

Abstract
BACKGROUND
The standard hormonal therapy with currently available antiandrogens and the leutinizing hormone releasing hormone (LHRH) analogs is not effective in the hormone-refractory stage of prostate cancer due to changes in androgen receptor (AR) signaling axis. In this refractory stage, AR continues to play a significant role in the growth of cancer cells even though the cancer cells are no longer dependent on the level of circulating androgens.
METHODS
A series of 11β-Δ9-19 nortestosterone compounds were designed through structure-based rationale and tested for their binding affinity against AR and glucocorticoid receptor (GR) using fluorescence polarization assays, their agonistic ability to induce AR dependent transcription using PSA-driven report gene assays, and their growth inhibitory affects against a series of AR positive (LAPC4, LNCap, and CWR22R) and negative human prostate cancer cell lines (PC3) using MTT cell proliferation assays.
RESULTS
This study proposes the design of novel bifunctional antiandrogens based on the conjugation of 11β and/or 7α-Δ9-19 nortestosterone class of steroidal compounds to the synthetic ligand for FK506-binding proteins. As a critical step towards the development of bifunctional antiandrogens, highly potent and AR-specific lead compounds were identified using in vitro data. The lead compounds identified in this study possessed low binding affinity for GR, indicating the absence of undesirable antiglucocorticoid activity.
CONCLUSIONS
The results of this study validate our drug discovery rationale based on the structural biology of AR and pave the pay for future development of bifunctional compounds in order to block AR function in hormone refractory stage of prostate cancer.
Keywords: prostate cancer, bifunctional, antiandrogens, FKBP, SLF
INTRODUCTION
Androgens are major growth factors for the normal prostate, and androgen receptor (AR) is fundamental for androgen signaling within the prostate gland [1]. Recently, there has been a major shift in the thinking concerning the role of AR in prostate cancer progression to its lethal stage. Since aggressive forms of androgen ablation (i.e., leutinizing hormone releasing hormone, LHRH, analogs plus antiandrogens plus chemical inhibition of adrenal androgen production) do not substantially increase the survival of prostate cancer patients above that produced by LHRH analogs alone, it had been assumed that this therapeutic failure meant that AR is no longer engaged in the lethal phase of the disease. A series of correlative and experimental data, however, do not support such a conclusion [2–6] and our attention is now refocused on how cancer cells engage AR to stimulate proliferation and survival in androgen ablation failing prostate cancer patients. A large number of studies have been conducted to resolve the potential mechanism(s) for such AR dependent responses in a physiological androgen depleted environment [7–9]. Some of these studies have implicated the role of “cross talk” with other signaling pathways (i.e., MAPK kinase cascade, IL-6, Stat3, etc.) under such androgen ablated conditions [10]. Other studies have reported the presence of point mutations in AR protein (T877A, W741C, etc.) that allow binding of non-androgenic ligands to AR and conversion of antiandrogens into partial agonists [11]. Due to these hardwiring changes in AR signaling axis, currently marketed antiandrogens (e.g., casodex, hydroxyflutamide, nilutamide, etc.) are ineffective in androgen ablation failing prostate cancer patients therefore, novel agents are critically needed in order to decrease the current mortality rate of ~30,000 men a year dying from prostate cancer.
AR is a member of the steroid/nuclear receptor super family of ligand-dependent transcription factors [12]. AR contains a central DNA binding domain, which separates the amino terminus from the carboxy terminus. The amino terminus contains an activation function (AF-1) domain while the carboxy terminus harbors the ligand-dependent activation function (AF-2) domain within the ligand binding domain (LBD) of AR. It has been demonstrated that these two terminus must interact so that co-activator proteins, such as SRC-1 and TIF2 can bind and thus activate functional transcription [13]. The X-ray crystallographic studies [14] indicate that AR can adopt a structural fold in the LBD with either an “agonist” conformation which binds co-activators or an “antagonistic fold” which binds co-repressors. In androgen ablation conditions, abnormal phosphorylation of these co-activators and/or AR, induced by “cross talk” with other signaling cascades, allows these phosphorylated co-activators to bind to the AF-2 domain and force the AR into an agonist state either without ligand or when bound by low molecular weight antagonists.
A novel strategy to block growth stimulatory pathways induced by AR in androgen ablation failing patients is to prevent co-activator binding and subsequent re-modeling of AR by structurally locking the AF-2 domain of the AR surface in an antagonist conformation. Following this rationale, structural studies have been conducted by Fletterick group [15] towards designing small molecule inhibitors of co-activator binding surface. However, protein–protein interactions are notoriously difficult to inhibit using small molecules [16] and utility of these efforts remains to be proven. Since current small molecule antiandrogens lack sufficient steric bulk to lock AR in an inactive configuration, we have developed a bifunctional approach to design novel bulky antiandrogens which can recruit FK506-binding chaperone proteins (FKBPs) to the co-activator binding site of AF-2 domain, thereby sterically preventing binding of any co-activator proteins to AR (Fig. 1). FKBPs are encoded by multiple genes, are ubiquitously expressed in all mammalian cells [17] and are ideal candidates for recruitment to protein surfaces [18,19]. Previous studies on SH2 domain [18], ErbB receptors [20] and β-amyloid [21] have demonstrated that such a bifunctional approach based on the recruitment of FKBPs to the surface of cellular targets can be efficient in preventing protein–protein interactions, thereby inhibiting corresponding signaling pathways driven by these proteins. Following a similar rationale, we have designed novel bifunctional antiandrogens through coupling of AR ligands to the synthetic ligand for FKBPs (SLF) (Fig. 1C). Such a bifunctional molecule should bind to and project from the AR-LBD ligand binding pocket and allow FKBP proteins to bind to SLF ligand on the other end exposed to the solvent (Fig. 1B). Since the binding constant (Kd) value for SLF binding to FKBP is ≈20 nM and since the size of the FKBP proteins are >10 kDa the recruitment of FKBPs to the AF-2 domain of AR should be highly efficient and, based upon our analysis of crystal structures of AR, provide sufficient bulk to “mask” the entire surface of the co-activator groove formed by helices 3,4,5, and 12 of LBD, thus sterically preventing binding of any co-activators.
Fig. 1.

Bifunctional approach towards designing novel antiandrogens. A: Androgen binding to the ligand binding domain (LBD) of AR induces agonist conformation which allows binding of co-activators and machinery and subsequent binding to DNA. B: Bifunctional molecule recruits FKBP proteins to the co-activator binding site of AR, thereby locking androgen receptor in an antagonist conformation. This conformation does not result in gene transcription. C: Chemical structures of proposed bifunctional compounds based on the chemical coupling of 11β/7α/Δ9-19-nortestosterone steroidal core via a linker to the synthetic ligand (SLF)for FKBP proteins. D: Chemical structure of mifepristone.
To successfully design and develop bifunctional antiandrogens, it is critical to determine ideal chemical functionality and biological behavior of AR ligands before they can be conjugated to the SLF moiety. The biophysical characteristics of AR ligand are crucial for the final potency of the bifunctional compound as well as its ability to recruit FKBP to the AR surface. For example, a previously described bifunctional molecule composed of SLF and methotrexate was unable to bring together FKBPs and dihydrofolate reductase due to a short length linker distance between the two ligands [22,23]. In this article, we set out to design and identify lead AR ligands containing ideal chemical functionality for their coupling to carboxyl moiety of SLF to produce novel bifunctional antiandrogens that will have the potential to neutralize AR function in androgen-ablation failing prostate cancer patients. Towards this goal, we have utilized mechanistic insights gained from the crystal structure analysis of AR LBD to design and synthesize a series of 11β-Δ9-19 nortestosterone steroidal compounds via chemical modification of Δ9-19 nortestosterone steroidal core at the 11β-position. In order to identify the best compounds for further development into the bifunctional molecules via SLF conjugation, a series of in vitro assays were conducted, including ligand binding assays, MTT cell proliferation assays, PSA-promoter based luciferase reporter gene assays, and glucocorticoid binding assays.
MATERIALS AND METHODS
Androgen Receptor Ligand Binding Assays
The fluorescence polarization (FP) technique was used to determine the binding affinity of all nortestosterone analogs using PolarScreen™ Androgen Receptor Competitor Assays kit (Catalog # P3018) purchased from Invitrogen. All reagents including the purified AR and fluorescent probe (Fluormone AL green) were supplied with the Invitrogen kit and protocol developed by the manufacturer was used to conduct the competitive binding assays with few modifications. The recombinant un-liganded AR-LBD was supplied as a GST fusion protein. Essentially, the protocol involved titration of nortestosterone analogs against the pre-formed complex of Fluormone AL green (2 nM) and the AR-LBD (50 nM) with the concentration of competitive ligand ranging from nM to μM. The assay mixture was allowed to equilibrate at room temperature in 96-well half area well solid black plates for 4 hr after which the polarization values are measured at room temperature using DTX-880 machine. The excitation and emission wavelength values for the Fluormone were 485 and 535 nm respectively. The data analysis for the ligand binding assays was done using SigmaPlot v8.0 (Systat Software, Inc.) and equilibrium inhibition constant values (Ki) for each ligand were obtained by fitting the data with the one-site competition model.
Glucocorticoid Ligand Binding Assays
The FP technique was used to evaluate the non-specific binding affinity of nortestosterone analogs using PolarScreen™ Glucocorticoid Receptor Competitor Assays kit (Catalog # P2816) purchased from Invitrogen. The protocol supplied by manufacturer was used for these assays and was essentially the same as AR Ligand Binding Assays except that a stabilizing peptide was added in the assay mixtures.
MTT Cell Proliferation Assays
The effect of nortestosterone compounds on prostate cancer cell growth was measured by a 3-(4,5-dime-thylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay; Promega, Madison, WI) as described previously [24]. Four different prostate cancer cell lines (LNCaP, LAPC4, CWR22R, and PC3) were used in these assays. Cell numbers for each sample were extrapolated from standard curves (absorbance vs. cell number) prepared for each cell line. Briefly, in these assays, 10,000 viable cells were plated in a 96-well format in phenol red media containing 10% Dextran coated charcoal (DCC) stripped fetal calf serum. After allowing overnight attachment, the starting number of viable cells at day 0 was determined. Then 8 wells per dose were exposed to different concentrations of the test compound. At days 3, 6, and 9, the number of viable cells was determined as described previously. This assay allows documentation of whether a test compound decreases the rate of proliferation, detectable as a reduction in cell proliferation with time compared to untreated control cells, or whether it can induce the death of cells as detectable by an actual decrease in the number of viable cells over time vs. the starting number of cells at day 0. The results are used to calculate the IC50 values for each compound.
Luciferase Reporter Gene Assays
To evaluate the transcription activity of nortestosterone analogs, reporter gene assays were conducted using transfected PC3 cell lines as described previously [25]. PC3 cell lines lack endogenous expression of AR gene and are best suited for these assays. Essentially, PC3 cell lines were grown in RPMI 1640 media supplanted with 10% DCC FBS and after 24 hr incubation, cells were transfected with PSA promoter containing pGL3PSA plasmid and co-transfected with plasmids cloned with wild-type or mutant AR (pSG5AR, pSG5AR-W741C, pSG5AR-T877A, or pSG5AR-W741C-T877A) using FuGene transfection reagent (Roche Applied Science). Two hours later, 0.1 nM of DHT and 1 μM of nortestosterone compounds were added in the culture media. After incubation for another 24 hr, cells were lysed and luciferase activity was measured with Bright-GloTM Luciferase Assay system (Promega) using a GENE Light 55 luminometer. The activity of β-galactosidase was measured using a mammalian β-gal assay kit and MT Max microplate reader. The results were expressed as the ratio of luciferase activity to β-galactosidase activity.
Crystal Structure Analysis
The crystal structures of the AR LBD were downloaded from the RCSB protein data bank (www.rcsb.org) and analyzed using Molecular Operating Environment software (Chemical Computing Group, Montreal). The molecular graphics were generated using Yasara package (www.yasara.org). The specific crystal structures analyzed were: AR LBD bound with DHT (PDB code: 1AMA), mutant AR LBD bound with casodex (PDB code: 1Z95), mutant AR LBD bound with cyproterone acetate (PDB code: 2OZ7), estrogen receptor alpha ligand binding domain bound with tamoxifen (PDB code: 3ERT), estrogen receptor alpha ligand binding domain bound with ICI-164, 384 compound (PDB code: 1HJ1).
Chemical Synthesis
The exact details of the reaction schemes are provided as supplemental information. All reagents and solvents were obtained from Aldrich. Anhydrous solvents were used as received. Reaction progress was monitored with analytical thin-layer chromatography (TLC) plates on 0.25 mm Merck F-254 silica gel glass plates. Visualization was achieved using UV illumination. 1H NMR spectra were obtained at 400 MHz on a Bruker Avance spectrometer and are reported in parts per million downfield relative to tetramethylsilane (TMS). EI-MS profiles were obtained using a Bruker Esquire 3000 plus. HRMS (FAB-MS) were run by the Johns Hopkins University Mass Spectrometry Facility on a VG-70S Magnetic Sector Mass Spectrometer.
RESULTS AND DISCUSSION
Rationale Design Through Crystal Structure Analysis
Crystal structure analysis of AR LBD bound with DHT reveal that AR ligand binding site is deeply buried in the hydrophobic core of the protein (Fig. 2). Recent structural studies [26,27] on antiandrogens and our crystal structure analysis suggest that when ligands like mifepristone (Fig. 1D) bind to the AR LBD, helix 12 of the AF-2 subdomain is displaced by the bulky sidechain (dimethylaniline) of mifepristone at the 11β-position of the C-ring steroid nucleus. This displacement of helix 12 of the receptor results in an “antagonistic form” of AR LBD which binds nuclear co-repressors (i.e., NCoR), thus antagonizing AR function. Similarly, crystal structure analysis of estrogen receptor-β [28], a homolog of AR, in complex with antiestrogen ICI-164, 384 suggests that sidechains at 7α-position in the B-ring of steroid nucleus can antagonize helix 12 through an altered binding mode in which the ligand is flipped 180° long steroid axis leading to positioning of 7-position in the place of 11-position. Thus, 11β and 7α positions of steroidal AR ligands are ideal locations for the placement of a chemical linker which can displace helix 12 and get exposed to solvent where the potential recruitment of FKBPs can take place (Fig. 2).
Fig. 2.

Crystal structure of androgen receptor ligand binding domain in complex with dihydrotestosterone (DHT). The chemical structure of DHT is shown on right side. Binding of agonistic ligands to the AR results in the agonist conformation of helix 12 (shown in magenta) which allows the co-activator binding at the AF-2 region indicated by dotted circle. Chemical modification at the 11-position of steroidal ring results in the displacement of helix 12 from its agonistic conformation and allow the sidechain at 11-position to be exposed to solvent. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
As a potential candidate for incorporation into bifunctional antiandrogens, mifepristone is problematic because it has unwanted antiglucocorticoid activity (i.e., Ki of 15 nM for glucocorticoid receptor vs. 55 nM for AR). Studies by Muddana et al. [29] have demonstrated that instead of using the steroidal core of mifepristone towards antiandrogen design, Δ9-19-nortestosterone chemical structure can be used as the core backbone, thereby eliminating antiglucocorticoid activity of the derivatives (Fig. 1D). Hence, Δ9-19-nortestosterone nucleus was incorporated into the design of bifunctional antiandrogen and the 11β-position of steroidal nucleus was identified as the ideal location for the placement of linker connecting SLF moiety to the AR ligand core.
Chemical Synthesis of 11β-Δ9-19 Nortestosterone Chemical Library
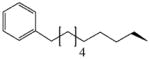
As a critical step towards the validation of our bifunctional approach, we designed a focused library of 11β-Δ9-19 nortestosterone compounds containing linkers of varying chemical nature and length at the 11β-position of the steroidal core (Tables I and II). The goal was to provide sufficient chemical diversity at the 11β-position that would allow the evaluation of impact of the chemical nature and the length of linker at the 11β-position on the AR binding affinity and agonist/antagonist ability against AR-dependent transcription, thereby permitting the identification of lead compounds with the ideal chemical functionality for their ultimate development into bifunctional antiandrogens. While designing the 11β analogs, we insured that both the planar (i.e., benzyl, compounds S1–3) and flexible (i.e., alkyl, compounds S6–9) sidechains of varying lengths were incorporated. Additionally, as a mimic of bifunctional molecule approach, compound S11 was designed that contained two Δ9-19-nortestosterone moieties connected by a 12-carbon aliphatic sidechain at 11β-position. To a lesser extent, an aromatic group at the end of 11β-sidechain in compound S8 serves similar purpose. Lastly, included in the library is a compound S9 that has an opposite stereochemistry at 11-position (11α rather than 11β). The binding site analysis of AR LBD suggests that the sidechain at 11α sterically clash with the stable hydrophobic core of AR LBD, potentially leading to a significant decrease in the binding potency. Inclusion of this compound in the library will allow the evaluation of this structural prediction as well as our design strategy based on the crystal structure analysis. Lastly, compounds S4, S5, and S10 added in the library lacked a 3-keto group that is shown to be critical for binding affinity. These compounds were included as negative controls for our in vitro assays.
TABLE I.
Chemical Structures of Novel 11-Sidechain-Δ9-19-Nortestosterone Compounds and Their Ki Values Obtained From Ligand Binding Assays Using Fluorescence Polarization Technique
 | ||
|---|---|---|
| R | Ki | |
| S1 |

|
79 nM ± 11 |
| S2 |

|
182 nM ± 7 |
| S3 |
|
1 μM ± 80 nM |
| S6 |
|
180 nM ± 30 |
| S7 |

|
170 nM ± 30 |
| S8 |
|
166 nM ± 21 |
| S9 |

|
825 nM ± 90 |
TABLE II.
Chemical Structures of Novel 11-Sidechain-Δ9-19-Nortestosterone Compounds and Their Ki Values Obtained From Ligand Binding Assays Using Fluorescence Polarization Technique
| Compound | Ki | |
|---|---|---|
| S4 |

|
1.2 μM ± 120 nM |
| S5 |

|
1.8 μM ± 110 nM |
| S10 |

|
1.1 μM ± 135 nM |
| S11 |

|
59 nM ± 10 |
In Vitro Binding Potencies
Towards the identification of the lead compounds among newly designed 11β-Δ9-19 nortestosterone compounds, in vitro competitive binding assays were performed using FP technique. Tables I and II present the results in the form of equilibrium inhibition constant (Ki) values. For the bifunctional approach to be effective, it is critical that the steroidal core of the bifunctional molecule with the linker at the 11β-position retain adequate binding affinity with the AR LBD. As presented in Tables I and II, for compounds with aromatic sidechain analogs (i.e., S1, S2, and S3) the binding affinity decreases drastically as the length of sidechain is increased from 1 benzyl group to 2 and 3. The effect of sidechain length on the binding affinity is less drastic in the case of compounds with aliphatic sidechains (i.e., S6, S7, and S8). For compound S8 which has one of the largest aliphatic sidechains, binding affinity still remains lower than 200 nM. Based on these results, it can be concluded that aliphatic sidechains preserve the binding affinity even as the sidechain length is drastically increased, thereby making aliphatic sidechains an ideal chemical functionality for bifunctional antiandrogens. This argument is further strengthened by high binding potency (59 nM) of the dimer compound S11 which contains a 12-carbon long aliphatic sidechain at 11β-position. High binding affinity of this bifunctional-mimic suggests that in spite of the presence of a large group such as SLF moiety at the end of 12-carbon long aliphatic sidechain, the bifunctional compounds would still retain high affinity towards AR. Ki value for 11α-isomer (S9) (825 nM) is highest among all compounds. The poor affinity of this compound is in agreement with our structural rationale, thereby validating our design strategy based on the crystal structure analysis.
AR-Dependent Transcriptional Activity
The aliphatic 11β-sidechain compounds, which possessed high affinity towards purified AR LBD, were subsequently tested using reporter gene assays in order to assess their effect on full length AR in the physiological setting of a prostate cancer cell. The transcriptional read-out from reporter gene assays would indicate how well a compound could cross the cell membrane, bind to AR inside the cell and induce AR-dependent transcriptional activity in a cellular milieu composed of numerous interfering factors. Different forms of AR (wild-type, T877A and W741C) were used in these assays to evaluate robustness of response across disease relevant genetic variants of AR [30,31].
Figure 3 presents the results of reporter gene assays conducted with or without 0.1 nM DHT, a potent agonist of AR (i.e., Kd ~20 nM). The reporter gene assays without DHT were conducted in order to measure the absolute agonistic ability of compounds while assays with 0.1 nM were performed to assess whether compounds can compete and exhibit enhanced agonistic response in the presence of DHT. This is relevant to the physiological situation where small amount of androgens are always present in the circulation.
Fig. 3.

Reporter gene assays using PC3 cell lines transfected with PSA-promoter driven luciferase gene and wild-type or mutant androgen receptor. The compounds were tested at 1 μM concentration in the presence of 100 nM of DHT. Symbols, D(−) and D(+), represent control samples without and with 100 nM DHT. Other symbols represent: casodex (CX), hydroxyflutamide (HF), nilutamide (NL), and cyproterone acetate(CPA).
In the absence of DHT (Fig. 3A), all compounds induced AR (wild-type) dependent transcription. Particularly, compounds S5 and S8 showed higher agonistic activity compared to 0.1 nM DHT. Surprisingly, compound S11, which had the highest binding affinity among all 11β-nortestosterone compounds, exhibited a low transcriptional activity, only comparable to low affinity (825 nM) compound S9. This suggests that compound S11 may not be bioavailable at the site of AR transcription, potentially due to its high hydrophobicity which may lead to high non-specific binding of this compound to cell membrane and other cellular targets. Overall, it can be concluded that most nortestosterone compounds are able to get inside the cells and specifically bind AR in the complex cellular environment. When these compounds were tested in the presence of 0.1 nM DHT (Fig. 3B), most of the newly nortestosterone analogs, except S8, did not enhance the transcriptional activity of wild-type AR compared to DHT alone. Only Compound S8 produced enhanced transcriptional activity compared to the DHT alone.
Against PC3 cell lines transfected with T877A mutant AR (Fig. 3C), the overall response of newly designed compounds was same as before, that is, nortestosterone analogs did not show any significant enhancement in AR transcriptional activity in the presence of 0.1 nM DHT. Similarly, S8 was the only compound with enhanced agonistic response. These results are in agreement with the fact that T877 residue does not play any differential role in modulating binding of 11β-sidechain compounds since steroidal core which interacts with T877 core is the same for all compounds. Past structural studies [25,32] have suggested that T877 residue is critical for AR specificity toward androgens and T877A mutation leads to a promiscuous AR which binds to non-androgens with steroidal cores different than typical androgens. Since all of our compounds have the same steroidal core, T877A mutation does not have any impact on the AR-dependent transcription activity.
Against PC3 cell lines transfected with W741C mutant AR (Fig. 3D) most of the nortestosterone compounds show a drastic increase in their potential for inducing AR transcriptional activity. The crystal structure studies [26] on W741C mutant AR shows that W741 residue sterically clashes with the sulphonyl moiety of casodex and mutation at this position converts casodex into an agonist molecule due to enhanced binding. A similar rationale can also be applied to explain the enhanced transcriptional activity of 11-sidechain nortestosterone compounds, and these findings suggests the crucial role of W741 residue in modulating the binding potencies of nortestosterone compounds in our library.
The results of reporter gene assays demonstrated that most steroidal 11-sidechain compounds were bioavailable within the cell and could bind to full length AR. Additionally, only compound S8 produced the enhanced induction of transcriptional activity for wild-type and mutant AR in the presence of 0.1 nM DHT. These results coupled with high in vitro binding affinity suggested that compound S8 was the best candidate for development into a bifunctional antiandrogen.
Cell Proliferation Assays
Subsequent to the reporter gene assays, we conducted MTT cell proliferation assays in order to explore growth stimulatory/inhibitory effects of 11-sidechain nortestosterone compounds on prostate cancer cells. Four different prostate cancer cell lines were used in our cell proliferation assays: LAPC4 (wild-type AR), LNCaP (T877A mutant AR), CWR22R (truncated AR), and PC3 cell lines (AR negative). A unique advantage of including AR negative PC3 cell lines was that they would allow the determination of off target effects. A positive or a negative effect on the cell growth combined with no effect on PC3 cell growth would demonstrate the specificity of response. On the other hand, a simultaneous effect on AR-positive cell lines and PC3 cell lines would indicate the off target activity of our compounds.
Figure 4 presents the percentage growth values (normalized with respect to control) for nortestosterone compounds with aliphatic sidechains. As shown, none of the compounds tested at 1 μM concentration showed growth inhibitory effects against AR-negative PC3 cell lines indicating the absence of off target effects for these compounds (Fig. 4A). These results indirectly suggest the high selectivity of these compounds for AR. Against LAPC4 cell lines that express wild-type AR, compounds S11 and S6 showed ~50% growth inhibition (Fig. 4B). Against LNCaP cell lines (T877A mutant AR) and CWR22R cell lines, all compounds show growth inhibitory effect in the range of 40–60% (Fig. 4C, D).
Fig. 4.

MTT cell proliferation assays. LNCaP, PC3, and CWR22R cell lines were grown in RPMI 1640 media supplanted with 10% FBS. The LAPC4 cell lines were grown in Iscove’s media supplanted with 10% FBS and 0.1 nM R1881. All compounds were tested at 1 μM concentration. Casodex (CX) served as a control in these assays.
Presence of growth inhibitory activity among 11-sidechain is counterintuitive given the fact that these compounds behaved as agonists in the reporter gene assays. This can be explained by the results of recent studies exploring the disconnect between AR-driven malignant growth and its transcription activity [33]. These studies suggest that cancer cells engage AR to drive their growth independent of AR mediated transcriptional expression of differentiation markers such as PSA. Thus, agonistic ability of 11-sidechain analogs in the reporter gene assays does not predict the effect of newly synthesized compounds on the growth of cancer cells. Nonetheless, growth inhibitory effects of 11-sidechain analogs are a positive result considering their eventual development into bifunctional antiandrogens.
In conclusion, MTT cell proliferation assays suggest that 11-sidechain analogs are selective for AR-positive cell lines and show growth inhibitory response with minimal off target effects. Similar to the findings of reporter assays, these compounds have variable response on the AR-positive cell lines depending upon the mutant status of AR.
Binding Affinity Against Glucocorticoid Receptor
For the bifunctional approach to have any therapeutic potential, it is critical that lead 11-sidechain analogs described in our study do not possess high binding potency for the glucocorticoid receptor (GR). GR is a homolog of AR and part of the nuclear hormone receptor family. The GR is expressed in almost every cell in the body and involved in a wide variety of cellular processes including development, glucogenesis and metabolism, and immune response. In order to evaluate precise binding affinity of our lead compounds against GR, we conducted in vitro ligand binding assays against purified GR LBD using FP technique. Figure 5 presents the Ki values for S11, S8, S9 and cyproterone acetate (CPA). The Ki values for all three 11-sidechain compounds are higher than 1 μM. When compared with the Ki value of currently approved antiandrogen cyproterone acetate (260 nM) and glucocorticoid ligand mifepristone (Ki ~20 nM), the 11β-Δ9-19 nortestosterone compounds possess extremely low affinity for GR. These results confirm that the lead compounds do not interfere with the GR signaling and validate our original rationale of using Δ9-19 nortestosterone moiety as the steroidal core for bifunctional antiandrogens.
Fig. 5.

Glucocorticoid ligand binding assays using fluorescence polarization technique. The steroidal antiandrogen cyproterone acetate (CPA) was used as a benchmark.
CONCLUSIONS
The results of this study validate our drug design rationale based on the structural biology of AR and pave the way for the design and development of bifunctional antiandrogens through chemical conjugation of lead compounds discovered in this study to the SLF moiety. The compound S8 was identified as the best lead compound in the rationally designed library of 11β-Δ9-19 nortestosterone compounds based on the results of a series of in vitro assays which included AR ligand binding assays, reporter gene assays and cell proliferation assays. The ligand binding assays revealed that aliphatic chemical functionality at the 11β-position was ideal for incorporation into bifunctional antiandrogens since aliphatic sidechain compounds retained modest binding affinity with the AR even when a large phenyl group (in S8) or a second steroidal group (in S11) was added at the end of a large 11β-sidechain. The compounds with aliphatic linkers were subsequently tested for their ability to cross cell membrane and induce transcriptional activation of AR using reporter gene assays. These assays indicated that the compound S8 produced the maximum induction of AR transcription activity and this was true even in the case of AR mutants. To further characterize this and other aliphatic linker containing compounds, cell proliferation assays were conducted against multiple cell lines to identify their off target effects. The MTT assays confirmed that these compounds were specific towards AR-positive cell lines and did not show any physiological response against AR-negative PC3 cell lines. Additionally, S8 and a subset of 11-sidechain compounds (i.e., S11, S9) did not cross-interfere with GR signaling, thus eliminating the possibility of any antiglucocorticoid activity of these compounds. The results of these assays taken together suggest that 11β-Δ9-19 nortestosterone containing 12-carbon aliphatic linker at the 11-position are ideally suited for subsequent transformation into bifunctional antiandrogens.
The work described in this report allows the completion of a critical step towards the future development of bifunctional antiandrogens as novel prostate cancer therapeutics. Having identified compound S8 as the lead candidate for transformation into a bifunctional compound, we are currently pursuing the chemical synthesis of bifunctional compounds via conjugation of carboxyl group of SLF to the amine-containing 12-carbon long aliphatic sidechain at the 11β-position of steroidal core. Resulting bifunctional compounds will be subsequently tested for AR binding, their ability to recruit FKBP chaperone proteins, and in vitro and in vivo potency towards inhibiting AR function in hormone refractory cell lines and in appropriate animal models. Additionally, we are in the process of conducting similar studies on 7-side-chain analogs as well. The lead compounds from the 7α-Δ9-19 nortestosterone family will provide additional candidates for subsequent development into bifunctional antiandrogens.
Acknowledgments
This work was funded by NIH grant CA091409. We sincerely thank Lizamma Antony for her valuable input on the experimental protocols.
Footnotes
Additional supporting information may be found in the online version of this article.
References
- 1.Mooradian AD, Morley JE, Korenman SG. Biological actions of androgens. Endocr Rev. 1987;8(1):1–28. doi: 10.1210/edrv-8-1-1. [DOI] [PubMed] [Google Scholar]
- 2.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 3.Eder IE, Hoffmann J, Rogatsch H, Schafer G, Zopf D, Bartsch G, Klocker H. Inhibition of LNCaP prostate tumor growth in vivo by an antisense oligonucleotide directed against the human androgen receptor. Cancer Gene Ther. 2002;9(2):117–125. doi: 10.1038/sj.cgt.7700416. [DOI] [PubMed] [Google Scholar]
- 4.Solit DB, Zheng FF, Drobnjak M, Munster PN, Higgins B, Verbel D, Heller G, Tong W, Cordon-Cardo C, Agus DB, Scher HI, Rosen N. 17-Allylamino-17-demethoxygeldanamycin induces the degradation of androgen receptor and HER-2/neu and inhibits the growth of prostate cancer xenografts. Clin Cancer Res. 2002;8(5):986–993. [PubMed] [Google Scholar]
- 5.Yang Q, Fung KM, Day WV, Kropp BP, Lin HK. Androgen receptor signaling is required for androgen-sensitive human prostate cancer cell proliferation and survival. Cancer Cell Int. 2005;5(1):8. doi: 10.1186/1475-2867-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62(4):1008–1013. [PubMed] [Google Scholar]
- 7.Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Res. 2001;61(13):5038–5044. [PubMed] [Google Scholar]
- 8.Gao J, Isaacs JT. Development of an androgen receptor-null model for identifying the initiation site for androgen stimulation of proliferation and suppression of programmed (apoptotic) death of PC-82 human prostate cancer cells. Cancer Res. 1998;58(15):3299–3306. [PubMed] [Google Scholar]
- 9.Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling? J Clin Endocrinol Metab. 2003;88(7):2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 10.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, Kim R, Rubin MA, Pienta KJ. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004;64(24):9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 11.Gottlieb B, Beitel LK, Wu JH, Trifiro M. The androgen receptor gene mutations database (ARDB): 2004 update. Hum Mutat. 2004;23(6):527–533. doi: 10.1002/humu.20044. [DOI] [PubMed] [Google Scholar]
- 12.Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3(11):950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- 13.He B, Gampe RT, Jr, Kole AJ, Hnat AT, Stanley TB, An G, Stewart EL, Kalman RI, Minges JT, Wilson EM. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell. 2004;16(3):425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 14.Estebanez-Perpina E, Moore JM, Mar E, Delgado-Rodrigues E, Nguyen P, Baxter JD, Buehrer BM, Webb P, Fletterick RJ, Guy RK. The molecular mechanisms of coactivator utilization in ligand-dependent transactivation by the androgen receptor. J Biol Chem. 2005;280(9):8060–8068. doi: 10.1074/jbc.M407046200. [DOI] [PubMed] [Google Scholar]
- 15.Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol. 2004;2(9):E274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao L, Chmielewski J. Inhibiting protein-protein interactions using designed molecules. Curr Opin Struct Biol. 2005;15(1):31–34. doi: 10.1016/j.sbi.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Standaert RF, Galat A, Verdine GL, Schreiber SL. Molecular cloning and overexpression of the human FK506-binding protein FKBP. Nature. 1990;346(6285):671–674. doi: 10.1038/346671a0. [DOI] [PubMed] [Google Scholar]
- 18.Briesewitz R, Ray GT, Wandless TJ, Crabtree GR. Affinity modulation of small-molecule ligands by borrowing endogenous protein surfaces. Proc Natl Acad Sci USA. 1999;96(5):1953–1958. doi: 10.1073/pnas.96.5.1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science. 1993;262(5136):1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 20.Muthuswamy SK, Gilman M, Brugge JS. Controlled dimerization of ErbB receptors provides evidence for differential signaling by homo- and heterodimers. Mol Cell Biol. 1999;19(10):6845–6857. doi: 10.1128/mcb.19.10.6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gestwicki JE, Crabtree GR, Graef IA. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science. 2004;306(5697):865–869. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]
- 22.Braun PD, Barglow KT, Lin YM, Akompong T, Briesewitz R, Ray GT, Haldar K, Wandless TJ. A bifunctional molecule that displays context-dependent cellular activity. J Am Chem Soc. 2003;125(25):7575–7580. doi: 10.1021/ja035176q. [DOI] [PubMed] [Google Scholar]
- 23.Sellmyer MA, Stankunas K, Briesewitz R, Crabtree GR, Wandless TJ. Engineering small molecule specificity in nearly identical cellular environments. Bioorg Med Chem Lett. 2007;17(10):2703–2705. doi: 10.1016/j.bmcl.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Litvinov IV, Antony L, Isaacs JT. Molecular characterization of an improved vector for evaluation of the tumor suppressor versus oncogene abilities of the androgen receptor. Prostate. 2004;61(4):299–304. doi: 10.1002/pros.20187. [DOI] [PubMed] [Google Scholar]
- 25.Urushibara M, Ishioka J, Hyochi N, Kihara K, Hara S, Singh P, Isaacs JT, Kageyama Y. Effects of steroidal and non-steroidal antiandrogens on wild-type and mutant androgen receptors. Prostate. 2007;67(8):799–807. doi: 10.1002/pros.20542. [DOI] [PubMed] [Google Scholar]
- 26.Bohl CE, Gao W, Miller DD, Bell CE, Dalton JT. Structural basis for antagonism and resistance of bicalutamide in prostate cancer. Proc Natl Acad Sci USA. 2005;102(17):6201–6206. doi: 10.1073/pnas.0500381102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bohl CE, Wu Z, Miller DD, Bell CE, Dalton JT. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J Biol Chem. 2007;282(18):13648–13655. doi: 10.1074/jbc.M611711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, Li YL, Gustafsson JA, Carlquist M. Structural insights into the mode of action of a pure antiestrogen. Structure. 2001;9(2):145–153. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 29.Muddana SS, Price AM, MacBride MM, Peterson BR. 11beta-alkyl-Delta9-19-nortestosterone derivatives: High-affinity ligands and potent partial agonists of the androgen receptor. J Med Chem. 2004;47(21):4985–4988. doi: 10.1021/jm0342515. [DOI] [PubMed] [Google Scholar]
- 30.Taplin ME, Rajeshkumar B, Halabi S, Werner CP, Woda BA, Picus J, Stadler W, Hayes DF, Kantoff PW, Vogelzang NJ, Small EJ. Androgen receptor mutations in androgen-independent prostate cancer: Cancer and Leukemia Group B Study 9663. J Clin Oncol. 2003;21(14):2673–2678. doi: 10.1200/JCO.2003.11.102. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki H, Akakura K, Komiya A, Aida S, Akimoto S, Shimazaki J. Codon 877 mutation in the androgen receptor gene in advanced prostate cancer: Relation to antiandrogen withdrawal syndrome. Prostate. 1996;29(3):153–158. doi: 10.1002/1097-0045(199609)29:3<153::aid-pros2990290303>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 32.Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J Biol Chem. 2005;280(45):37747–37754. doi: 10.1074/jbc.M507464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denmeade SR, Sokoll LJ, Dalrymple S, Rosen DM, Gady AM, Bruzek D, Ricklis RM, Isaacs JT. Dissociation between androgen responsiveness for malignant growth vs. expression of prostate specific differentiation markers PSA, hK2, and PSMA in human prostate cancer models. Prostate. 2003;54(4):249–257. doi: 10.1002/pros.10199. [DOI] [PubMed] [Google Scholar]