

Abstract
In this issue of Cancer Cell, Finley and coworkers report that genetic loss of the deacetylase SIRT3 leads to metabolic re-programming toward glycolysis. This shift is mediated by an increase in cellular reactive oxygen species (ROS) generation that amplifies HIF-α stabilization and HIF-dependent gene expression, thereby driving the tumor phenotype.
In cancer cells, reprogramming of cellular metabolism drives substrate utilization toward a dependence on glucose. First described by Otto Warburg (Warburg, 1956), the significance of this response for tumor growth has been controversial. However, it appears that this glycolytic shift is necessary to provide a source of substrates for the synthesis of amino acid, lipids, and nucleic acids that are needed for proliferation (Vander Heiden et al., 2009). Indeed, enhanced glucose uptake by tumor cells forms the basis for the clinical detection of tumors by imaging regions exhibiting increased uptake of the glucose analog, 18F-fluordeoxyglucose. While the association between cancer and the Warburg metabolic shift is well established, the cellular mechanisms regulating this response are not fully understood.
Post-translational modifications of proteins are important for regulating their function in health and disease. Critical roles for protein deacetylases are also emerging in cancer. For example, Kim et al. identified a role for SIRT3, a member of the seven-member sirtuin family, as a tumor suppressor (Kim et al., 2010). They showed that genetic deletion of SIRT3 pushes the cell in the direction of oncogenic transformation. While activation of two oncogenes (such as Myc and Ras) is needed to transform an immortalized fibroblast into a tumor-forming cell, genetic deletion of SIRT3 reduced that number to one. Thus, SIRT3 functions as a tumor suppressor (Schumacker, 2010). The mechanistic basis for SIRT3's tumor-suppressive role seems to reside in its ability to regulate reactive oxygen species (ROS) generation or clearance by the cell. Kim et al. noted that ROS levels were increased in SIRT3-/- cells, as a consequence of a decreased expression of antioxidant enzymes such as catalase and MnSOD. The transcription factor FOXO3a plays an important role in regulating the expression of MnSOD and other antioxidants, and SIRT3-mediated deacetylation of FOXO3a promotes its nuclear localization (Jacobs et al., 2008). Thus, loss of SIRT3 activity suppresses FOXO3a, leading to an increase in cellular ROS signaling. Enhanced ROS levels have been linked to cancer, and Kim et al. observed an increase in the incidence of mammary tumors in the SIRT3 knockout mice. They suggested that the chronic increase in mitochondrial ROS stress might result in mitochondrial or genomic DNA damage, but that mechanism was not directly tested. Nevertheless, their study identified an important pathway by which SIRT3 suppresses tumor cell survival and proliferation through its effects on cellular ROS regulation.
But even the best studies leave many questions unanswered. The principal issue left in the wake of the Kim et al. study related to how the increase in ROS (caused by loss of SIRT3) mediates the enhanced tumor phenotype of cells. The answer to that question arrives in the article by Finley and coworkers in this issue of Cancer Cell (Finley et al., 2011). While confirming that genetic loss of SIRT3 leads to an enhanced tumor phenotype in a ROS-dependent manner, they move the field forward in a profound way by showing that this occurs because the ROS enhance HIF-1α stabilization. HIF-1 and HIF-2 are hypoxia-responsive transcription factors that regulate the altered expression of more than 200 genes in responses to hypoxia (Semenza, 2010a). Up-regulation of HIF activity is strongly linked to the survival and proliferation of tumor cells because some HIF-dependent genes such as glucose transporters and glycolytic enzymes, and Vascular Endothelial Growth Factor (VEGF) are critical for cell survival when chronically exposed to a hypoxic microenvironment in the tumor. Other HIF-dependent genes affect metabolic reprogramming, apoptosis, cell migration, remodeling of the extracellular matrix, iron metabolism, pH regulation, vascular reactivity, and other functions that are intimately involved in tumor progression and metastasis (Semenza, 2010b). Finley et al. made this connection by comparing the gene expression profile in brown adipose tissue of wild type and SIRT3 knockout mice, and noting that the loss of SIRT3 led to a response that mimicked the change when cells were exposed to low oxygen (Finley et al., 2011). The mechanism underlying the response to SIRT3 deletion appears to involve an increase in ROS-dependent inhibition of prolyl hydroxylase (PHD) (Fig. 1). This mechanism is consistent with previous studies demonstrating that mitochondrial ROS signals regulate HIF-α stability in hypoxia (Chandel et al., 1998). Interestingly, the loss of SIRT3, via enhanced HIF expression, mediates the Warburg shift in metabolism in fibroblasts, driving them toward enhanced glucose utilization that is required for the enhanced growth properties they exhibit.
Figure 1.
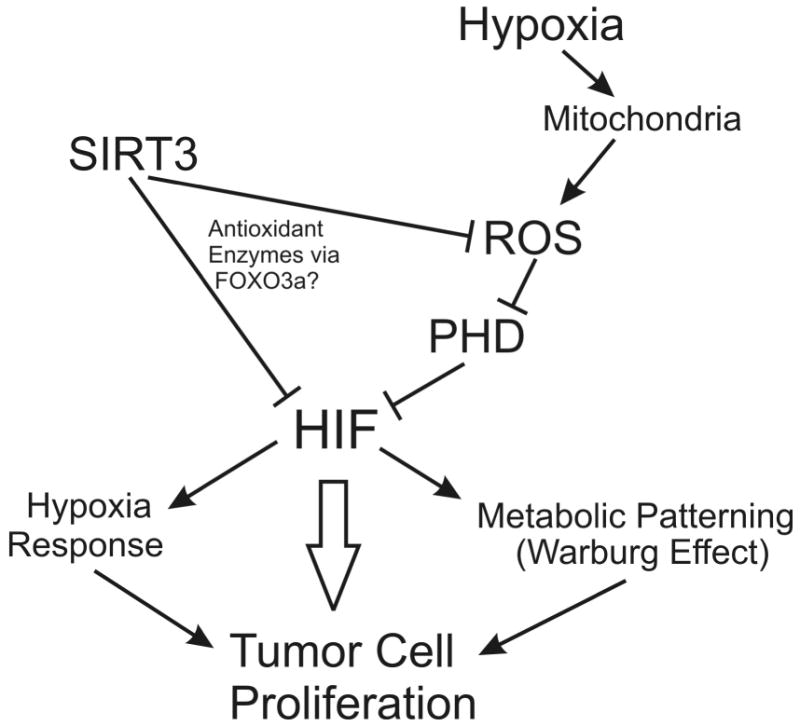
Regulation of tumor cell phenotype by SIRT3.
The loss of SIRT3 appears to drive HIF activation even under basal normoxic conditions, and to enhance the HIF response to hypoxia through a ROS-dependent mechanism. They report that treatment of SIRT3 knockout cells with the antioxidant, N-acetyl cysteine (NAC), returns the normoxic HIF to wild type levels, while it abolishes the increased rate of proliferation and metabolic reprogramming. In vivo, NAC treatment repressed the hypoxia-associated gene expression profile in brown adipose tissue, without affecting expression patterns in wild type animals. By contrast, over-expression of SIRT3 shifts the cell away from the Warburg effect and suppresses the hypoxic response, although the role of ROS in that response was not directly investigated.
Impressively, Finley et al. go on to provide correlative evidence showing an association between the loss of SIRT3 in human breast cancers and the enhanced expression of HIF-1-dependent genes. Whether or not the loss of SIRT3 is a causative agent in cancer initiation, or whether subsequent loss of SIRT3 in an existing tumor cell helps to drive its glycolytic and proliferative phenotype, are questions that still need to be addressed. It seems likely that both events may occur in different conditions. In either case, the finding that SIRT3 loss can enhance the HIF-dependent response in cancer opens therapeutic opportunities for drug targets, while it underscores the potential importance of identifying agents that can selectively inhibit ROS signaling and HIF activity in cancer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley LWS, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigney MCP. SIRT3 opposes reprogramming of cancer cell metabolism through HIF1α destabilization. Cancer Cell. 2011 doi: 10.1016/j.ccr.2011.02.014. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs KM, Pennington JD, Bisht KS, ykin-Burns N, Kim HS, Mishra M, Sun L, Nguyen P, Ahn BH, Leclerc J, Deng CX, Spitz DR, Gius D. SIRT3 interacts with the daf-16 homolog FOXO3a in the mitochondria, as well as increases FOXO3a dependent gene expression. Int J Biol Sci. 2008;4:291–299. doi: 10.7150/ijbs.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Patel K, Muldoon-Jacobs K, Bisht KS, ykin-Burns N, Pennington JD, van der MR, Nguyen P, Savage J, Owens KM, Vassilopoulos A, Ozden O, Park SH, Singh KK, Abdulkadir SA, Spitz DR, Deng CX, Gius D. SIRT3 is a mitochondria-localized tumor suppressor required for maintenance of mitochondrial integrity and metabolism during stress. Cancer Cell. 2010;17:41–52. doi: 10.1016/j.ccr.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacker PT. A tumor suppressor SIRTainty. Cancer Cell. 2010;17:5–6. doi: 10.1016/j.ccr.2009.12.032. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010a;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010b;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]