynes AE, Frost GS, Seal LJ, et al. Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. Am J Physiol Endocrinol Metab. 2001;281:155–161. doi: 10.1152/ajpendo.2001.281.1.E155. [DOI] [PubMed] [Google Scholar]
- Egan JM, Clocquet AR, Elahi D. The insulinotropic effect of acute exendin-4 administered to humans: comparison of nondiabetic state to type 2 diabetes. J Clin Endocrinol Metab. 2002;87:1282–1290. doi: 10.1210/jcem.87.3.8337. [DOI] [PubMed] [Google Scholar]
- Egan JM, Meneilly GS, Elahi D. Effects of 1-mo bolus subcutaneous administration of exendin-4 in type 2 diabetes. Am J Physiol Endocrinol Metab. 2003;284:E1072–E1079. doi: 10.1152/ajpendo.00315.2002. [DOI] [PubMed] [Google Scholar]
- Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267:7402–7405. [PubMed] [Google Scholar]
- Fabunmi R, Nielsen LL, Quimbo R, Schroeder B, Misurski D, Wintle M, et al. Patient characteristics, drug adherence patterns, and hypoglycemia costs for patients with type 2 diabetes mellitus newly initiated on exenatide or insulin glarging. Curr Med Res Opin. 2009;25:777–786. doi: 10.1185/03007990802715199. [DOI] [PubMed] [Google Scholar]
- Farilla L, Hui H, Bertolotto C, Kang E, Bulotta A, Di Mario U, et al. Gluagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. 2002;143:4397–4408. doi: 10.1210/en.2002-220405. [DOI] [PubMed] [Google Scholar]
- Fishburn CS. The pharmacology of PEGylation: balancing PD with PK to generate Novel Therapeutics. J Pharm Sci. 2008;97:4167–4183. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- Fontana A, Spolaore B, Mero A, Veronese FM. Site-specific modification and PEGylation of pharmaceutical proteins mediated by transglutaminase. Adv Drug Deliv Rev. 2008;60:13–28. doi: 10.1016/j.addr.2007.06.015. [DOI] [PubMed] [Google Scholar]
- French AC, Thompson AL, Davis BG. High-purity discrete PEG-oligomer crystals allow structural insight. Angew Chem Int Ed Engl. 2009;48:1248–1252. doi: 10.1002/anie.200804623. [DOI] [PubMed] [Google Scholar]
- Gong N, Zhang LJ, Ma AN, Huang JL, Xu M, Wang YX. Pharmacokinetic and pharmacodynamic evaluations of site-specific PEGylated exenatide analogs as long-acting glucoregulatory agents. Chin Pharmacol Bull. 2009;25:140. [Google Scholar]
- Greig NH, Holloway HW, De Ore KA, Jani D, Wang Y, Zhou J, et al. Once daily injection of exendin-4 to diabetic mice achieves long-term beneficial effects on blood glucose concentrations. Diabetologia. 1999;42:45–50. doi: 10.1007/s001250051111. [DOI] [PubMed] [Google Scholar]
- Hargrove DM, Kendall ES, Reynolds JM, Lwin AN, Herich JP, Smith PA, et al. Biological activity of AC3174, a peptide analog of exendin-4. Regul Pept. 2007;141:113–119. doi: 10.1016/j.regpep.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;87:1409–1439. doi: 10.1152/physrev.00034.2006. [DOI] [PubMed] [Google Scholar]
- Kendall DM, Riddle MC, Rosenstock J, Zhuang D, Kim DD, Fineman MS, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care. 2005;28:1083–1091. doi: 10.2337/diacare.28.5.1083. [DOI] [PubMed] [Google Scholar]
- Kim D, MacConell L, Zhuang D, Kothare PA, Trautmann M, Fineman M, et al. Effects of once-weekly dosing of a long-acting release formulation of exenatide on glucose control and body weight in subjects with type 2 diabetes. Diabetes Care. 2007;30:1487–1493. doi: 10.2337/dc06-2375. [DOI] [PubMed] [Google Scholar]
- Kolterman OG, Buse JB, Fineman MS, Gaines E, Heintz S, Bicsak TA, et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. 2003;88:3082–3089. doi: 10.1210/jc.2002-021545. [DOI] [PubMed] [Google Scholar]
- Kolterman OG, Kim DD, Shen L, Ruggles JA, Nielsen LL, Fineman M, et al. Pharmacokinetics, pharmacodynamics, and safety of a range of subcutaneous exenatide (exendin-4) doses in subjects with type 2 diabetes. Am J Health Syst Pharm. 2005;62:173–181. doi: 10.1093/ajhp/62.2.173. [DOI] [PubMed] [Google Scholar]
- Lopez de Maturana R, Willshaw A, Kuntzsch A, Rudolph R, Donnelly D. The isolated N-terminal domain of the glucagon-like peptide-1 (GLP-1) receptor binds exendin peptides with much higher affinity than GLP-1. J Biol Chem. 2003;278:10195–101200. doi: 10.1074/jbc.M212147200. [DOI] [PubMed] [Google Scholar]
- Malendowicz LK, Nowak KW, Zyterska A, Nussdorfer GG, Macchi C, Nowak M. Exendin-4, a GLP-1 receptor agonist, stimulates entero-insular axis in the rat, through a mechanism involving adrenal medulla. Biomed Res. 2001;22:295–297. [Google Scholar]
- Manjula BN, Tsai A, Upadhya R, Perumalsamy K, Smith PK, Malavalli A, et al. Site-specific PEGylation of hemoglobin at Cys-93(beta): correlation between the colligative properties of the PEGylated protein and the length of the conjugated PEG chain. Bioconjug Chem. 2003;14:464–472. doi: 10.1021/bc0200733. [DOI] [PubMed] [Google Scholar]
- Mann R, Nasr N, Hadden D, Sinfield J, Abidi F, Al-Sabah S, et al. Peptide binding at the GLP-1 receptor. Biochem Soc Trans. 2007;35:713–716. doi: 10.1042/BST0350713. [DOI] [PubMed] [Google Scholar]
- Mann RJ, Nasr NE, Sinfield JK, Paci E, Donnelly D. The major determinant of exendin-4/glucagon-like peptide 1 differential affinity at the rat glucagon-like peptide 1 receptor N-terminal domain is a hydrogen bond from SER-32 of exendin-4. Br J Pharmacol. 2010;160:1973–1984. doi: 10.1111/j.1476-5381.2010.00834.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montrose-Rafizadeh C, Yang H, Rodgers BD, Beday A, Pritchette LA, Eng J. High potency antagonists of the pancreatic glucagon-like peptide-1 receptor. J Biol Chem. 1997;272:21201–21206. doi: 10.1074/jbc.272.34.21201. [DOI] [PubMed] [Google Scholar]
- Nauck MA, Kleine N, Orskov C, Holst JJ, Willms B, Creutzfeldt W. Normalization of fasting hyperlycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in Type-2 (non-insulin-dependent) diabetic patients. Diabetologia. 1993;36:741–744. doi: 10.1007/BF00401145. [DOI] [PubMed] [Google Scholar]
- Neidigh JW, Fesinmeyer RM, Prickett KS, Andersen NH. Exendin-4 and glucagon-like-peptide-1: NMR structural comparisons in the solution and micelle-associated states. Biochemistry. 2001;40:13188–13200. doi: 10.1021/bi010902s. [DOI] [PubMed] [Google Scholar]
- Parkes D, Jodka C, Smith P, Nayak S, Rinehart L, Gingerich R, et al. Pharmacokinetic actions of exendin-4 in the rat: comparison with glucagon-like peptide-1. Drug Dev Res. 2001;53:260–267. [Google Scholar]
- Perez-Tilve D, Gonzalez-matias L, Aulinger BA, Alvarez-Crespo M, Gil-Lozano M, Alvarez E, et al. Exendin-4 increases blood glucose levels acutely in rats by activation of the sympathetic nervous system. Am J Physiol Endocrinol Metab. 2010;298:E1088–E1096. doi: 10.1152/ajpendo.00464.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry T, Lahiri DK, Chen D, Zhou J, Shaw KTY, Egan JM, et al. A novel neurotrophic property of glucagon-like peptide 1: a promoter of nerve growth factor-mediated differentiation in PC12 cells. J Pharmacol Exp Ther. 2002;300:958–966. doi: 10.1124/jpet.300.3.958. [DOI] [PubMed] [Google Scholar]
- Qualmann C, Nauck MA, Holst JJ, Qrskov C, Creutzfeldt W. Insulinotropic actions of intravenous glucagon-like peptide-1 (GLP-1) [7–36 amide] in the fasting state in healthy subjects. Acta Diabetol. 1995;32:13–16. doi: 10.1007/BF00581038. [DOI] [PubMed] [Google Scholar]
- Rosendahl MS, Doherty DH, Smith DJ, Carlson SJ, Chlipala EA, Cox GN. A long-acting, highly potent interferon-2 conjugate created using site-specific PEGylation. Bioconjugate Chem. 2005;16:200–207. doi: 10.1021/bc049713n. [DOI] [PubMed] [Google Scholar]
- Runge S, Schimmer S, Oschmann J, Schiodt CB, Knudsen SM, Jeppesen CB, et al. Differential structural properties of GLP-1 and exendin-4 determine their relative affinity for the GLP-1 receptor N-terminal extracellular domain. Biochemistry. 2007;46:5830–5840. doi: 10.1021/bi062309m. [DOI] [PubMed] [Google Scholar]
- Runge S, Thogersen H, Madsen K, Lau J, Rudolph R. Crystal structure of the ligand-bound glucagon-like peptide-1 receptor extracellular domain. J Biol Chem. 2008;283:11340–11347. doi: 10.1074/jbc.M708740200. [DOI] [PubMed] [Google Scholar]
- Szayna M, Doyle ME, Betkey JA, Holloway HW, Spencer RG, Greig NH, et al. Exendin-4 decelerates food intake, weight gain, and fat deposition in Zucker rats. Endocrinology. 2000;141:1936–1941. doi: 10.1210/endo.141.6.7490. [DOI] [PubMed] [Google Scholar]
- Triplitt C, Defronzo RA. Exenatide: first-in-class incretin mimetic for the treatment of type 2 diabetes mellitus. Expert Rev Endocrinol Metab. 2006;3:329–341. doi: 10.1586/17446651.1.3.329. [DOI] [PubMed] [Google Scholar]
- Yamamoto Y, Tsutsumi Y, Yoshioka Y, Nishibata T, Kobahashi K, Okamoto T, et al. Site-specific PEGylation of a lysine-deficient TNF-α with full bioactivity. Nat Biotechnol. 2003;21:546–552. doi: 10.1038/nbt812. [DOI] [PubMed] [Google Scholar]
- Youn YS, Jung JY, Oh SH, Yoo SD, Lee KC. Improved intestinal delivery of salmon calcitonin by Lys18-amino specific PEGylation, Stability, permeability, pharmacokinetic behavior and in vivo hypocalcemic efficacy. J Control Rel. 2006;114:334–342. doi: 10.1016/j.jconrel.2006.06.007. [DOI] [PubMed] [Google Scholar]
- Young AA, Gedulin BR, Bhavsar S, Bodkin N, Jodka C, Hansen B, et al. Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta) Diabetes. 1999;48:1026–1034. doi: 10.2337/diabetes.48.5.1026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.