

Abstract
In this report we highlight the significant potential of ethylene as a reagent for the introduction of a vinyl group with excellent stereoselectivity at three different stages in the synthesis of a broad class of natural products, best exemplified by syntheses of pseudopterosins. The late-stage applications of the asymmetric hydrovinylation reaction further illustrate the compatibility of the catalyst with complex functional groups. We also show that depending on the choice of the catalyst, it is possible to either enhance or even completely reverse the inherent diastereoselectivity in the reactions of advanced chiral intermediates. This should enable the synthesis of diastereomeric analogs of several classes of medicinally relevant compounds that are not readily accessible by the existing methods, which depend on ‘substrate-control’ for the installation of many of the chiral centers, especially in molecules of this class.
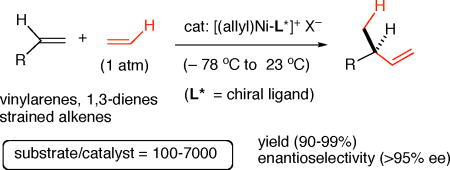
Ethylene is one of the most abundantly available feedstocks, providing up to 30% of all petroleum-based commodity chemicals, yet its applications in complex organic synthesis are just beginning to emerge.1 Among the handful of catalyzed asymmetric C-C bond-forming reactions that use a feedstock carbon source, is the asymmetric hydrovinylation (HV), a reaction in which ethylene is added, as a vinyl group and a hydrogen, to common organic precursors such as an activated olefin, a 1,3- diene or a strained alkene, to produce an enantiomerically pure product (Eq 1).2
 |
(1) |
While contemplating applications of this reaction in complex molecule synthesis, we envisioned that three of the four chiral centers (each marked with an asterisk in 1, Figure 1) of pseudopterosin aglycones could be installed independently by repeated use of the asymmetric hydrovinylation reaction. Any synthesis of the core skeleton might also provide facile entry into other more complex molecules shown in Figure 1. Late-stage applications of the asymmetric hydrovinylation in such complex settings would rigorously test the limits of this reaction in two important ways: will the Ni(II)-catalyst system be compatible with substrate with several Lewis-basic centers? Perhaps more importantly, is it possible to enhance or even override, by ligand tuning, any inherent diastereoselectivity dictated by the resident chirality already present in an advanced intermediate? It can be convincingly argued that even in the most developed catalyzed reactions, absolute control of diastereoselectivity in reactions of chiral substrates can be a more formidable challenge than achieving useful levels of asymmetric induction in a prochiral substrate. In the event, we have completed a highly stereoselective total synthesis of the aglycone of pseudopterosins G-J. Since an advanced enantiopure intermediate synthesized during this study has already been converted into pseudopterosins A-F aglycone3 this report also represents a formal stereoselective synthesis of this molecule and its enantiomer, pseudopterosins K-L aglycone. Among the sequence of reactions employed, the asymmetric hydrovinylation was used three times with excellent yield and stereoselectivity (yields/selectivity as expressed in enantiomeric or diastereomeric ratio: 99%/er = 96:4; 92%/dr = 92:8 and 99%/dr >99:1). We also show that the configuration of the asymmetric centers can be independently controlled by the judicious choice of catalysts and/or reaction conditions. This report discloses the details of this study.
Figure 1.

Structures of Pseudopterosins and Related Compounds
West Indian and Caribbean sea whip Pseudopterogorgia elisabethae has been a rich source of biologically active compounds of varying structural complexity.4 These include (Figure 1) anti-inflammatory pseudopterosins,5a and compounds with significant anti-mycobacterial properties such as pseudopteroxazole,5b colombiasin A,5c elisapterosin B5d and a key biogenic precursor, elisabethin A.5e Even in cases where the structures are not very complex, the major challenge associated with the synthesis of these compounds has been the control of the stereocenters at locations with no neighboring controlling functionalities. An exocyclic methyl-bearing carbon such as C3 or C7 in pseudopterosins is a very common structural motif in many of these structures, and a chain carrying this carbon is often attached at a stereogenic center in a ring. Even though several creative solutions to tackle this so-called ‘exocyclic stereochemistry’ problem have been advanced, no broadly applicable method that uses an easily accessible precursor has emerged.6a,b In pseudopterosins there is the additional challenge of stereoselectively introducing the 2-methylpropenyl group at the C1 carbon. An examination of the various published procedures3,7–10 for the syntheses of pseudopterosins exposes the limitations of the current methodology to address the synthetic problems associated with the installation of these chiral centers. The best stereoselective syntheses solve these problems by starting with naturally occurring monoterpenes which already possess one or two of the chiral centers of the final products, and installing the others via diastereoselective transformations, with varying degrees of success. Such approaches always depend on the availability of both enantiomers of the starting materials and, often, involve difficult skeletal modifications to reach the eventual target. For example, in earlier work (−)- limonene7,8 and (+)-menthol,9 served as the starting materials for the syntheses of the aglycone of pseudopterosins A-F (1). Kocienski later reported10a the use of (−)-citronellal and (−)-isopulegol for the syntheses of both pseudopterosin A-F, and the enantiomeric pseudopterosin K-L (3) aglycones. While the Kocienski approach is better in overall stereoselectivity, his scheme, like the previous syntheses, also relies on a cumbersome aromatic annulation, which consumes several avoidable steps if one were to start with a more logical aromatic precursor. Because of the ‘substrate-control’ nature of the key reactions by which the chiral centers are introduced, all these syntheses have serious limitations if stereoisomeric congeners of these compounds are needed. Asymmetric hydrovinylation allows a high degree of reagent control in the installation of three of the four chiral centers of the pseudopterosin core. Since both enantiomers of the ligands are readily available, either enantiomer of the natural product maybe synthesized from the same prochiral precursor.
Enantioselective Synthesis of Pseudopterosin Core
In our synthesis we targeted the tricyclic ketone 16 as a common key intermediate from which the aglycones of all pseudopterosins can be synthesized. The synthesis (Scheme 1)11 started with an exceptionally efficient Ni-catalyzed asymmetric hydrovinylation of 2,3-dimethoxy-4-methylstyrene (6) using the ligand L1, which was chosen from a collection of ligands we prepared for optimization of the hydrovinylation reaction.12 This reaction gives a quantitative yield of 7, which incorporates the correct configuration (S) at the C7 center for the pseudopterosin A-J series. This compound was converted into the ketone 10 in a series of simple steps, starting with the hydroboration/oxidation of 7,13 iodide formation and displacement with CN− resulting in the chain-extended product 9. The cyanide 9 was hydrolyzed to an acid and subsequently converted into an acid chloride, which was cyclized under Friedel-Crafts conditions to form 10. The diene 11 was synthesized in 86% yield from 10 via a Stille reaction14 of a vinyltriflate generated in situ from the ketone. The compound 11 is set up for the installation of the chiral center at C3 via a second hydrovinylation, this time that of a 1,3-diene. Even though the starting material is a nearly enantiopure diene, the resident chirality (at the benzylic position, C7) in the molecule has little influence on the stereoselectivity of the second hydrovinylation. Indeed, the reaction using an achiral ligand L3 shows only a slight preference [C3(S):C3(R) = 59:41] for the desired compound 12a. However, this preference for 12a can be reinforced and a dr of 92:8 (12a:12b) can be obtained with the use of the phosphoramidite L2 in the Ni-catalyzed asymmetric hydrovinylation reaction. In a powerful demonstration of the versatility of asymmetric catalysis, it can be shown that an enantiomeric catalyst generated from the ligand ent-L2 overcame the inherent preference for the C3(S) isomer and gave a ratio of 3:97 for 12a:12b, with C3(R) predominating. Thus, the configuration of the C3 and C7 centers can be controlled independently, an option not available in any of the previous syntheses of these molecules or its congeners. The ensuing installation of the correct configuration at C4 turned out to be a challenge. After much experimentation, the correct C4(R)-center (dr: >20:1) was established by a lithium/NH3 reduction15 of a primary alcohol that was prepared from the alkene 12a by hydroboration/oxidation procedure. Various directed hydrogenations of 12a or 13 using either Rh(I) or Ir(I), including Crabtree16 or Pfaltz17 hydrogenation catalysts were either sluggish or led to mixtures of products. Incidentally, catalytic hydrogenation using Pd/C/H2 gave the C4 epimeric compound 14b as the major product (14a:14b =1:13), once again demonstrating the flexibility of a reagent-based strategy for the control of stereoselectivity. The alcohol 14a was readily converted into the tricyclic ketone 16 in a series of high-yielding reactions (Swern oxidation to an aldehyde, 15, Lindgen-Kraus-Pinnick oxidation to an acid, acid chloride formation with oxalyl chloride, and, finally intramolecular Friedel-Crafts cyclization). This sequence involved just one chromatography to purify the final product 16. Recrystallization of the crude product gave an overall yield of 47% of a highly crystalline tricyclic ketone from the alcohol 14a as a single enantiomer. The solid-state structure of this ketone as revealed by X-ray crystallographic analysis confirmed the correct configurations at the various chiral centers. As described below the intermediate 16 is useful for stereoselective syntheses of aglycones of pseudopterosin A-F and G-J.
Scheme 1.

Stereoselective Installation the C1 Side-chain of Pseudopterosins G-J via Asymmetric Hydrovinylation
While the synthesis of pseudopterosin A-F aglycone with the C1- isobutenyl group in the more stable quasi-axial orientation is quite straight forward as previously reported3,9,18 (Scheme 2), approach to the C1-epimeric G-J aglycone is less apparent. Again we find that hydrovinylation provides a very attractive solution for the installation of the last of the stereogenic centers in pseudopterosins G-J. The requisite substrate for this process was readily synthesized from the ketone 16 as shown in Scheme 3. Sodium borohydride reduction of 16 gave a mixture of alchols (1:1), which was quantitatively converted into the tetrahydrophenalene 17 upon treatment with camphor sulfonic acid in CH2Cl2. Hydrovinylation of 17 with Ni(allyl)(L2)/BARF gave 18 in nearly quantitative yield with complete stereoselectivity (dr >99.5:0.5). Conversion of the vinyl group into the aldehyde and a subsequent Wittig reaction proceeded without incident giving the pseudopterosin G-J aglycone dimethyl ether 2B. Depending on the reaction conditions, varying amounts (up to 13%) of isomerization of the intermediate aldehyde was observed during the Wittig reaction, giving the aglycone of pseudopterosin A-F as a byproduct. Incidentally, a more direct route involving Ru-catalyzed cross-metathesis,19 in spite of considerable effort, only gave unacceptable (<10%) yields.
Scheme 2.

Stereoselective Synthesis of Pseudopterosin A-F Aglycone
Scheme 3.

Stereoselective Synthesis of Pseudopterosin G-J Aglycone via Installation of the C1 Side-chain Using Asymmetric Hydrovinylation
In conclusion, we demonstrate the utility of ethylene for highly enantioselective syntheses of aglycones of all known pseudopterosins. This approach provides unprecedented flexibility with regard to the preparation of other diastereomeric analogs of these molecules. In addition, the stereochemical features associated with some of the advanced intermediates (e. g., alcohol 14a) are common to a number of important diterpenes including pseudopteroxazole,5b,20 colombiasin A,5c,21 elisabethin A,5e,22a elisapterosin B5d,22b and helioporins.23 With the stereochemical issues largely resolved, it should now be possible to design expeditious entry into these molecules and their congeners. Such studies are currently in progress.
Supplementary Material
Acknowledgments
Financial assistance for this research by NIH (General Medical Sciences, R01 GM075107) and a fellowship provided by the Chemistry-Biology Interface Training Program (NIH) to G. A. Cox are gratefully acknowledged. We thank Dr. A. Zhang for several initial experiments and Dr. Judith Gallucci for determination of the crystal structure of 16, shown in Scheme 1.
Footnotes
Supporting Information Available: Full experimental details for the preparation of the intermediates and hydrovinylation reactions, spectroscopic and chromatographic data for characterization of all compounds, Crystallographic Information File for the ketone 16. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.For examples of the use of unactivated alkenes including ethylene as reagents in catalytic reactions, see: Jolly PW, Wilke G. Hydrovinylation. In: Cornils B, Herrmann WA, editors. Applied Homogeneous Catalysis with Organometallic Compounds. Vol. 2. VCH; New York: 1996. pp. 1024–1048.RajanBabu TV. Chem Rev. 2003;103:2845. doi: 10.1021/cr020040g.Bower JF, Kim IS, Patman RL, Krische MJ. Angew Chem Int Ed Engl. 2009;48:34. doi: 10.1002/anie.200802938.Ho CY, Schleicher KD, Chan CW, Jamison TF. Synlett. 2009:2565. doi: 10.1055/s-0029-1217747.Hess W, Treutwein J, Hilt G. Synthesis. 2008:3537.For recent, representative examples of use of ethylene in fine chemical synthesis: Mori M, Saito N, Tanaka D, Takimoto M, Sato Y. J Am Chem Soc. 2003;125:5606. doi: 10.1021/ja029747a.Ogoshi S, Haba T, Ohashi M. J Am Chem Soc. 2009;131:10350. doi: 10.1021/ja903510u.Smith CR, RajanBabu TV. Tetrahedron. 2010;66:1102. doi: 10.1016/j.tet.2009.11.017.Sharma RK, RajanBabu TV. J Am Chem Soc. 2010;132:3295. doi: 10.1021/ja1004703.Matsubara R, Jamison TF. J Am Chem Soc. 2010;132:6880. doi: 10.1021/ja101186p.An outstanding example of iterative use of an asymmetric C-C bond-forming reaction in natural product synthesis, see: Han SB, Hassan A, Kim IS, Krische MJ. J Am Chem Soc. 2010;132:15559. doi: 10.1021/ja1082798.For a review of asymmetric catalysis in complex molecule synthesis, see: Taylor MS, Jacobsen EN. Proc Natl Acad Sci U S A. 2004;101:5368. doi: 10.1073/pnas.0307893101.
- 2.RajanBabu TV. Synlett. 2009:853. doi: 10.1055/s-0028-1088213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buszek KR, Bixby DL. Tetrahedron Lett. 1995;36:9129.(b) see also ref (9).
- 4.Rodríguez AD. Tetrahedron. 1995;51:4571. doi: 10.1016/0040-4020(95)00216-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Look SA, Fenical W, Jacobs RS, Clardy J. Proc Natl Acad Sci U S A. 1986;83:6238. doi: 10.1073/pnas.83.17.6238. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rodríguez AD, Ramírez C, Rodríguez II, González E. Org Lett. 1999;1:527. doi: 10.1021/ol9907116. [DOI] [PubMed] [Google Scholar]; (c) Rodríguez AD, Ramírez C. Org Lett. 2000;2:507. doi: 10.1021/ol991362i. [DOI] [PubMed] [Google Scholar]; (d) Rodríguez AD, Ramírez C, Rodríguez II, Barnes CL. J Org Chem. 2000;65:1390. doi: 10.1021/jo9914869. [DOI] [PubMed] [Google Scholar]; (e) Rodríguez AD, González E, Huang SD. J Org Chem. 1998;63:7083. doi: 10.1021/jo981385v. [DOI] [PubMed] [Google Scholar]
- 6.(a) Guevel AC, Hart DJ. J Org Chem. 1996;61:465. doi: 10.1021/jo951531m. [DOI] [PubMed] [Google Scholar]; (b) Saha B, Smith CR, RajanBabu TV. J Am Chem Soc. 2008;130:9000. doi: 10.1021/ja711475f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Broka CA, Chan S, Peterson B. J Org Chem. 1988;53:1584. [Google Scholar]
- 8.Corey EJ, Lazerwith SE. J Am Chem Soc. 1998;120:12777. [Google Scholar]
- 9.Corey EJ, Carpino P. J Am Chem Soc. 1989;111:5472. [Google Scholar]
- 10.Pseudopterosin A-F and K-L: Kocienski PJ, Pontiroli A, Qun L. J Chem Soc, Perkin Trans. 2001;1:2356.Other syntheses of pseudopterosins Majdalani A, Schmalz H-G. Synlett. 1997:1303.Harrowven DC, Tyte MJ. Tetrahedron Lett. 2004;45:2089.Synthesis of related pseudopteroxazole: Harmata M, Hong X. Org Lett. 2005;7:3581. doi: 10.1021/ol0515412.
- 11.Details of the experimental procedures, and spectroscopic and chromatographic data for all compounds are included in the Supporting Information.
- 12.Smith CR, Mans DJ, RajanBabu TV. Org Synth. 2008;85:238.Smith CR, RajanBabu TV. Org Lett. 2008;10:1657. doi: 10.1021/ol800395m.Phosphoramidites as ligands for asymmetric hydrovinylation of vinylarenes, see: Leitner: Francio G, Faraone F, Leitner W. J Am Chem Soc. 2002;124:736. doi: 10.1021/ja012099v.
- 13.For hydroboration/oxidation of hydrovinylation products, see: Zhang A, RajanBabu TV. Org Lett. 2004;6:3159. doi: 10.1021/ol048790v.Böing C, Hahne J, Francio G, Leitner W. Adv Synth Catal. 2008;350:1073.
- 14.De Riccardis F, Meo D, Izzo I, Di Filippo M, Casapullo A. Eur J Org Chem. 1998:1965. [Google Scholar]
- 15.Cesati IRR, de Armas J, Hoveyda AH. J Am Chem Soc. 2003;126:96. doi: 10.1021/ja0305407. [DOI] [PubMed] [Google Scholar]
- 16.Crabtree RH, Davis MW. J Org Chem. 1986;51:2655. [Google Scholar]
- 17.Bell S, Wustenberg B, Kaiser S, Menges F, Netscher T, Pfaltz A. Science. 2006;311:642. doi: 10.1126/science.1121977.We thank Professor Pfaltz for providing samples of these catalysts.
- 18.A mixture of 1B and 2B (1:2) can be readily synthesized from the ketone 16 via the corresponding enol triflate formation, a Stille reaction with 2-methyl-1- tributylstannylpropene, followed by Li/NH3 reduction of the resulting 1,3-diene. See Supporting Information for details.
- 19.Stewart IC, Douglas CJ, Grubbs RH. Org Lett. 2008;10:441. doi: 10.1021/ol702624n. [DOI] [PubMed] [Google Scholar]
- 20.Davidson JP, Corey EJ. J Am Chem Soc. 2003;125:13486. doi: 10.1021/ja0378916. [DOI] [PubMed] [Google Scholar]
- 21.Syntheses of colombiasin A: Nicolaou KC, Vassilikogiannakis G, Magerlein W, Kranich R. Angew Chem Int Ed. 2001;40:2482. doi: 10.1002/1521-3773(20010702)40:13<2482::AID-ANIE2482>3.0.CO;2-A.Full paper: Nicolaou KC, Vassilikogiannakis G, Magerlein W, Kranich R. Chem Eur J. 2001;7:5359. doi: 10.1002/1521-3765(20011217)7:24<5359::aid-chem5359>3.0.co;2-z.Kim AI, Rychnovsky SD. Angew Chem Int Ed. 2003;42:1267. doi: 10.1002/anie.200390325.Boezio AA, Jarvo ER, Lawrence BM, Jacobsen EN. Angew Chem Int Ed. 2005;44:6046. doi: 10.1002/anie.200502178.Harrowven DC, Pascoe DD, Demurtas D, Bourne HO. Angew Chem Int Ed. 2005;44:1221. doi: 10.1002/anie.200462268.Davies HML, Dai X, Long MS. J Am Chem Soc. 2006;128:2485. doi: 10.1021/ja056877l.
- 22.Synthesis of elisabethin A: Heckrodt TJ, Mulzer J. J Am Chem Soc. 2003;125:4680. doi: 10.1021/ja034397t.The conclusions of this paper has since been questioned: Zanoni G, Franzini M. Angew Chem Int Ed. 2004;43:4837. doi: 10.1002/anie.200460570.For a synthesis of elisapterosin B and of ent-2-epi-elisabethin A, see: Waizumi N, Stankovic AR, Rawal VH. J Am Chem Soc. 2003;125:13022. doi: 10.1021/ja035898h.See also references 21(b)-(e) for other syntheses of elisapterosin B.
- 23.Tanaka J, Ogawa N, Liang J, Higa T, Gravalos DG. Tetrahedron. 1993;49:811. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.