

Abstract
Alcohol-induced Wernicke-Korsakoff syndrome (WKS) culminates in bilateral diencephalic lesion and severe amnesia. Using the pyrithiamine-induced thiamine deficiency (PTD) animal paradigm of WKS, our laboratory has demonstrated hippocampal dysfunction in the absence of gross anatomical pathology. Extensive literature has revealed reduced hippocampal neurogenesis following a neuropathological insult, which might contribute to hippocampus-based learning and memory impairments. Thus, the current investigation was conducted to determine whether PTD treatment altered hippocampal neurogenesis in a stage-dependent fashion. Male Sprague-Dawley rats were assigned to one of 4 stages of thiamine deficiency based on behavioral symptoms: pre-symptomatic stage, ataxic stage, early post-opisthotonus stage, or the late post-opisthotonus stage. The S-phase mitotic marker 5′-bromo-2′-deoxyuridine (BrdU) was administered at the conclusion of each stage following thiamine restoration and subjects were perfused 24-hours or 28-days after BrdU to assess cellular proliferation or neurogenesis and survival, respectively. Dorsal hippocampal sections were immunostained for BrdU (proliferating cell marker), NeuN (neurons), GFAP (astrocytes), Iba-1 (microglia), and O4 (oligodendrocytes). The PTD treatment increased progenitor cell proliferation and survival during the early post-opisthotonus stage. However, levels of neurogenesis were reduced during this stage as well as the late post-opisthotonus stage where there was also an increase in astrocytogenesis. The diminished numbers of newly generated neurons (BrdU/NeuN co-localization) was paralleled by increased BrdU cells that did not co-localize with any of the phenotypic markers during these later stages. These data demonstrate that long-term alterations in neurogenesis and gliogenesis might contribute to the observed hippocampal dysfunction in the PTD model and human WKS.
Keywords: neurogenesis, cell proliferation, BrdU, pyrithiamine, Wernicke-Korsakoff syndrome
1. Introduction
Wernicke-Korsakoff syndrome (WKS) is an insidious neurodegenerative disorder caused by thiamine deficiency. It involves an acute Wernicke's encephalopathy phase and a chronic Korsakoff's syndrome phase (Butters, 1981; Kopelman et al. 2009; Victor et al. 1971, 1989). Although WKS is observed in disorders associated with malnutrition, it is most commonly diagnosed in chronic alcoholics (Harper and Kril, 1990; Kopelman, 1995; Kril, 1996; Langlais et al. 1996; Victor et al. 1989). The anatomical consequence of prolonged thiamine deficiency is a bilateral lesion of multiple thalamic nuclei (anterior n., midline n., mediodoral n., and posterior n.) and the mammillary bodies as well as degeneration of the fimbria/fornix and mammillothalamic tract that interconnect the limbic structures (Caulo et al. 2005; Gold and Squire, 2006; Kopelman et al. 2009; Kril, 1996; Langlais et al. 1996; Sullivan and Pfefferbaum, 2009; Victor et al. 1989). This neuropathological constellation culminates in temporally graded retrograde amnesia and severe anterograde amnesia characterized by impaired explicit memory, indicative of hippocampal dysfunction (Caulo et al. 2005; Fama et al. 2004; Victor et al. 1989). However, there is limited evidence of overt hippocampal pathology in the WKS brain (Caulo et al. 2005; Fama et al. 2004; Gold and Squire, 2006).
The pyrithiamine-induced thiamine deficiency (PTD) animal paradigm models both the diencephalic neuropathology and behavioral impairments observed in human WKS (Langlais and Savage, 1995; Langlais and Zhang, 1997; Langlais et al., 1996; Markowitsch, 1988; Troncoso et al. 1981; Witt, 1985). Similar to WKS, there is little evidence of gross hippocampal pathology in the PTD model (Langlais et al. 1992; Mair, 1994) despite functional impairment on several spatial memory tasks, including the delayed non-matching-to-position task (Langlais and Savage, 1995; Roland and Savage, 2007), Morris water maze (Langlais et al. 1992), and the spontaneous alternation task (Savage et al. 2003; Vetreno et al. 2008). The deficits on hippocampal-dependent spatial memory tasks are paralleled by blunted hippocampal cholinergic efflux assessed during maze learning (Roland and Savage, 2007; Vetreno et al. 2008).
In both human WKS and the PTD animal model, assessment of neural activity during memory testing revealed that the hippocampus is functionally impaired and likely contributes to the amnestic state (see Caulo et al. 2005; Savage et al. 2003). However, the precise physiological mechanisms underlying the hippocampal dysfunction remain to be elucidated. Recent work suggests that diminished neuroplasticity within the hippocampal network might be a contributing factor.
Neurogenesis involves the generation, differentiation, migration, and functional integration of newly generated neurons into mature brain tissue (Altman and Das, 1965; Zhao, Deng et al. 2008; Zhao et al. 2006), and occurs across mammalian species (Gould et al., 1999b; Kuhn et al., 1996), including humans (Eriksson et al. 1998). The subventricular zone of the lateral ventricles and the subgranular zone (SGZ) of the hippocampal dentate gyrus represent two distinct mitotically active microenvironments wherein neurogenesis continues to occur throughout life (Abrous et al. 2005). Neurogenesis within the hippocampus has been heavily implicated in hippocampal-mediated cognitive function (Ambrogini et al. 2000; Gould et al. 1999a; Kee et al. 2007; Kempermann et al. 2004; Shors et al. 2001). Experimental facilitation or inhibition of neurogenesis enhances and diminishes performance on spatial tasks, respectively (Fabel et al. 2009; Gould et al. 1999a; Madsen et al. 2003; Shors et al. 2001; van Praag et al. 1999b; Winocur et al. 2006). Furthermore, it is a highly dynamic process affected by many intrinsic and extrinsic factors, including neurotransmitters (e.g., acetylcholine [Cooper-Kuhn et al. 2004]), drug abuse (e.g., alcoholism [Crews et al. 2004; He et al. 2009; Klintsova et al. 2007]), environmental enrichment (Cotman and Berchtold, 2002; Fabel et al. 2009), and pathological insults (Richardson et al. 2007; Rola et al. 2006; Zhao, Zhong et al. 2008).
There is also evidence indicating that altered neurogenesis plays a role in behavioral abnormalities observed during an episode of thiamine deficiency. Indeed, work assessing the effects of thiamine deficiency on neurogenesis in mice demonstrated reductions beginning on the 9th day of exposure to a thiamine deficient diet, which were paralleled by behavioral impairments on a Y-maze avoidance task (Zhao, Zhong et al. 2008). However, the animals in this experiment were only exposed to dietary restriction of thiamine, a paradigm that does not completely model the brain pathology associated with the chronic condition of WKS (see Langlais et al. 1996).
Thus, the present study sought to determine whether neurogenesis and/or gliogenesis were altered as thiamine deficiency progressed from the early acute Wernicke encephalopathy phase to the more chronic WKS (as defined by Zhang et al., 2005). Levels of neurogenesis (BrdU/NeuN co-localization) and gliogenesis (astrocytes [BrdU/GFAP], microglia [BrdU/Iba-1], oligodendrocytes [BrdU/O4]) were assessed in separate groups of PTD and pair-fed (PF) control rats that were administered the S-phase mitotic marker 5′-bromo-2′-deoxyuridine (BrdU) at one of 4 stages of thiamine deficiency: (1) the pre-symptomatic stage (8 days after treatment onset), (2) the ataxic stage (15 days after treatment onset), (3) the early post-opisthotonus stage (24-hours after thiamine replacement therapy), or (4) the late post-opisthotonus stage (28 days after thiamine replacement therapy). The goal of this study was to document how progenitor cell proliferation and survival as well as neurogenesis and gliogenesis change as thiamine deficiency progresses to the culmination of neuropathology and the chronic post-opisthotonus state that defines WKS.
2. Results
2.1 PF treatment does not negatively affect progenitor cell proliferation or neurogenesis
The PF subjects were assessed across the disease progression stages to determine whether the control treatment altered measures of neurogenesis. The PF subgroups did not demonstrate any statistical differences in estimates of progenitor cell proliferation and survival, or measures of neurogenesis and gliogenesis (all p's>.07). Thus, the PF subgroups were aggregated into a single PF control group. The PF subject's evidenced 71.5% co-localization of BrdU-positive cells with NeuN, which complements previous research demonstrating 70 – 80% BrdU/NeuN co-expression in normal rats (Cameron et al. 1993; Hattiangady and Shetty, 2010). Only a fraction (0.25%) of BrdU-immunoreactive cells in the PF-treated animals were observed to co-express the astrocytic marker GFAP. The paucity of BrdU+/GFAP+ cells was similar to previous research demonstrating that there is little to no BrdU/GFAP co-localization within the hippocampal GCL in the healthy brain (see Bruel-Jungerman et al. 2005; Cameron and McKay, 1999; Cameron et al. 1993; Helfer et al. 2009a). Similarly, assessment of O4 and Iba-1, markers of oligodendrocytes and microglia (respectively), revealed no co-localize with BrdU in control animals, a finding supported by other research groups (see e.g., Chumley et al. 2007; He et al. 2009; Helfer et al. 2009a, 2009b; Nixon et al. 2008). Finally, the percentage of BrdU-positive cells that did not express any of the phenotypic markers in the PF subjects was similar to previous reports in control subjects (see Van der Borght et al. 2005).
2.2 Body weights across treatment
The stage-dependent effects of PTD and PF treatment on body weight are shown in Figure 2. Mean body weight differed as a function of STAGE (F[3,112]=257.3, p<0.01) as both PTD and PF food-restricted animals demonstrated weight loss during the initial 3 stages that was recovered during the late post-opisthotonus stage when free-feed was re-instituted. Importantly, there were no other main effects (both p's>0.6) or interactions (all p's>0.07) indicating that any differences in BrdU-immunoreactivity or neurogenesis between PTD and PF subjects were not due to differences in caloric intake.
Figure 2.

Comparison of the mean body weights (in grams) between PF- and PTD-treated subjects at different time points during and after PTD treatment (mean ± S.E.M.).
2.3 Stage-dependent reductions of IVDs in the PTD model
The overall analysis revealed a main effect of TREATMENT (F[4,118]=8.0, p<0.01), but not TIME POINT (p>0.1). Follow-up comparisons demonstrated a significant loss of thalamic mass in the PTD animals, relative to PF subjects, during the late post-opisthotonus stage (p<0.01; see Table 1).
Table 1. Intraventricular distances across stages of thiamine deficiency.
Intraventricular distances (in mm) between PF- and PTD-treated animals across stages of disease progression, which are reported as means (± S.E.M.).
| PF | PRE | ATAX | E. Post | L. Post | |
|---|---|---|---|---|---|
| IVD (in mm) | 3.02 ± 0.03 | 3.06 ± 0.06 | 2.88 ± 0.06 | 2.88 ± 0.09 | 2.56 ± 0.09 b |
p<0.01, relative to PF
Images of thalamus during the late post-opisthotonus stage (L. Post) 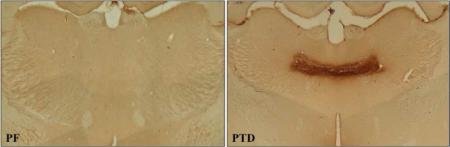
2.4 Thiamine deficiency increased proliferation and survival of hippocampal progenitor cells during the early post-opisthotonus stage
Stereological estimates of BrdU-immunoreactive cells in the hippocampal GCL/SGZ were conducted to ascertain whether proliferation and survival of newly generated cells was altered as a consequence of the stage of PTD treatment. A significant main effect of TREATMENT (F[4,118]=3.6, p<0.01) revealed that overall PTD treatment resulted in an increase in BrdU-immunopositive cells, relative to the PF subjects. However, follow-up comparisons of the TREATMENT effect revealed that the main effect was primarily driven by PTD subjects during the early post-opisthotonus stage evidencing a significant increase in the number of BrdU-immunopositive cells during the 24-hour proliferation phase (p<0.01; see Figure 3) and the 28-day survival phase (p≤0.05, see Figure 4).
Figure 3.

Stage-dependent estimates of progenitor cell proliferation in the hippocampal dentate gyrus of PF- and PTD-treated rats. Data expressed as mean ± S.E.M. # = p<0.01. Micrographs depict outline of GCL/SGZ of representative PF- and PTD-treated animals during the early post-opisthotonus stage. Images captured with a 4X objective.
Figure 4.

Stage-dependent estimates of progenitor cell survival in the hippocampal dentate gyrus of PFand PTD-treated rats. Data expressed as mean ± S.E.M. * = p≤0.05. Micrographs depict outline of GCL/SGZ of representative PF- and PTD-treated animals during the early post-opisthotonus stage. Images captured with a 4X objective.
Regardless of TREATMENT condition, all animals evidenced greater levels of BrdU-positive cells at the 24-hour time point after BrdU administration relative to subjects during the 28-day survival period (main effect of TIME POINT: F[1,118]=44.2, p<0.01). The observed 43% reduction in BrdU-positive cells at the 28-day time point is consistent with literature demonstrating an approximate 50% loss of BrdU-immunoreactive cells between proliferation and survival (see Christie and Cameron, 2006; Dayer et al. 2003). The interaction of TREATMENT X TIME POINT was not significant (all p's>0.5). The Gundersen-Jensen estimator of the error coefficient (with a smoothness factor = 1) for BrdU-immunoreactive cells was not different as a function of TREATMENT (PTD = 0.19; PF = 0.18; p>0.1).
2.5 Percentages of neurogenesis and gliogenesis are altered during the post-opisthotonus stages of PTD treatment
To establish whether the percentage of neurogenesis and/or gliogenesis was altered in the PTD model, co-localization of BrdU with NeuN, GFAP, O4, and Iba-1 was assessed at different stages of thiamine deficiency. The neurogenesis data revealed a significant main effect of TREATMENT (F[4,43]=4.9, p<0.01) as PTD-treated animals evidenced reduced BrdU/NeuN co-localization relative to PF control subjects in a stage-dependent manner (see Table 2). Although rates of neurogenesis in the PTD subjects were statistically similar to the PF animals during both the pre-symptomatic and ataxic stages (both p's>0.5), there were significant 13% and 23% reductions in the percentage of neurogenesis during the early post-opisthotonus (p<0.05) and late post-opisthotonus stages (p<0.01), respectively.
Table 2. Mean percentage of cellular phenotype across stages of thiamine deficiency.
Percent of BrdU+ cells that co-labeled with the neuronal marker NeuN, the astrocytic marker GFAP, or likely remained undifferentiated in the hippocampal dentate gyrus across stages (pre-symptomatic [PRE], ataxic [ATAX], early post-opisthotonus [E. Post], late post-opisthotonus [L. Post]) of PTD treatment relative to the PF control subjects. BrdU was administered at each stage and subjects were sacrificed 28 days later to assess co-localization. All data are represented as means (± S.E.M.). (A-C) Confocal images of BrdU+ cells (green) and NeuN+ granule cells (red) in the hippocampal dentate gyrus. (D-G) Confocal images of BrdU+ cells (green) and GFAP+ granule cells (blue) in the hippocampal dentate gyrus. (H-J) Confocal image of a BrdU+ cell (green) that did not co-localize with NeuN (red) in the hippocampal dentate gyrus. White arrow indicates cell of interest. Scale bar = 20 μm.
| Cellular Phenotype |
|||
|---|---|---|---|
| Neuron | Astrocyte | BrdU Only | |
| PF | 71.6 ± 2.2 | 0.26 ± 0.1 | 28.2 ± 2.2 |
| PRE | 75.0 ± 6.9 | 0 | 25.0 ± 6.9 |
| ATAX | 71.0 ± 1.8 | 0 | 29.0 ± 1.8 |
| E. Post | 58.1 ± 7.4 a | 0 | 41.9 ± 7.4 a |
| L. Post | 48.1 ± 7.8 b | 1.28 ± 0.5 a | 50.6 ± 7.7 b |
p<0.05, relative to PF
p<0.01, relative to PF
Images of BrdU co-localization at during the late post-opisthotonus stage 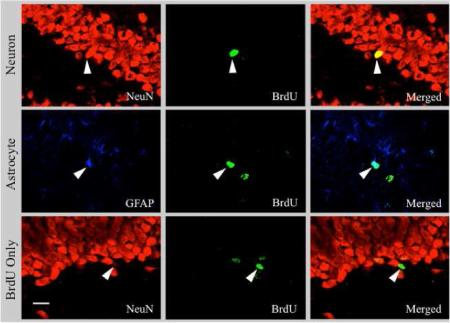
Assessment of gliogenesis also revealed a main effect of TREATMENT for BrdU/GFAP co-localization (F[4,43]=3.8, p<0.05): PTD treatment increased astrocytogenesis in relation to PF animals. The GFAP/BrdU co-labeling did not differ relative to PF subjects during the pre-symptomatic, ataxic, or early post-opisthotonus stages (all p's>0.3). However, an increase in the percentage of astrocytogenesis was observed during the late post-opisthotonus stage (p<0.05). None of the BrdU-immunoreactive cells in the GCL co-expressed either the oligodendrocyte or microglia marker at any stage of thiamine deficiency (data not shown).
The percentage of BrdU-positive cells that did not co-localize in the PTD animals was greater than the PF subjects (main effect of TREATMENT: F[4,43]=4.6, p<0.01). There were comparable percentages of BrdU-positive cells that did not co-localize during both the pre-symptomatic and ataxic stages relative to PF subjects (both p's>0.9). However, PTD treatment resulted in an increase in the percentage of BrdU-positive cells that did not co-express any of the phenotypic markers used during the early post-opisthotonus stage (p<0.05) and late post-opisthotonus stage (p<0.01).
2.6 Thiamine deficiency alters absolute estimates of neurogenesis and gliogenesis during the post-opisthotonus stages of PTD treatment
Since PTD treatment was observed to reduce the percentage of neurogenesis during the early and late post-opisthotonus stages, we examined whether the absolute estimates of neurogenesis and gliogenesis were decreased at these stages. There was a reduction in the absolute estimates of newborn neurons during the late post-opisthotonus stage of PTD treatment (F[1,30]=5.1, p<0.05), but not the early post-opisthotonus stage (p>0.5). Similarly, there was an increase in the number of newly generated astrocytes during the late post-opisthotonus stage (F[1,30]=4.4, p<0.05), but not the early post-opisthotonus stage (p>0.3). Finally, there were also significantly greater numbers of BrdU-positive cells that did not co-label during both the early post-opisthotonus (F[1,30]=11.5, p<0.01) and late post-opisthotonus stages (F[1,30]=6.2, p<0.01; see Table 3).
Table 3. Mean estimate of absolute phenotypes across stages of thiamine deficiency.
Absolute estimates of BrdU+ cells that co-localized with the neuronal marker NeuN, the astrocytic marker GFAP, or did not co-localize in the hippocampal dentate gyrus across stages (pre-symptomatic [PRE], ataxic [ATAX], early post-opisthotonus [E. Post], late post-opisthotonus [L. Post]) of PTD treatment relative to the PF subjects. BrdU was administered at each stage and subjects were sacrificed 28 days later to assess co-localization. All data are represented as means (± S.E.M.).
| Cellular Phenotype |
|||
|---|---|---|---|
| Neuron | Astrocyte | Undifferentiated | |
| PF | 903.3 ± 90.3 | 10.5 ± 2.1 | 356.5 ± 41.3 |
| PRE | 1117.9 ± 283.2 | 0 | 340.6 ± 71.7 |
| ATAX | 870.3 ± 141.4 | 0 | 347.9 ± 52.6 |
| E. Post | 1041.1 ± 226.1 | 0 | 733.5 ± 151.9 b |
| L. Post | 531.2 ± 79.4 a | 13.2 ± 4.7 a | 652.5 ± 168.3 a |
p< 0.05, relative to PF
p< 0.01, relative to PF
3. Discussion
This was the first experiment to demonstrate that (a) there is an initial increase in progenitor cell proliferation and survival after the excitotoxic insult produced by PTD treatment; however, (b) these effects were followed by a chronic reduction of hippocampal neurogenesis. These results occurred later in treatment than was reported by Zhao, Zhong, and colleagues (2008) who demonstrated reduced neurogenesis in mice with concomitant hippocampal impairment after only 9 days of exposure to diet-induced thiamine deficiency. However, methodological differences appear to underlie the disparate results between experiments. Unlike the diencephalic lesion produced in WKS or the PTD model (see image in Table 1), dietary restriction of thiamine alone typically results in a lesion that is primarily restricted to brainstem nuclei (Langlais and Savage, 1995; Langlais et al. 1996; Troncoso et al. 1981; Witt, 1985). Thus, differences in the pathological insult evoked via dietary restriction and PTD treatment might account for the variation in onset of altered neurogenesis. Furthermore, dietary restriction-induced thiamine deficiency in rats tends to be more protracted, often requiring 30 days to elicit both neurophysiological and behavioral effects (Pires et al. 2001, 2005). The fact that Zhao, Zhong et al. (2008) reported diencephalic and brainstem lesion as well as cholinergic cell loss beginning on day 14 of thiamine deficiency suggests that the consequences of dietary-induced thiamine deficiency in mice might be accelerated relative to what is commonly observed in rats.
Pyrithiamine-induced thiamine deficiency resulted in an increase in cellular proliferation (52.9%) at the 24-hour time point during the early post-opisthotonus stage. This increase in BrdU-immunoreactivity was countered by a 13% reduction of neurogenesis (BrdU/NeuN co-localization) and 13% increase unlabeled BrdU+ cells during this stage when assessed during the 28-day survival stage. However, assessment of the absolute estimates of neurogenesis revealed an increase in the number of BrdU-immunoreactive cells that did not co-express any of the cellular markers, but no reduction in the absolute numbers of newly generated neurons. This enhanced BrdU immunoreactivity and spared neurogenesis in the PTD-treated subjects during the early post-opisthotonus stage might be a consequence of several factors. First, the increased BrdU-immunoreactivity during the 24-hour proliferation time point might reflect dead or dying cells as increased cell death, either apoptotic or necrotic, is common following a pathophysiological brain insult. Indeed, Bauer and Patterson (2005) reported that approximately 73% of BrdU-immunoreactive cells co-localize with the cell death marker TUNEL following brain irradiation. Furthermore, experimental denervation of cholinergic inputs to the hippocampal dentate gyrus results in an approximate 77% increase in cell death (TUNEL-positive cells) in this region (Cooper-Kuhn et al. 2004). Thus, the increase in BrdU-immunoreactive cells observed during the early post-opisthotonus stage might be partially due to cells that have undergone apoptosis and/or necrosis. A second potential explanation for the increased BrdU-immunoreactivity might be seizure-induced increases in proliferation, survival, and neurogenesis (Jessberger et al. 2007; Parent, 2002; Parent et al. 1997). Although there was a reduction in the percentage of neurogenesis, absolute estimates of neurogenesis were not reduced. Thus, the thiamine deficiency-induced seizure activity might have contributed to the increased BrdU-immunoreactivity and consequent maintenance of hippocampal neurogenesis. A third potential explanation for the increased progenitor cell proliferation and neurogenesis might be compensatory attempts at maintaining homeostasis following a pathophysiological insult. The hippocampus is a highly plastic structure capable of initiating compensatory neuronal responses following certain brain injuries that are aimed at minimizing damage (Kuhn et al. 2001; Shetty et al. 2010). Pathological increases of cellular proliferation and neurogenesis have been reported in other animal models, including Alzheimer's disease (Chevallier et al. 2005), ischemic infarcts (Nakatomi et al. 2002; Takagi et al. 1999), and traumatic brain injury (Chirumamilla et al. 2002). Thus, the increased progenitor cell response observed during the early post-opisthotonus stage might reflect a compensatory response aimed at preserving hippocampal function and/or minimizing the impact of neuronal cell loss following a thiamine deficiency-induced neurodegenerative insult.
A long-term reduction of neurogenesis was observed during the late post-opisthotonus stage as PTD-treated animals demonstrated a 23% reduction in BrdU+/NeuN+ co-localization as well as an overall reduction of the absolute estimates of neurogenesis. In addition, a negligible 1.0% increase in astrocytogenesis and a 22% increase in unlabeled BrdU-positive cells was observed indicating that a significant proportion of proliferating cells in the thiamine deficient brain were unable to develop a neuronal phenotype at longer delays following neurotrauma. Taken together, these data demonstrate that thiamine deficiency disrupts the neurogenic process in the hippocampus and might account for some of the behavioral impairments observed in the PTD model.
The long-term reductions of hippocampal neurogenesis in the present study are likely due to a variety of factors that culminate in a compromised neurogenic microenvironment, including reduced bioavailability of thiamine-dependent enzymes, altered neurotrophic support, loss in inputs to the hippocampus, and diminished acetylcholine (ACh) innervation. Thiamine is converted to thiamine pyrophosphate and is a necessary co-factor for the production of transketolase, ∝-ketoglutarate dehydrogenase, and pyruvate dehydrogenase, which play key roles in the maintenance of normal cellular function (Gibson et al. 1984; Hazell and Butterworth, 2009; Karuppagounder and Gibson, 2009; Kopelman, 1995; Langlais et al. 1996; Leong and Butterworth, 1996; Vorhees et al. 1977) and might contribute to the observed decrease of neurogenesis. For instance, Zhao and colleagues (2009) reported that 9 days of dietary-induced thiamine deficiency resulted in reduced levels of hippocampal neurogenesis that were accompanied by diminished levels of transketolase. These researchers also demonstrated that transketolase silencing inhibited proliferation, growth, and migration of hippocampal progenitor cells. In further support for a role for diminished levels of thiamine-dependent enzymes in the reduction of neurogenesis, knockout of ∝-ketoglutarate dehydrogenase in mice decreased hippocampal neurogenesis (Calingasan et al. 2008). In addition, mitochondria are especially liable to damage during reduced levels of pyruvate dehydrogenase (Zell et al. 1997) and immature neurons are particularly vulnerable to mitochondrial inhibition resulting in reduced survival of newly generated neurons (Voloboueva et al. 2010). Thus, thiamine deficiency and the resultant downstream reductions of thiamine-dependent enzymes likely contribute to the diminished levels of neurogenesis.
Reductions in hippocampal neurotrophic support might represent another factor that contributes to a compromised neurogenic niche and the resultant diminution of neurogenesis in the PTD model. Our laboratory has recently demonstrated blunted brain-derived neurotrophic factor (BDNF) protein expression in the hippocampus of PTD-treated animals (Vetreno et al. In Press). This neurotrophin plays a vital role in neurogenesis by facilitating the survival, maintenance, and growth of newly generated neurons (Cotman and Berchtold, 2002; Lee et al. 2002; Nonner et al. 1996; Sairanen et al. 2005). Genetic knockout of BDNF in mice results in increased cell death and impaired hippocampal function (Choi et al. 2009; Linnarsson et al. 2000; Monteggia et al. 2004) whereas facilitation of BDNF expression via exposure to voluntary exercise increases neurogenesis and cell survival (Berchtold et al. 2002, 2005; Farmer et al. 2004). Although the precise pathway through which BDNF exerts its pro-survival effects remains to be fully elucidated, recent work suggests that it might regulate the surface expression of α7 nicotinic ACh receptors (α7nAChRs) located on newly generated neurons (Massey et al. 2006). These α7nAChRs are critical to the survival of newly generated neurons as transgenic α7nAChR knockout mice evidence reduced survival of newborn neurons and the cells that do survival remain immature and integrate poorly (Campbell et al. 2010). Thus, one way through which PTD treatment might hinder neurogenesis is through decreased pro-survival signaling involving BDNF and α7nAChRs. Furthermore, the newborn neurons that do survive may integrate aberrantly thereby producing hippocampal impairment. However, further research is necessary to determine the relation between these factors in the impaired neurogenesis observed in the PTD model.
A third contributing factor to the PTD-induced reduction of neurogenesis appears to be a loss of regulatory cholinergic input to the hippocampus. A hallmark pathophysiological consequence of thiamine deficiency is degeneration of the fornix, a fiber tract that connects the basal forebrain cholinergic system with the hippocampal dentate gyrus. This circuit, termed the septohippocampal pathway, is necessary for normal hippocampal functioning as experimental lesion of this pathway decreases BDNF expression (Berchtold et al. 2002; Gil-Bea et al. 2010; Lapchak et al. 1993; Wrenn and Wiley, 1998) as well as cholinergic innervation to the hippocampus (Anzalone et al. 2010). In a similar fashion, experimental ablation of this circuit reduces nerve growth factor function, and impairs plasticity and memory retention (Conner et al. 2009). Thus, the reduction of afferents to the hippocampus might contribute to impaired neurogenesis indirectly by diminishing neuronal or pro-survival signals necessary for proper differentiation, migration, or integration of newly generated neurons.
In addition to the diencephalic and fiber tract degeneration (Arendt et al. 1983; Langlais and Mair, 1990; Langlais and Savage, 1995; Langlais and Zhang, 1997; Langlais et al. 1996; Markowitsch, 1988; Zhang et al. 1995), cholinergic cells of the medial septum/diagonal band (MS/DB) are vulnerable to thiamine deficiency-induced neurodegeneration. Work from our laboratory has consistently demonstrated an approximate 30% reduction in the population of choline acetyltransferase (ChAT)-positive cells in the MS/DB (Pitkin and Savage, 2001, 2004; Roland and Savage, 2009). This region is of particular import as it provides the majority of ACh innervation to the hippocampus and a substantial proportion of the cholinergic cells project directly to the dentate gyrus where they synapse on the dendrites of granule cells (Amaral et al. 2007). Experimental ablation of ChAT-positive cells in the MS/DB decreases the survival of newly generated granule cells whereas pharmacological enhancement of ChAT inputs to the hippocampus facilitate the survival of newly generated neurons (Cooper-Kuhn et al. 2004; Kotani et al. 2008; Mohapel et al. 2005; Van der Borght et al. 2005). Thus, although the effects of thiamine deficiency on ACh in relation to diminished neurogenesis have not yet been elucidated, it seems plausible that the diminished cholinergic input to the hippocampus following PTD treatment might also be a contributing factor to the reductions in neurogenesis.
A significant proportion of the hippocampal BrdU+ cells did not co-localize with any of the cellular markers during the early and late post-opisthotonus stages of PTD treatment. However, caution must be exerted in interpreting this finding as exhaustive phenotypic labeling was not conducted and some of this population of BrdU-immunopositive cells may co-localize with other cellular markers. Indeed, these BrdU-positive cells could potentially express the immature neuronal marker doublecortin that were unable to mature and become fully integrated into the granule cell layer. Thus, this population of BrdU+ cells might reflect immature or undifferentiated cells that were unable to differentiate into neurons (Morris et al. 2010; Ngwenya et al. 2008) due to a compromised neurogenic microenvironment. Thiamine deficiency and PTD treatment result in reduced thiamine-dependent enzymes essential for neurogenesis (Calingasan et al. 2008; Voloboueva et al. 2010; Zell et al. 1997; Zhao et al. 2009) as well as diminished expression of pro-neurogenic signals (e.g., ACh, BDNF [Pitkin and Savage, 2001, 2004; Roland and Savage, 2009; Vetreno et al. 2008; Vetreno et al. In Press]). Thus, a therapeutic strategy for rescuing the diminished neurogenesis in the PTD model, and perhaps in human WKS, would be to facilitate the expression of pro-neurogenic factors in the compromised neurogenic microenvironment.
It remains to be determined whether the reduced neurogenesis might be involved in the hippocampal behavioral impairments commonly observed in the PTD model. However, given the substantial evidence implicating this process in learning and memory, it appears to be a plausible explanation. Indeed, hippocampal neurogenesis is highly correlated with performance on hippocampal-dependent tasks (e.g., Aimone et al. 2006; Ambrogini et al. 2000; Gould et al. 1999a; Kee et al. 2007; Kempermann, 1997; Kempermann et al. 2008; Sisti et al. 2007; van Praag et al. 1999a) whereas experimental ablation of neurogenesis, either by administration of methylazoxymethanol acetate or irradiation, impairs performance (Madsen et al. 2003; Shors et al. 2001, 2002; Snyder et al. 2005; Winocur et al. 2006; Wojtowicz et al. 2008). Furthermore, in animal models of neuropathology, enhancement of neurogenesis via exposure to voluntary exercise leads to recovery of function on hippocampal-dependent tasks (Griesbach et al. 2004, 2009; Luo et al. 2007; Redila et al. 2006). Thus, the beneficial effects of voluntary exercise might be applicable the PTD model and serve as a non-invasive therapy.
In summary, the results of the current experiment demonstrate short- and long-term alterations of neurogenesis following a thiamine deficiency episode. Furthermore, due to the involvement of neurogenesis in hippocampal learning and memory, these data suggest that hippocampal impairment observed in the PTD model, and perhaps in human WKS, might be partially due to diminished production of new neurons. Future studies aimed at re-instituting neurogenesis via exposure of PTD-treated rats to voluntary exercise would serve to demonstrate whether impaired neurogenesis was involved in the learning and memory impairment.
4. Experimental procedure
4.1 Subjects
One hundred and twenty-eight male Sprague-Dawley rats (275 – 325 g, 2 ½ months old, Harlan Corp., IN) served as subjects. The animals were pair-housed and maintained on a 12 hr light/dark cycle (onset at 06:00 hr; offset at 18:00 hr) in a temperature (20°C) controlled vivarium. All experiments were conducted according to the National Institute of Health guide for the care and use of laboratory animals. The Institutional Animal Care and Use Committee of the State University of New York at Binghamton approved the experimental procedures used in this study. Furthermore, every effort was made to minimize animal suffering and the number of animals used.
4.2 Pyrithiamine-induced thiamine deficiency and pair-fed treatment
Animals were randomly assigned to one of the following treatment conditions: (i) pyrithiamine-induced thiamine deficiency (PTD, n = 64) or (ii) pair-fed control (PF, n = 64). Within each individual treatment condition (PTD/PF), subjects were further randomly divided into 4 stages based on previous research (see Langlais and Mair, 1990; Langlais and Savage, 1995; Langlais and Zhang, 1997; Pfefferbaum et al. 2007; Zhang et al. 1995 [n = 16 per stage (pre-symptomatic, ataxic, early post-opisthotonus, late post-opisthotonus [see below])]), each with two separate time points (n = 8 per time point [24-hour, 28-day]).
Subjects in the PTD treatment group were free-fed thiamine-deficient chow (Teklad Diets, Madison, WI) and received daily injections of pyrithiamine hydrobromide (0.25 mg/kg, i.p. [Sigma-Aldrich, St. Louis, MO]). Pyrithiamine is an inhibitor of thiamine pyrophosphokinase, an enzyme that converts thiamine to thiamine pyrophosphate (Butterworth and Heroux, 1989). Subjects in the PF treatment group were fed an amount of thiamine deficient chow equivalent to the amount consumed by the PTD animals the previous day (to replicate the anorexic effects) and received daily injections of thiamine hydrochloride (0.4 mg/kg, i.p. [Sigma-Aldrich, St. Louis, MO]). Animals that receive PF treatment do not demonstrate any changes in brain levels of thiamine-dependent enzymes (Butterworth and Heroux, 1989).
Pyrithiamine- and PF-treated subjects assigned to the pre-symptomatic stage were placed back on vitamin fortified rat chow on Day 8 after the initiation of their respective treatments. Subjects in the ataxic stage were placed back on ad libitum vitamin fortified rat chow on Day 15 of their respective treatments and PTD animals received one reversal dose of thiamine hydrochloride (100 mg/kg, i.p. [Sigma-Aldrich, St. Louis, MO]) to halt the progression of thiamine deficiency. This stage was characterized by the presence of ataxia and an approximate 15% reduction of body weight, but occurred before loss of righting reflex. Subjects were then monitored for 24 hr to ensure the absence of opisthotonus development.
Subjects in the early and late post-opisthotonus stages were monitored until they developed neurological symptoms of ataxia, loss of righting reflex, and eventually opisthotonus activity (approximate Day 16 of PTD treatment). Following 4 hr 15 min of the onset of opisthotonus, PTD- and PF-treated animals were placed back on ad libitum access to vitamin fortified rat chow and PTD animals were administered a reversal injection of thiamine hydrochloride (100 mg/kg, i.p.). A second dose of thiamine hydrochloride (100 mg/kg, i.p.) was administered 24 hr later to ensure survival and recovery. Administration of thiamine and restoration of normal chow reversed the acute neurological symptoms within 8 hr and all animals were recovered within 24 – 48 hr of PTD treatment.
4.3 5′-bromo-2′-deoxyuridine (BrdU) administration
To label proliferating cells, the thymidine analog 5′-bromo-2′-deoxyuridine (BrdU), which is incorporated into DNA during the DNA synthesis phase, was administered in a single dose (300 mg/kg [in sterile 0.9% saline (30 mg/ml)], i.p., Sigma-Aldrich, St. Louis, MO). This dose was used because it fully saturates the brain and labels all cells that are proliferating at the time of administration (Cameron and McKay, 2001; Eadie et al. 2005). Subjects received BrdU at one of the 4 stages of PTD/PF treatment: (1) pre-symptomatic stage (Day 8 of PTD/PF treatment [n = 32 (PTD/PF = 16)]), (2) ataxic stage (Day 15 of PTD/PF treatment [n = 32 (PTD/PF = 16)]), (3) early post-opisthotonus stage (24 hr following thiamine reversal [n = 32 (PTD/PF = 16)]), or (4) late post-opisthotonus stage (28 days following thiamine reversal [n = 32 (PTD/PF = 16)]). Subjects at each stage were then sacrificed at one of two time points: (1) 24-hours after BrdU administration to assess cell proliferation (n = 16 [PTD/PF = 8]) or (2) 28-days following BrdU administration to assess cell survival and differentiation (n = 16 [PTD/PF = 8]; see Figure 1 for experimental outline).
Figure 1.

Schematic of the experimental design. On Day 1, all subjects were started on either PF or PTD treatment. Subjects that were assigned to the pre-symptomatic stage (PRE; n=32) were administered BrdU on Day 8 and their respective treatments were terminated. Subjects that were assigned to the ataxic stage (ATAX; n=32) were administered BrdU on Day 15 and their respective treatments were terminated. Subjects that were assigned to the early post-opisthotonus stage (E. Post; n=32) were administered BrdU 24 hr after thiamine reversal whereas subjects that were assigned to the late post-opisthotonus stage (L. Post; n=32) were administered BrdU 28 days after thiamine reversal. Subjects in each group were then sacrificed either 24-hours or 28-days after BrdU administration.
4.4 Tissue preparation
At the time of tissue collection, all animals were administered a lethal dose of Sleep Away (0.5 mg/kg, i.p. [26.0% sodium pentobarbital in 7.8% isopropyl alcohol and 20.7% propylene glycol solution], Fort Dodge Animal Health, Fort Dodge, IA) and transcardially perfused with phosphate-buffered saline (PBS) followed by 4.0% paraformaldehyde (Electron Microscopy Services, Hatfield, PA) in PBS. Brains were extracted and post-fixed in 4.0% paraformaldehyde/PBS solution for 24 hr followed by 4 days of fixation in a 30% sucrose solution. Frozen coronal sections (40 μm [see Gould et al. 1999a; Klintsova et al. 2007]) were cut on a sliding microtome (Sm2000r; Lecia Instruments, Wetzlar, Germany) from the level of the anterior commissure to the level of the posterior pontine tegmentum. Sections were then sequentially collected into individual wells and stored at −20°C in a cryoprotectant solution consisting of 30% glycerol and 30% ethylene glycol for later immunohistological processing.
4.5 BrdU immunohistochemistry
A systematic random sampling procedure similar to a previously published protocol (see Klintsova et al. 2007) was used to select tissue from the hippocampus. Sampling was confined to the dorsal hippocampus with the first section in series selected randomly within the first 5 sections containing the hippocampal dentate gyrus. Every 5th section was subsequently placed into an individual well resulting in 8 sections per animal with approximately 200 μm between each section.
To assess proliferation and survival of newly generated cells, immunohistochemical staining of BrdU-immunoreactive cells was performed as follows: Free-floating sections were rinsed in 0.1 M Tris-buffered saline (TBS; pH = 7.4) and endogenous peroxidase activity was quenched via a 30 min wash in a 0.3% H2O2 solution. Sections were then incubated for 2 hr in a 50% formamide/2X saline-sodium citrate buffer (SSC) solution at 65°C and washed in a 2X SSC buffer. DNA was denatured via incubation in 2N HCl at 37°C for 30 min followed by a rinse in 0.1 M boric acid in TBS (pH=8.6) for 10 min. Sections were rinsed in TBS and blocked in a solution containing 0.2% TritonX-100/3.0% normal goat serum (NGS; Vector Laboratories, Burlingame, CA) in TBS (NGS-TBS) for 60 min. Sections were then incubated for 48 hr in NGS-TBS containing the rat monoclonal anti-BrdU antibody (1:200; Accurate Chemical, Westbury, NY) at 4°C on an orbital shaker. Negative control for BrdU-immunoreactive specificity was conducted on sections randomly selected from PTD- and PF-treated animals employing the above procedures with the exception that the primary antibody was omitted from the 48 hr incubation step. The sections were then rinsed in TBS and incubated for 2 hr in the secondary antibody (NGS-TBS + biotinylated goat anti-rat IgG [1:200; Vector Laboratories, Burlingame, CA]) for 2 hr. The sections were rinsed in TBS and incubated for 2 hr in an avidin-biotin complex solution (ABC; Vectastain ABC Kit; Vector Laboratories, Burlingame, CA), followed by submersion in the chromogen 3,3′-diaminobenzidine (DAB Fast Tablet, Sigma-Aldrich, St. Louis, MO) to help visualize staining. Sections were rinsed, mounded on gelatinized slides, and cover slipped.
4.6 Phenotype analysis and fluorescent immunohistochemistry
A similar random sampling procedure was used for cell phenotype analyses, except that every 10th section was selected for a total of 4 section per animal and co-localization was determined for BrdU/NeuN (NEUronal Nuclei; neuron phenotype), BrdU/GFAP (Glial Fibrillary Acidic Protein; astrocyte phenotype), BrdU/O4 (Oligodendrocyte marker O4, oligodendrocyte phenotype), and BrdU/Iba-1 (Ionized calcium-Binding Adaptor molecule-1; microglia phenotype [see Helfer et al. 2009a, 2009b]). All 8 subjects from each PTD stage and a subset (n = 6 per group) of PF subjects from each stage were randomly selected to represent each group.
The following primary antibodies and dilutions were used for fluorescent immunohistochemistry: rat monoclonal anti-BrdU (1:200; Accurate Chemical, Westbury, NY), mouse monoclonal anti-NeuN (1:500; Millipore, Temecula, CA), rabbit polyclonal anti-GFAP (1:1000; Millipore, Temecula, CA), mouse monoclonal anti-O4 (1:500; Millipore, Temecula, CA), and rabbit anti-Iba-1 (1:500; Wako, Richmond, VA). The secondary antibodies were ordered from Jackson ImmunoResearch (West Grove, PA) and the following dilutions were used: biotin-SP-conjugated AffiniPure donkey anti-rat IgG (1:200), aminomethylcoumarin acetate (AMCA)-conjugated AffiniPure donkey anti-rabbit IgG (1:200), cyanine (Cy)2-conjugated streptavidin (1.7 μL/mL), and Cy3-conjugated AffiniPure donkey anti-mouse IgG (1:200).
For triple labeling, sections were first rinsed in 0.1 M TBS and were then incubated for 2 hr in a 50% formamide/2X SSC solution. After a wash in 2X SSC, DNA was denatured via a 30 min rinse in 2N HCl at 37°C followed by a rinse in a solution of 0.1 M boric acid in TBS (pH=8.6) for 10 min. Sections were rinsed in TBS and blocked with 0.2% Trition X-100/3.0% normal donkey serum (NDS; Jackson ImmunoResearch, West Grove, PA) in TBS for 60 min. Tissue was incubated for 48 hr in a primary antibody cocktail at 4°C on an orbital shaker and then rinsed in TBS and immersed in a secondary antibody cocktail for 2 hr. The sections were subsequently rinsed in TBS and incubated for 3 hr in Cy2-conjugated streptavidin to amplify the BrdU signal. Following further TBS rinses, the tissue was mounted onto non-gelatinized slides and cover slipped with an anti-fade medium (ProLong Antifade Media, Molecular Probes/Invitrogen, Carlsbad, CA). The edges of each slide were sealed with clear nail polish after the mounting medium had dried for 24 hr and the slides were stored at −20°C. For each protocol, control for specificity was conducted on sections randomly selected from PTD- and PF-treated animals employing the above procedures with the exception that the primary antibody cocktail was omitted.
4.7 Unbiased stereology
Unbiased stereology was used to estimate BrdU immunopositive cell counts throughout the SGZ and GCL of the dorsal hippocampal dentate gyrus. The GCL/SGZ of the dorsal hippocampal was sampled between −2.12 mm and −3.80 mm from Bregma based on the atlas of Paxinos and Watson (1986). Parameters for sampling the region of interest were modified from a previously published study assessing the number of BrdU-immunoreactive cells in the GCL (Helfer et al. 2009b). All cell counts were conducted on coded slides to ensure that the investigator was blind to the treatment and stage conditions. A low power image (Zeiss Plan-NEOFLUAR 5×) was captured with a digital camera (DVC-1310; DVC Company, Austin, TX) on a Zeiss microscope (Axioscope 2-Plus) with an attached 3-axis motorized stage. Using StereoInvestigator 8.0 software (MicroBrightField, Williston, VT), a contour of the region of interest was traced. The GCL/SGZ was defined as the band of tissue between the molecular layer and hilus of the dentate gyrus. The BrdU-immunopositive cells were counted with a Zeiss Plan-NEOFLUAR 40× objective (see Clark et al. 2009; Herrera et al. 2003) using the Optical Fractionator approach. The fractionator sampling formula consisted of a section sampling fraction (ssf = 1/5), an area sampling fraction representing a ratio between sampling grid size and counting frame size (asf = 50 μm × 50 μm/100 μm × 75 μm) and a height sampling fraction (hsf = 32μm/40 μm). The equation for determining cell estimates using unbiased stereology was: N = ΣQ− 1/ssf × 1/asf × 1/hsf, where Q constitutes the actual number of cells counted in a specimen and N represents the total cell estimate. A guard zone of 2 μm and a dissector height of 28 μm were used. The frozen tissue sections were cut at 40 μm, but immunostaining and mounting resulted in a reduction in the thickness of the tissue.
4.8 Confocal microscopy
Confocal microscopy was conducted to determine cellular phenotype rates of BrdU-immunoreactive cells in the GCL of animals sacrificed 28 days after BrdU administration. All phenotyping was conducted on coded slides to ensure that the investigator was blind to the treatment and stage conditions. The sections were assessed using a Zeiss LSM 510 laser scanning confocal microscope (Carl Zeiss MicroImaging, Thornwood, NY) equipped with standard high-power objectives and corresponding software (LSM 510 META). Every 10th section in series from subjects in each group and stage was assessed using a 40× and 63× oil-immersion lens (see Chevallier et al. 2005; Clark et al. 2009; Cooper-Kuhn et al. 2004; Guo et al. 2009). Single optical sections (Z-stacks [1 μm]; 1-airy unit) were collected throughout the section using lasers with excitation/emission wavelengths of 350 nm/450 nm (AMCA, blue), 492 nm/510 nm (Cy2, green), and 550 nm/570 nm (Cy3, orange). A minimum of 30 BrdU-immunoreactive cells per animal (see Chevallier et al. 2005) for each triple labeling procedure was randomly sampled and an experimenter blind to both treatment and stage conditions performed quantification of cellular phenotype.
4.9 Intraventricular distance
The intraventricular distance (IVD) was used to quantify thalamic tissue loss following PTD treatment (Anzalone et al. 2010; Robinson and Mair, 1992; Roland and Savage, 2009). A researcher blind to treatment determined it by measuring the distance (in mm) between the floor of the 3rd ventricle and the roof of the 3rd ventricle at the following approximate locations based on the atlas of Paxinos and Watson (1986 [from Bregma: −1.80 mm to −3.30 mm]). Quantitative measures were collected on a Nikon light microscope (Nikon Eclipse E400; Nikon Instruments, Melville, NY, USA) using a Scion 1394 camera JAVA module (Scion Corp; Fredrick, MD, USA) and analyzed using an image analyzer program (IMAGE-J, v.1.34, NIH, Bethesda, MD, USA) on a Macintosh G4 computer.
4.10 Data analyses
All analyses were conducted using Statview (SAS Institute, Cary, NC). The first analyses performed were to assess whether there were differences in PF brain measures as a function of stage of treatment on brain integrity (thalamic mass and neurogenesis). Two between-subjects factorial ANOVAs (STAGE: pre-symptomatic, ataxic, early post-opisthotonus, late post-opisthotonus; TIME POINT: 24-hour, 28-day) were performed to assess differences in IVDs and BrdU-immunoreactive cell estimates across PF subgroups. One between-subject factor ANOVAs (STAGE) were performed to assess percent co-localization and absolute estimates of co-localization across PF treatment. None of the neurogenesis analyses yielded significant results and the PF data was pooled into a single control group for further comparisons (see below).
A three between-subjects factorial ANOVA (GROUP: PF, PTD; STAGE: pre-symptomatic, ataxic, early post-opisthotonus, late post-opisthotonus; TIME POINT: 24-hour, 28-day) was conducted to assess body weight. Two between-subjects factorial ANOVAs (TREATMENT: PF, pre-symptomatic, ataxic, early post-opisthotonus, late post-opisthotonus; TIME POINT: 24-hour, 28-day) were conducted to assess IVD measures and estimates of BrdU-immunopositive cells. Both the percent co-localization and absolute co-localization estimates were analyzed with one between-subject factor ANOVAs (TREATMENT). The mean estimate of absolute phenotype was generated by multiplying BrdU-positive cell estimates by the co-localization percentage obtained for neurogenesis, gliogenesis, and undifferentiated BrdU+ cells. A one between-subject factor ANOVA (TREATMENT) was also performed to assess the unbiased stereology coefficient of error. Fisher's PLSD was used for post-hoc testing when applicable. All data are expressed as mean values ± the standard error of the mean (S.E.M.) and significance was defined at a level of p≤0.05.
Acknowledgements
This work was supported by a research grant NINDS 054272 to LMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrous DN, Koehl M, Le Moal M. Adult neurogenesis: From precursors to network and physiology. Physiol. Rev. 2005;85:523–569. doi: 10.1152/physrev.00055.2003. [DOI] [PubMed] [Google Scholar]
- Aimone JB, Wiles J, Gage FH. Potential role for adult neurogenesis in the encoding of tie in new memories. Nat. Neurosci. 2006;9:723–727. doi: 10.1038/nn1707. [DOI] [PubMed] [Google Scholar]
- Altman J, Das GD. Post-natal origin of microneurones in the rat brain. Nature. 1965;207:953–956. doi: 10.1038/207953a0. [DOI] [PubMed] [Google Scholar]
- Amaral DG, Scharfman HE, Lavenex P. The dentate gyrus: Fundamental neuroanatomical organization. Prog. Brain Res. 2007;163:3–22. doi: 10.1016/S0079-6123(07)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrogini P, Cuppini R, Cuppini C, Ciaroni S, Cecchini T, Ferri P, Sartini S, Del Grande P. Spatial learning affects immature granule cell survival in adult rat dentate gyrus. Neurosci. Lett. 2000;296:21–24. doi: 10.1016/s0304-3940(00)01074-0. [DOI] [PubMed] [Google Scholar]
- Anzalone S, Vetreno RP, Ramos RL, Savage LM. Cortical cholinergic abnormalities contribute to the amnestic state induced by pyrithiamine-induced thiamine deficiency in the rat. Eur. J. Neurosci. 2010;32:847–858. doi: 10.1111/j.1460-9568.2010.07358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt T, Bigl V, Arendt A, Tennstedt A. Loss of neurons in the nucleus basalis of Meynert in Alzheimer's disease, paralysis agitans and Korsakoff's Disease. Acta Neuropathol. 1983;61:101–108. doi: 10.1007/BF00697388. [DOI] [PubMed] [Google Scholar]
- Bauer S, Patterson PH. The cell cycle-apoptosis connection revisited in the adult brain. J. Cell Biol. 2005;171:641–650. doi: 10.1083/jcb.200505072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchtold NC, Chinn G, Chou M, Kesslak JP, Cotman CW. Exercise primes a molecular memory for brain-derived neurotrophic factor protein induction in the rat hippocampus. Neurosci. 2005;133:859–861. doi: 10.1016/j.neuroscience.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Kesslak JP, Cotman CW. Hippocampal brain-derived neurotrophic factor gene regulation by exercise and the medial septum. J. Neurosci. Res. 2002;68:511–521. doi: 10.1002/jnr.10256. [DOI] [PubMed] [Google Scholar]
- Bruel-Jungerman E, Laroche S, Rampon C. New neurons in the dentate gyrus are involved in the expression of enhanced long-term memory following environmental enrichment. Eur. J. Neurosci. 2005;21:513–521. doi: 10.1111/j.1460-9568.2005.03875.x. [DOI] [PubMed] [Google Scholar]
- Butters N. The Wernicke-Korsakoff syndrome: A review of psychological, neuropathological and etiological factors. Curr. Alcohol. 1981;8:205–232. [PubMed] [Google Scholar]
- Butterworth RF, Heroux M. Effect of pyrithiamine treatment and subsequent thiamine rehabilitation on regional cerebral amino acids and thiamine-dependent enzymes. J. Neurochem. 1989;52:1079–1084. doi: 10.1111/j.1471-4159.1989.tb01850.x. [DOI] [PubMed] [Google Scholar]
- Calingasan NY, Ho DJ, Willie EJ, Campagna MV, Ruan J, Dumont M, Yang L, Shi Q, Gibson GE, Beal MF. Influence of mitochondrial enzyme deficiency on adult neurogenesis in mouse models of neurodegenerative diseases. Neurosci. 2008;153:986–996. doi: 10.1016/j.neuroscience.2008.02.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron HA, McKay R. Restoring production of hippocampal neurons in old age. Nat. Neurosci. 1999;2:894–897. doi: 10.1038/13197. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay R. Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J. Comp. Neurol. 2001;435:406–417. doi: 10.1002/cne.1040. [DOI] [PubMed] [Google Scholar]
- Cameron HA, Woolley CS, McEwen BS, Gould E. Differentiation of newly born neurons and glia in the dentate gyrus of the adult rat. Neuroscience. 1993;36:337–344. doi: 10.1016/0306-4522(93)90335-d. [DOI] [PubMed] [Google Scholar]
- Campbell NR, Fernandes CC, Halff AW, Berg DK. Endogenous signaling through α7-containing nicotinic receptors promotes maturation and integration of adult-born neurons in the hippocampus. J. Neurosci. 2010;30:8734–8744. doi: 10.1523/JNEUROSCI.0931-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caulo M, Van Hecke J, Toma L, Ferretti A, Tartaro A, Colosimo C, Romani GL, Uncic A. Functional MRI study of the diencephalic amnesia in Wernicke-Korsakoff syndrome. Brain. 2005;128:1584–1594. doi: 10.1093/brain/awh496. [DOI] [PubMed] [Google Scholar]
- Chevallier NL, Soriano S, Kand DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am. J. Pathol. 2005;167:151–159. doi: 10.1016/S0002-9440(10)62962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chirumamilla S, Sun D, Bullock MR, Colello RJ. Traumatic brain injury induced cell proliferation in the adult mammalian central nervous system. J. Neurotrauma. 2002;19:693–703. doi: 10.1089/08977150260139084. [DOI] [PubMed] [Google Scholar]
- Choi SH, Li Y, Parada LF, Sisodia SS. Regulation of hippocampal progenitor cell survival, proliferation and dendritic development by BDNF. Mol. Neurodegener. 2009;4:1–12. doi: 10.1186/1750-1326-4-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie BR, Cameron HA. Neurogenesis in the adult hippocampus. Hippocampus. 2006;16:199–207. doi: 10.1002/hipo.20151. [DOI] [PubMed] [Google Scholar]
- Chumley MJ, Catchpole T, Silvany RE, Kernie SG, Henkemeyer M. EphB receptors regulate stem/progenitor cell proliferation, migration, and polarity during hippocampal neurogenesis. J. Neurosci. 2007;27:13481–13490. doi: 10.1523/JNEUROSCI.4158-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark PJ, Brzezinska WJ, Puchalski EK, Krone DA, Rhodes JS. Functional analysis of neurovascular adaptations to exercise in the dentate gyrus of young adult mice associated with cognitive gain. Hippocampus. 2009;19:937–950. doi: 10.1002/hipo.20543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Franks KM, Titterness AK, Russell K, Merrill DA, Christie BR, Sejnowski TJ, Tuszynski MH. NGF is essential for hippocampal plasticity and learning. J. Neurosci. 2009;29:10883–10889. doi: 10.1523/JNEUROSCI.2594-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper-Kuhn CM, Winkler J, Kuhn HG. Decreased neurogenesis after cholinergic forebrain lesion in the adult rat. J. Neurosci. Res. 2004;77:155–165. doi: 10.1002/jnr.20116. [DOI] [PubMed] [Google Scholar]
- Cotman CW, Berchtold NC. Exercise: A behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25:295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- Crews FT, Nixon K, Wilkie ME. Exercise reverses ethanol inhibition of neural stem cell proliferation. Alcohol. 2004;33:63–71. doi: 10.1016/j.alcohol.2004.04.005. [DOI] [PubMed] [Google Scholar]
- Dayer AG, Ford AA, Cleaver KM, Yassaee M, Cameron HA. Short-term and long-term survival of new neurons in the rat dentate gyrus. J. Comp. Neurol. 2003;460:563–572. doi: 10.1002/cne.10675. [DOI] [PubMed] [Google Scholar]
- Eadie BD, Redila VA, Christie BR. Voluntary exercise alters the cytoarchitecture of the adult dentate gyrus by increasing cellular proliferation, dendritic complexity, and spine density. J. Comp. Neurol. 2005;486:39–47. doi: 10.1002/cne.20493. [DOI] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Bjork-Eriksson T, Alborn A, Nordborg C, Peterson DA, Gage FH. Neurogenesis in the adult human hippocampus. Nat. Med. 1998;4:1313–1317. doi: 10.1038/3305. [DOI] [PubMed] [Google Scholar]
- Fabel K, Wolf SA, Ehninger D, Babu H, Leal-Galicia P, Kempermann G. Additive effects of physical exercise and environmental enrichment on adult hippocampal neurogenesis in mice. Front. Neurosci. 2009;3:1–7. doi: 10.3389/neuro.22.002.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fama R, Marsh L, Sullivan EV. Dissociation of remote and anterograde memory impairment and neural correlates in alcoholic Korsakoff syndrome. J. Int. Neuropsychol Soc. 2004;10:427–441. doi: 10.1017/S135561770410310X. [DOI] [PubMed] [Google Scholar]
- Farmer J, Zhao X, van Praag H, Wodtke K, Gage FH, Christie BR. Effects of voluntary exercise on synaptic plasticity and gene expression in the dentate gyrus of adult male Sprague-Dawley rats in vivo. Neurosci. 2004;124:71–79. doi: 10.1016/j.neuroscience.2003.09.029. [DOI] [PubMed] [Google Scholar]
- Gibson GE, Ksiezak-Reding H, Sheu KF, Mykytyn V, Blass JP. Correlation of enzymatic, metabolic, and behavioral deficits in thiamin deficiency and its reversal. Neurochem. Res. 1984;9:803–814. doi: 10.1007/BF00965667. [DOI] [PubMed] [Google Scholar]
- Gil-Bea FJ, Solas M, Mateos L, Winblad B, Ramirez MJ, Cedazo-Minguez A. Cholinergic hypofunction impairs memory acquisition possibly through hippocampal Arc and BDNF downregulation. Hippocampus. 2010 doi: 10.1002/hipo.20812. doi: 10.1002/hipo.20812. [DOI] [PubMed] [Google Scholar]
- Gold JJ, Squire LR. The anatomy of amnesia: Neurohistological analysis of three new cases. Learn. Mem. 2006;13:699–710. doi: 10.1101/lm.357406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould E, Beylin A, Tanapat P, Reeves A, Shors TJ. Learning enhances adult neurogenesis in the hippocampal formation. Nat. Neurosci. 1999a;2:260–265. doi: 10.1038/6365. [DOI] [PubMed] [Google Scholar]
- Gould E, Tanapat P, Hastings NB, Shors TJ. Hippocampal neurogenesis in adult Old World primates. Proc. Natl. Acad. Sci. USA. 1999b;96:5263–5267. doi: 10.1073/pnas.96.9.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Gomez-Pinilla F. Exercise-induced improvement in cognitive performance after traumatic brain injury in rats is dependent on BDNF activation. Brain Res. 2009;1288:105–115. doi: 10.1016/j.brainres.2009.06.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Hovda DA, Molteni R, Wu A, Gomez-Pinilla F. Voluntary exercise following traumatic brain injury: Brain-derived neurotrophic factor upregulation and recovery of function. Neurosci. 2004;125:129–139. doi: 10.1016/j.neuroscience.2004.01.030. [DOI] [PubMed] [Google Scholar]
- Guo Y, Zhang Z, Wang S, Sui Y, Sun Y. Notch1 signaling, hippocampal neurogenesis and behavioral responses to chronic unpredicted mild stress in adult ischemic rats. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2009;33:688–694. doi: 10.1016/j.pnpbp.2009.03.022. [DOI] [PubMed] [Google Scholar]
- Harper CG, Kril JJ. Neuropathology of alcoholism. Alcohol Alcohol. 1990;25:207–216. doi: 10.1093/oxfordjournals.alcalc.a044994. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Shetty AK. Decreased neuronal differentiation of newly generated cells underlies reduced hippocampal neurogenesis in chronic temporal lobe epilepsy. Hippocampus. 2010;20:97–112. doi: 10.1002/hipo.20594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazell AS, Butterworth RF. Update of cell damage mechanisms in thiamine deficiency: Focus on oxidative stress, excitotoxicity and inflammation. Alcohol Alcohol. 2009;44:141–147. doi: 10.1093/alcalc/agn120. [DOI] [PubMed] [Google Scholar]
- He J, Overstreet DH, Crews FT. Abstinence from moderate alcohol self-administration alters progenitor cell proliferation and differentiation in multiple brain regions of male and female P rats. Alcohol. Clin. Exp. Res. 2009;33:129–138. doi: 10.1111/j.1530-0277.2008.00823.x. [DOI] [PubMed] [Google Scholar]
- Helfer JL, Calizo LH, Dong WK, Goodlett CR, Greenough WT, Klintsova AY. Binge-like postnatal alcohol triggers cortical gliogenesis in adolescent rats. J. Comp. Neurol. 2009a;514:259–271. doi: 10.1002/cne.22018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helfer JL, Goodlett CR, Greenough WT, Klintsova AY. The effects of exercise on adolescent hippocampal neurogenesis in a rat model of binge alcohol exposure during the brain growth spurt. Brain Res. 2009b;1294:1–11. doi: 10.1016/j.brainres.2009.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera DG, Yague AG, Johnsen-Soriano S, Bosch-Morell F, Collado-Morente L, Muriach M, Romero FJ, Garcia-Verdugo J. Selective impairment of hippocampal neurogenesis by chronic alcoholism: Protective effects of an antioxidant. Proc. Natl. Acad. Sci. USA. 2003;100:7919–7924. doi: 10.1073/pnas.1230907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Nakashima K, Clemenson GD, Jr, Mejia E, Mathews E, Ure K, Ogawa S, Sinton CM, Gage FH, Hsieh J. Epigenetic modulation of seizure-induced neurogenesis and cognitive decline. J. Neurosci. 2007;27:5967–5975. doi: 10.1523/JNEUROSCI.0110-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder S, Gibson GE. Thiamine deficiency: A model of metabolic encephalopathy and of selective neuronal vulnerability. In: McCandless DW, editor. Metabolic Encephalopathy. Springer Science + Business Media, LLC; New York, NY: 2009. pp. 235–260. [Google Scholar]
- Kee N, Teixeira CM, Wang AH, Frankland PW. Preferential incorporation of adult-generated granule cells into spatial memory networks in the dentate gyrus. Nat. Neurosci. 2007;10:355–362. doi: 10.1038/nn1847. [DOI] [PubMed] [Google Scholar]
- Kempermann G. The neurogenic reserve hypothesis: What is adult hippocampal neurogenesis good for? Trends Neurosci. 2008;31:163–169. doi: 10.1016/j.tins.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature. 1997;86:493–496. doi: 10.1038/386493a0. [DOI] [PubMed] [Google Scholar]
- Kempermann G, Wiskott L, Gage FH. Functional significance of adult neurogenesis. Curr. Opin. Neurobiol. 2004;14:186–191. doi: 10.1016/j.conb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Klintsova AY, Helfer JL, Calizo LH, Dong WK, Goodlett CR, Greenough WT. Persistent impairment of hippocampal neurogenesis in young adult rats following early postnatal alcohol exposure. Alcohol. Clin. Exp. Res. 2007;31:2073–2082. doi: 10.1111/j.1530-0277.2007.00528.x. [DOI] [PubMed] [Google Scholar]
- Kopelman MD. The Korsakoff syndrome. Br. J. Psychiatry. 1995;166:154–173. doi: 10.1192/bjp.166.2.154. [DOI] [PubMed] [Google Scholar]
- Kopelman MD, Thomson AD, Guerrini I, Marshall E. The Korsakoff syndrome: Clinical aspects, psychology and treatment. Alcohol Alcohol. 2009;44:148–154. doi: 10.1093/alcalc/agn118. [DOI] [PubMed] [Google Scholar]
- Kotani S, Yamauchi T, Teramoto T, Ogura H. Donepezil, an acetylcholinesterase inhibitor, enhances adult hippocampal neurogenesis. Chem. Biol. Inter. 2008;175:227–230. doi: 10.1016/j.cbi.2008.04.004. [DOI] [PubMed] [Google Scholar]
- Kril JJ. Neuropathology of thiamine deficiency disorders. Metab. Brain Dis. 1996;11:9–17. doi: 10.1007/BF02080928. [DOI] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: Age-related decrease of neuronal progenitor proliferation. J. Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HG, Palmer TD, Fuchs E. Adult neurogenesis: A compensatory mechanism for neuronal damage. Eur. Arch. Psychiatry Clin. Neurosci. 2001;251:152–158. doi: 10.1007/s004060170035. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Mair RG. Protective effects of the glutamate antagonist MK-801 on pyrithiamine-induced lesions and amino acid changes in rat brain. J. Neurosci. 1990;10:1664–1674. doi: 10.1523/JNEUROSCI.10-05-01664.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlais PJ, Savage LM. Thiamine deficiency in rats produces cognitive and memory deficits on spatial tasks that correlate with tissue loss in diencephalon, cortex and white matter. Behav. Brain Res. 1995;68:75–89. doi: 10.1016/0166-4328(94)00162-9. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang S. Cortical and subcortical white matter damage without Wernicke's Encephalopathy after recovery from thiamine deficiency in the rat. Alcohol. Clin. Exp. Res. 1997;21:434–443. doi: 10.1111/j.1530-0277.1997.tb03788.x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Mandel RJ, Mair RG. Diencephalic lesions, learning impairments, and intact retrograde memory following acute thiamine deficiency in the rat. Behav. Brain Res. 1992;48:177–185. doi: 10.1016/s0166-4328(05)80155-x. [DOI] [PubMed] [Google Scholar]
- Langlais PJ, Zhang S, Savage LM. Neuropathology of thiamine deficiency: An update on the comparative analysis on human disorders and experimental models. Metab. Brain Dis. 1996;11:19–37. doi: 10.1007/BF02080929. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Araujo DM, Hefti F. Cholinergic regulation of hippocampal brain-derived neurotrophic factor mRNA expression: Evidence from lesion and chronic cholinergic drug treatment studies. Neurosci. 1993;52:575–585. doi: 10.1016/0306-4522(93)90407-7. [DOI] [PubMed] [Google Scholar]
- Lee J, Duan W, Mattson MP. Evidence that brain-derived neurotrophic factor is required for basal neurogenesis and mediates, in part, the enhancement of neurogenesis by dietary restriction in the hippocampus of adult mice. J. Neurochem. 2002;82:367–375. doi: 10.1046/j.1471-4159.2002.01085.x. [DOI] [PubMed] [Google Scholar]
- Leong DK, Butterworth RF. Neuronal cell death in Wernicke's Encephalopathy: Pathophysiologic mechanisms and implications for PET imaging. Metab. Brain Dis. 1996;11:71–79. doi: 10.1007/BF02080932. [DOI] [PubMed] [Google Scholar]
- Linnarsson S, Willsion CA, Ernfors P. Cell death in regenerating populations of neurons in BDNF mutant mice. Mol. Brain Res. 2000;75:61–69. doi: 10.1016/s0169-328x(99)00295-8. [DOI] [PubMed] [Google Scholar]
- Luo CX, Jiang J, Zhou QG, Zhu XJ, Wang W, Zhang ZJ, Han X, Zhu DY. Voluntary exercise-induced neurogenesis in the post-ischemic dentate gyrus is associated with spatial memory recovery from stroke. J. Neurosci. Res. 2007;85:1637–1646. doi: 10.1002/jnr.21317. [DOI] [PubMed] [Google Scholar]
- Madsen TM, Kristjansen PEG, Bolwig TG, Wortwein G. Arrested neuronal proliferation and impaired hippocampal function following fractionated brain irradiation in the adult rat. Neurosci. 2003;119:635–642. doi: 10.1016/s0306-4522(03)00199-4. [DOI] [PubMed] [Google Scholar]
- Mair RG. On the role of thalamic pathology in diencephalic amnesia. Rev. Neurosci. 1994;5:105–140. doi: 10.1515/revneuro.1994.5.2.105. [DOI] [PubMed] [Google Scholar]
- Markowitsch HJ. Diencephalic amnesia: A reorientation towards tracts? Brain Res. Rev. 1988;13:351–370. doi: 10.1016/0006-8993(88)91226-7. [DOI] [PubMed] [Google Scholar]
- Massey KA, Zago WM, Berg DK. BDNF up-regulates α7 nicotinic acetylcholine receptor levels on subpopulations of hippocampal interneurons. Mol. Cell. Neurosci. 2006;33:381–388. doi: 10.1016/j.mcn.2006.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapel P, Leanza G, Kokaia M, Lindvall O. Forebrain acetylcholine regulates adult hippocampal neurogenesis and learning. Neurobiol. Aging. 2005;26:939–946. doi: 10.1016/j.neurobiolaging.2004.07.015. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Barrot M, Powell CM, Berton O, Galanis V, Gemelli T, Meuth S, Nagy A, Greene RW, Nestler EJ. Essential role of brain-derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. USA. 2004;101:10827–10832. doi: 10.1073/pnas.0402141101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SA, Eaves DW, Smith AR, Nixon K. Alcohol inhibition of neurogenesis: A mechanism of hippocampal neurodegeneration in an adolescent alcohol abuse model. Hippocampus. 2010;20:596–607. doi: 10.1002/hipo.20665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatomi H, Kuriu T, Okabe S, Yamamoto S, Hatano O, Kawahara N, Tamura A, Kirino T, Nakafuku M. Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell. 2002;110:429–441. doi: 10.1016/s0092-8674(02)00862-0. [DOI] [PubMed] [Google Scholar]
- Ngwenya LB, Rosene DL, Peters A. An ultrastructural characterization of the newly generated cells in the adult monkey dentate gyrus. Hippocampus. 2008;18:210–220. doi: 10.1002/hipo.20384. [DOI] [PubMed] [Google Scholar]
- Nixon K, Kim DH, Potts EN, He J, Crews FT. Distinct cell proliferation events during abstinence after alcohol dependence: Microglia proliferation precedes neurogenesis. Neurobiol. Dis. 2008;31:218–229. doi: 10.1016/j.nbd.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonner D, Barrett EF, Barrett JN. Neurotrophin effects on survival and expression of cholinergic properties in cultured rat septal neurons under normal and stress conditions. J. Neurosci. 1996;16:6665–6675. doi: 10.1523/JNEUROSCI.16-21-06665.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM. The role of seizure-induced neurogenesis in epileptogenesis and brain repair. Epilepsy Res. 2002;50:179–189. doi: 10.1016/s0920-1211(02)00078-5. [DOI] [PubMed] [Google Scholar]
- Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J. Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic Press; San Diego, CA: 1986. [Google Scholar]
- Pfefferbaum A, Adalsteinsson E, Bell RL, Sullivan EV. Development and resolution of brain lesions caused by pyrithiamine- and dietary-induced thiamine deficiency and alcohol exposure in the alcohol-preferring rat: A longitudinal magnetic resonance imaging and spectroscopy study. Neuropsychopharmacol. 2007;32:1159–1177. doi: 10.1038/sj.npp.1301107. [DOI] [PubMed] [Google Scholar]
- Pires R, Pereira S, Oliveira-Silva IF, Franco GC, Ribeiro AM. Cholinergic parameters and the retrieval of learned and re-learned spatial information: A study using a model of Wernicke-Korsakoff syndrome. Behav. Brain Res. 2005;162:11–21. doi: 10.1016/j.bbr.2005.02.032. [DOI] [PubMed] [Google Scholar]
- Pires R, Pereira S, Pittella J, Franco GC, Ferreiro C, Fernandes PA, Ribeiro AM. The contribution of mild thiamine deficiency and ethanol consumption to central cholinergic parameter dysfunction and rats' open-field performance impairment. Pharmacol. Biochem. Behav. 2001;70:227–235. doi: 10.1016/s0091-3057(01)00593-7. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Aging potentiates the acute and chronic neurological symptoms of pyrithiamine-induced thiamine deficiency in the rodent. Behav. Brain Res. 2001;119:167–177. doi: 10.1016/s0166-4328(00)00350-8. [DOI] [PubMed] [Google Scholar]
- Pitkin SR, Savage LM. Age-related vulnerability to diencephalic amnesia produced by thiamine deficiency: The role of time of insult. Behav. Brain Res. 2004;148:93–105. doi: 10.1016/s0166-4328(03)00208-0. [DOI] [PubMed] [Google Scholar]
- Redila VA, Olson AK, Swann SE, Mohades G, Webber AJ, Weinberg J, Christie BR. Hippocampal cell proliferation is reduced following prenatal ethanol exposure but can be rescued with voluntary exercise. Hippocampus. 2006;16:305–311. doi: 10.1002/hipo.20164. [DOI] [PubMed] [Google Scholar]
- Richardson RM, Sun D, Bullock MR. Neurogenesis after traumatic brain injury. Neurosurg. Clin. N. Am. 2007;18:169–181. doi: 10.1016/j.nec.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Robinson JK, Mair RG. MK-801 prevents brain lesions and delayed non-matching-to-sample deficits produced by pyrithiamine-induced encephalopathy in rats. Behav. Neurosci. 1992;106:623–633. [PubMed] [Google Scholar]
- Rola R, Mizumatsu S, Otsuka S, Morhardt DR, Noble-Haeusslein LJ, Fishman K, Potts MB, Fike JR. Alternations in hippocampal neurogenesis following traumatic brain injury in mice. Exp. Neurol. 2006;202:189–199. doi: 10.1016/j.expneurol.2006.05.034. [DOI] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. Blunted hippocampal, but not striatal, acetylcholine efflux parallels learning impairment in diencephalic-lesioned rats. Neurobiol. Learn. Mem. 2007;87:123–132. doi: 10.1016/j.nlm.2006.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland JJ, Savage LM. The role of cholinergic and GABAergic medial septal/diagonal band cell populations in the emergence of diencephalic amnesia. Neuroscience. 2009;160:32–41. doi: 10.1016/j.neuroscience.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sairanen M, Lucas G, Ernfors P, Castren M, Castren E. Brain-derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 2005;25:1089–1094. doi: 10.1523/JNEUROSCI.3741-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage LM, Chang Q, Gold PE. Diencephalic damage decreases hippocampal acetylcholine release during spontaneous alternation testing. Learn. Mem. 2003;10:242–246. doi: 10.1101/lm.60003. [DOI] [PubMed] [Google Scholar]
- Shetty AK, Hattiangady B, Rao MS, Shuai B. Deafferentation enhances neurogenesis in the young and middle-aged hippocampus but not the aged hippocampus. Hippocampus. 2010 doi: 10.1002/hipo.20776. doi: 10.1002/hipo.20776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shors TJ, Miesegaes G, Beylin A, Zhao M, Rydel T, Gould E. Neurogenesis in the adult is involved in the formation of trace memories. Nature. 2001;410:372–376. doi: 10.1038/35066584. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Townsend DA, Zhao M, Kozorovitskiy Y, Gould E. Neurogenesis may relate to some but not all types of hippocampal-dependent learning. Hippocampus. 2002;12:578–584. doi: 10.1002/hipo.10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisti HM, Glass AL, Shors TJ. Neurogenesis and the spacing effect: Learning over time enhances memory and the survival of new neurons. Learn. Mem. 2007;14:368–375. doi: 10.1101/lm.488707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JS, Hong NS, McDonald RJ, Wojtowicz JM. A role for adult neurogenesis in spatial long-term memory. Neurosci. 2005;130:843–852. doi: 10.1016/j.neuroscience.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Sullivan EV, Pfefferbaum A. Neuroimaging of the Wernicke-Korsakoff syndrome. Alcohol Alcohol. 2009;44:155–165. doi: 10.1093/alcalc/agn103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi Y, Nozaki K, Takahashi J, Yodoi J, Ishikawa M, Hashimoto N. Proliferation of neuronal precursor cells in the dentate gyrus is accelerated after transient forebrain ischemia in mice. Brain Res. 1999;831:283–287. doi: 10.1016/s0006-8993(99)01411-0. [DOI] [PubMed] [Google Scholar]
- Troncoso JC, Johnston MV, Hess KM, Griffin JW, Price DL. Model of Wernicke's Encephalopathy. Arch. Neurol. 1981;38:350–354. doi: 10.1001/archneur.1981.00510060052007. [DOI] [PubMed] [Google Scholar]
- Van der Borght K, Mulder J, Keijser JN, Eggen B, Luiten P, Van der Zee EA. Input from the medial septum regulates adult hippocampal neurogenesis. Brain Res. Bull. 2005;67:117–125. doi: 10.1016/j.brainresbull.2005.06.018. [DOI] [PubMed] [Google Scholar]
- van Praag H, Christie BR, Sejnowski TJ, Gage FH. Running enhances neurogenesis, learning, and long-term potentiation in mice. Proc. Natl. Acad. Sci, USA. 1999a;96:13427–13431. doi: 10.1073/pnas.96.23.13427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Praag H, Kempermann G, Gage FH. Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nat. Neurosci. 1999b;2:266–270. doi: 10.1038/6368. [DOI] [PubMed] [Google Scholar]
- Vetreno RP, Anzalone SJ, Savage LM. Impaired, spared, and enhanced ACh efflux across the hippocampus and striatum in diencephalic amnesia is dependent on task demands. Neurobiol. Learn. Mem. 2008;90:237–244. doi: 10.1016/j.nlm.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetreno RP, Hall J, Savage LM. Alcohol-related amnesia and dementia: What animal models have told us about the relationships between etiological factors, neuropathology and cognitive impairments. Neurobiol. Learn. Mem. doi: 10.1016/j.nlm.2011.01.003. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor M, Adams RC, Collins GH. The Wernicke-Korsakoff syndrome. F.A. Davis; Philadelphia, PA: 1971. [Google Scholar]
- Victor M, Adams RC, Collins GH. The Wernicke-Korsakoff syndrome and related neurological disorders due to alcoholism and malnutrition. F. A. Davis; Philadelphia, P.A.: 1989. [Google Scholar]
- Voloboueva LA, Lee SW, Emery JF, Palmer TD, Giffard RG. Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J. Neurosci. 2010;30:12242–12251. doi: 10.1523/JNEUROSCI.1752-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorhees CV, Schmidt DE, Barrett RJ, Schenker S. Effects of thiamin deficiency on acetylcholine levels and utilization in vivo in rat brain. J. Nutr. 1977;107:1902–1908. doi: 10.1093/jn/107.10.1902. [DOI] [PubMed] [Google Scholar]
- Winocur G, Wojtowicz JM, Sekeres M, Snyder JS, Wang S. Inhibition of neurogenesis interferes with hippocampus-dependent memory function. Hippocampus. 2006;16:296–304. doi: 10.1002/hipo.20163. [DOI] [PubMed] [Google Scholar]
- Witt ED. Neuroanatomical consequences of thiamine deficiency: A comparative analysis. Alcohol. Alcohol. 1985;2:201–221. [PubMed] [Google Scholar]
- Wojtowicz JM, Askew ML, Winocur G. The effects of running and of inhibiting adult neurogenesis on learning and memory in rats. Eur. J. Neurosci. 2008;27:1494–1502. doi: 10.1111/j.1460-9568.2008.06128.x. [DOI] [PubMed] [Google Scholar]
- Wrenn CC, Wiley RG. The behavioral functions of the cholinergic basal forebrain: Lesions from 192 IgG-saporin. Int. J. Dev. Neurosci. 1998;16:595–602. doi: 10.1016/s0736-5748(98)00071-9. [DOI] [PubMed] [Google Scholar]
- Zell R, Geck P, Werdan K, Boekstegers P. TNF-alpha and IL-1 alpha inhibit both pyruvate dehydrogenase activity and mitochondrial function in cardiomyocytes: Evidence for primary impairment of mitochondrial function. Mol. Cell. Biochem. 1997;177:61–67. doi: 10.1023/a:1006896832582. [DOI] [PubMed] [Google Scholar]
- Zhang SX, Weilersbacher GS, Henderson SW, Corso T, Olney JW, Langlais PJ. Excitotoxic cytopathology, progression, and reversibility of thiamine deficiency-induced diencephalic lesions. J. Neuropathol. Exp. Neurol. 1995;54:255–267. doi: 10.1097/00005072-199503000-00012. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]
- Zhao C, Teng EM, Summers RG, Jr, Ming G, Gage FH. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci. 2006;26:3–11. doi: 10.1523/JNEUROSCI.3648-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao N, Zhong C, Wang Y, Zhao Y, Gong N, Zhou G, Xu T, Hong Z. Impaired hippocampal neurogenesis is involved in cognitive dysfunction induced by thiamine deficiency at early pre-pathological lesion stage. Neurobiol. Dis. 2008;29:176–185. doi: 10.1016/j.nbd.2007.08.014. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Pan X, Zhao J, Wang Y, Peng Y, Zhong C. Decreased transketolase activity contributes to impaired hippocampal neurogenesis induced by thiamine deficiency. J. Neurochem. 2009;111:537–546. doi: 10.1111/j.1471-4159.2009.06341.x. [DOI] [PubMed] [Google Scholar]