

Abstract
We demonstrate the application of fluorescence lifetime optical projection tomography (FLIM-OPT) to in vivo imaging of lysC:GFP transgenic zebrafish embryos (Danio rerio). This method has been applied to unambiguously distinguish between the fluorescent protein (GFP) signal in myeloid cells from background autofluorescence based on the fluorescence lifetime. The combination of FLIM, an inherently ratiometric method, in conjunction with OPT results in a quantitative 3-D tomographic technique that could be used as a robust method for in vivo biological and pharmaceutical research, for example as a readout of Förster resonance energy transfer based interactions.
OCIS codes: (170.3650) Lifetime-based sensing, (170.6900) Three-dimensional microscopy, (170.6920) Time-resolved imaging, (170.6960) Tomography
1. Introduction
Research into biomolecular signaling and disease mechanisms has progressed from average measurements on cell populations and extracts to “high content” imaging of single cells, both fixed and viable. This provides morphological information that can be related to physiology and phenotype and allows spatial and/or temporal analysis of variations in the intracellular distribution of molecules and their interactions. Increasingly, such high content analysis (HCA) is being implemented in automated imaging systems capable of assaying sample arrays at sufficient rates to screen against gene libraries. In conjunction with advances in labeling technology, particularly genetically expressed fluorescent proteins [1], this progression to HCA has driven the development of quantitative fluorescence microscopy techniques. Of particular interest is Förster resonance energy transfer (FRET) [2,3], which can be used to detect and monitor interactions between proteins labeled with appropriate (donor and acceptor) fluorophores. FRET can also be used to measure conformational changes of large molecules and is increasingly used to read out genetically expressed biosensors for signaling molecules such as calcium [4], potassium [5], chloride [6], GTP [7], IP3 [8], PIP2 [9] and others [10]. FRET is an example of fluorescence emission reporting variations in the local fluorophore environment (specifically the proximity of another fluorophore) and there are many other applications where such functional information can be obtained by exploiting fluorophores as sensors. In principle the fluorophore environment can be assayed by measurements of intensity through its dependence on quantum efficiency, but such measurements require knowledge of the probe concentration and the excitation and detection efficiencies, which is often not available in biological experiments and can be compromised by sample attenuation and scattering. This issue may be addressed by ratiometric imaging techniques, of which fluorescence lifetime imaging (FLIM) [11–13], measuring the average relaxation time of excited fluorescent molecules, is one of the most robust.
While FRET microscopy is in widespread academic use, it has yet to see significant uptake for drug discovery owing to the difficulties associated with acquiring sufficiently robust quantitative data. This might be mitigated using FLIM, but while there are now several commercially available FLIM microscope systems there are few reports of FLIM implemented for HCA in multiwell plate readers. This is partly due to the relatively low imaging speeds of the most common implementations of FLIM that are realized in laser scanning microscopes. We have recently reported a high-speed optically sectioning FLIM multiwell plate reader [14] able to acquire images in less than a second and believe that this illustrates the potential of FLIM as a tool to screen for protein-protein interactions and assay cell signaling networks. Unfortunately, however, the results of multiwell plate assays do not always translate along the drug discovery pipeline to clinical efficacy and there is a growing appreciation that in vitro cell mono-layers may exhibit non-physiological behavior due to their highly-artificial environment. This has led to increasing interest in assays implemented in three-dimensional (3-D) cell cultures [15] and in more physiologically realistic engineered or in vivo environments [16,17]. For drug discovery and many biological studies, there is increasing interest in live disease models that can be developed in organisms such as Drosophila, Caenorhabditis elegans and zebrafish embryos, which are optically accessible and can be genetically manipulated. Our vision is to develop high content assays of biomolecular interactions that can be translated from in vitro microscopy and analysis of cultured cells to 3-D analysis of disease models and in live organisms from embryonic to adult stages. Accordingly we are working to extend FLIM from microscopes and plate readers to in vivo imaging of model organisms and small animals. For larger animals this can be realized through intravital microscopy, which has been applied to in vivo FRET [18] and FLIM [19]. The limited depth penetration of microscopy, however, restricts this approach to image superficial tissues or implementation in conjunction with highly invasive surgical procedures. For larger animals, internal tissues could be accessed using FLIM endoscopy. While this has not yet been demonstrated, we have reported the first confocal FLIM microendoscope [20], which has recently been shown to be able to read out FRET in live cells in less than 2 seconds [21].
While optically sectioned imaging of relatively thin samples (<1 mm) is readily achievable with confocal/multiphoton microscopy, particularly for transparent samples, full 3-D imaging can be very time consuming and may not be possible for larger samples. To address this challenge for samples in the 1-10 mm range, often referred to as the “mesoscopic” regime, various tomographic techniques have been developed such as selective plane illumination microscopy (SPIM) [22], ultramicroscopy [23] and optical projection tomography (OPT) [24]. The first two of these techniques use orthogonal illumination and imaging axes, with the sample being illuminated by a “sheet” of light, defining the sectioning strength, and imaged using a standard wide-field system, defining the lateral resolution. This orthogonal geometry can provide significantly reduced photobleaching and improved axial resolution compared to confocal microscopy. SPIM and ultramicroscopy are limited to fluorescence imaging and typically require a different optical configuration to a standard microscope, although some implementations can be retrofitted to conventional inverted microscopes [25]. In contrast OPT, which is the optical equivalent of X-ray computed tomography, can be applied to reconstruct the absorption and/or fluorescence distributions in optically transparent samples and can be applied over sub-mm to ~cm scales. For small samples, OPT can be implemented on a standard wide-field microscope with only minor modifications.
The requirement of optical transparency for mesoscopic imaging techniques is, however, a major limitation. For less transparent and larger organisms chemical clearing techniques are required, which not only precludes the possibility of in vivo imaging, but can also have an adverse effect on the fluorescence properties of genetically expressed fluorescent proteins [26,27]. Some small organisms are sufficiently transparent to be imaged in vivo such as D. melanogaster [22] and Danio rerio [28] imaged using SPIM, and D. melanogaster [29] and C. elegans [30] imaged using OPT. We demonstrate here that it is possible to apply OPT to live zebrafish embryos labeled with genetically expressed fluorescent proteins up to at least ~3 days post-fertilization. We believe that this presents a significant advance since zebrafish are an attractive vertebrate animal model for biological and pharmacological research [31] due to their relatively easy maintenance and housing, short reproductive cycle, extensively sequenced genome, the potential for manipulation using well established genetic and molecular approaches and easy drug administration. Zebrafish have successfully applied in toxicology bioassays for on and off target effects [32–34], organ function assays [35] and to model diseases [36].
To date mesoscopic imaging techniques have not yet accessed most of the quantitative imaging methods implemented in microscopy, but have largely been limited to mapping intensity. This can provide 3-D reconstructions of fluorescent label distribution (e.g. probe localization and sample structure) and time-lapse (motion) data when applied to live specimens [22,37]. Extracting quantitative information beyond localization, however, is difficult due to the spatial variation in excitation and collection efficiency of fluorescence light, the variation in staining efficiency of chemical labels or expression levels of genetic fluorophores and variations in sample attenuation (“inner filter” effect). FLIM can provide a robust approach to quantitative readouts and we previously demonstrated the extension of OPT to FLIM-OPT of fixed and optically cleared samples [38], showing how the ratiometric nature of FLIM makes it insensitive to intensity variations. In that experiment, FLIM was able to distinguish between Alexa-488-labeled neurofilament and the autofluorescence from the heart and dorsal aorta of a fixed and cleared mouse embryo.
In this paper we report the first application of OPT to live transgenic zebrafish embryos and the first full body 3-D fluorescence lifetime imaging of a live animal model for biological research and drug discovery, which we believe can be applied in primary screening before the expensive mammal testing phase, providing functional readouts at higher resolution than is possible in mammals.
2. Materials and methods
2.1. Sample preparation and mounting
A transgenic zebrafish line (lysC:GFP, lysC-GFP were produced by Philip S. Crosier, University of Auckland 1142, NZ [39,40]) which express GFP in myeloid cells, was used as an exemplar model for in vivo imaging. Embryos were raised in embryo medium (dechlorinated system water containing 0.0003% (v/v) methylene blue and 30µg/ml N-phenylthiourea (Sigma) to prevent melanization). Embryos at 3 days post-fertilization were immobilized in 1% low melt point agarose (Flowgen, Lichfield, UK), made from embryo media, containing 0.3 mM MS-222 (Sigma) as anesthetic. They were then drawn into short lengths of translucent FEP tubing (06406-60, Cole-Palmer) with inner and outer diameters of 0.8 and 1.6 mm respectively. The tubing has a refractive index similar to that of water and so can be used in an index matched chamber for imaging applications [41]. Agarose was added to increase the viscosity of the water and prevent movement of the anaesthetized zebrafish under the action of gravity. Figure 1(a) shows a live zebrafish embryo (demonstrated by beating heart) imaged at 20 frames per second mounted in the FEP tubing.
Fig. 1.

Single frames from videos showing (a) a live zebrafish embryo (Media 1 (852.1KB, AVI) ) mounted in the OPT system and (b) an intensity-based in vivo OPT acquisition (Media 2 (591.6KB, AVI) ) of a lysC:GFP zebrafish embryo. Scale bar 500 μm.
2.2. Experimental set-up and reconstruction
A custom-built chamber was fabricated to hold the tube-mounted zebrafish embryos in a refractive index-matched environment and to allow stable sample rotation without lateral movement, as is required for OPT (see Figs. 1(b) and 2 ). The samples were imaged on a standard inverted wide-field microscope (IX-71, Olympus UK Ltd), utilizing both epifluorescence and transmitted light imaging with a 4x objective (UPLFLN4X, Olympus UK Ltd) and a filter cube for GFP (GFP-3035B-OMF, Laser 2000 Ltd). An aperture positioned directly behind the objective limited its numerical aperture to 0.07. During a standard OPT acquisition, fluorescence and/or transmitted light images are acquired at equal angular intervals as the sample rotated (e.g. acquiring images every 1° over a full rotation). The transmitted light images were acquired using the incandescent lamp and images were acquired through the GFP filter that has a spectral pass band of 525 ± 25 nm.
Fig. 2.
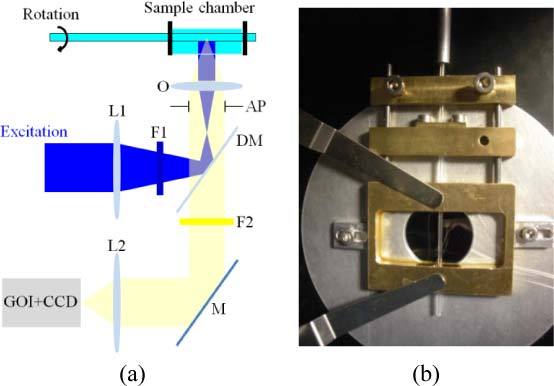
(a) Schematic of OPT system. O – objective, AP – aperture, L1 – condenser lens, F1 – excitation filter, DM – dichroic mirror, L2 – tube lens, F2 – emission filter, M – mirror. (b) Photograph of custom built chamber.
To implement FLIM we employed wide-field time-gated imaging using a gated optical intensifier (GOI). Wide-field excitation was provided by a spectrally-filtered (472 ± 15 nm) ultrafast fiber-laser-pumped super-continuum source (SC-400-2, Fianium Ltd) [42]. The emitted fluorescence was imaged onto the photocathode of the GOI (HRI, Kentech Instruments Ltd), which could be gated with an adjustable delay relative to the excitation pulses, for which the gate-width was set to 1 ns. The GOI phosphor was imaged onto a CCD camera (Clara, Andor Technology plc). Intensifier gate delays were adjusted using a computer-controlled electronic delay line (HDG, Kentech Instruments Ltd) that was synchronized to the excitation laser. During a typical acquisition, 5 time-gated images were recorded at 1 ns relative delay positions every 4° (i.e. a total of 450 images were recorded). In addition to the time-gated fluorescence images, a single transmitted light image was acquired every 1° through the same filter. The total acquisition time for the data set was ~20 minutes. Each frame had an integration time of 1 s and 0.5 s for the time-gated and transmitted light acquisitions respectively, leading to a total camera integration time of 630 s. The remaining time was due to the rotational and temporal delay scanning, camera readout time and switching between fluorescence and transmission modes and can be significantly reduced. The GOI has an effective pixel size of ~26 μm and used in this optical configuration results in an in-focus lateral spatial resolution of ~13 μm.
3-D image reconstruction was realized using a back-projection algorithm. Assuming a parallel projection regime, each row of pixels in an acquired image can be considered a 1-D projection, i.e. a sum along the direction of the optical axis, of a 2-D slice through the sample. As the sample rotates, these individual 1-D projections, Pθ(r), make up the sinogram (see Fig. 3(a) ). From the sinogram the signal (e.g. the fluorescence intensity) at each point in a 2-D slice through the sample can be reconstructed using an inverse Radon transform, as described by Eq. (1), where Sθ(w) is the Fourier transform of Pθ(r), w is the spatial frequency and θ is the angle at which the projection was acquired. During this “filtered back-projection” reconstruction process, the measured projection is Fourier transformed, filtered by |w| to account for the spatial frequency sampling as the object rotates, inverse Fourier transformed and back-projected or smeared across a 2-D plane at the corresponding acquisition angle, then repeated for every projection [24,43] (see Fig. 3(b)).
Fig. 3.

(a) An OPT raw data set comprises a set of wide-field images as a function of rotation. Reconstruction is performed using the sinograms (r-θ planes) for each row of pixels. (b) Single frame showing the reconstruction of a single slice from 90 fluorescence projections (Media 3 (468.8KB, AVI) ). Scale bar 500 μm.
| (1) |
This reconstruction approach is repeated for each set of time-gated measurements corresponding to each time delay. In this case, five time-gated 3-D intensity reconstructions of the fluorescence signal were obtained. The intensity decay in corresponding voxels of the time-gated reconstructions was then determined, assuming a single exponential fluorescence decay model, using an in-house fitting algorithm based on non-linear least squares optimization. This produced 3-D reconstructions of both the integrated fluorescence intensity and lifetime. For visualization purposes the lifetime was represented on a false color scale and merged with the integrated intensity to suppress noise in regions with little or no signal.
3. Results
Figures 4(a) and (b) show a 3-D fluorescent intensity reconstruction and an animation of the combined absorption (grayscale) and fluorescence (red) reconstruction respectively of a transgenic zebrafish (lysC:GFP), for which the expression of GFP is limited to myeloid cells (e.g. neutrophils and macrophages). We would therefore expect the fluorescence reconstruction to show a distribution of these cells throughout the embryo. In the raw data and reconstruction, however, a significant autofluorescence signal can also be observed, particularly in the region of the remaining yolk-sac. Without a priori knowledge of the expected fluorescent protein distribution, it is difficult to distinguish this signal from induced background fluorescence, since both signals will vary spatially in intensity due to uneven expression level, illumination and collection efficiency and artifacts in the reconstruction process. The GFP signal can, however, be clearly distinguished from autofluorescence via the fluorescence lifetime. Figure 5(a) shows the corresponding fluorescence lifetime reconstruction, for which the fluorescence lifetime is encoded in the color scale. The separation of lifetimes is such that the reconstruction can be viewed on a discrete color scale (Fig. 5(b)), showing the GFP signal in green and autofluorescence in blue. This contrast is confirmed by the fluorescence lifetime histogram (Fig. 5(c)), which shows these two distinct lifetime populations and confirms that the correct lifetime of ~2.5ns has been determined for GFP.
Fig. 4.

(a) A 3-D fluorescence intensity reconstruction and (b) a single frame showing combined fluorescence (red) and transmitted light (grey) intensity reconstructions of a live lysC:GFP transgenic zebrafish embryo 3 days post-fertilization (Media 4 (299.5KB, AVI) ). Scale bar 500 μm.
Fig. 5.
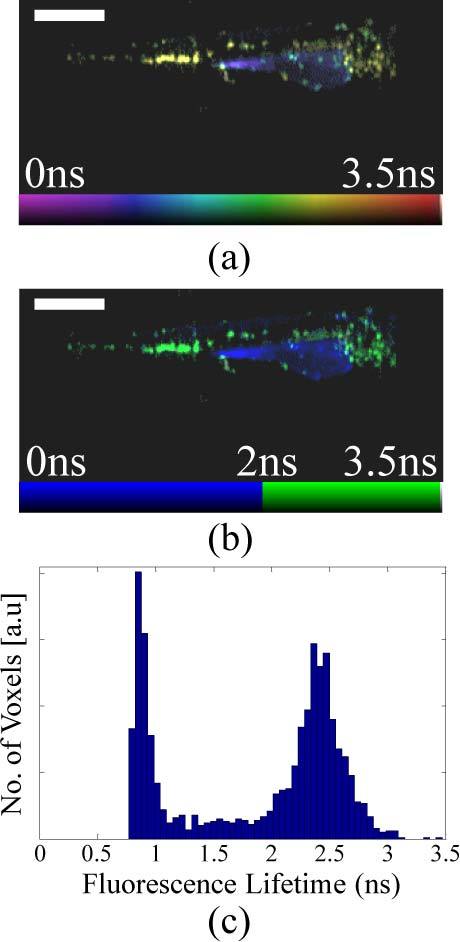
Single frames showing 3-D fluorescence lifetime reconstructions on a (a) continuous (Media 5 (328.7KB, AVI) ) and (b) discrete color scale (Media 6 (328.9KB, AVI) ) of a live lysC:GFP transgenic zebrafish embryo 3 days post-fertilization. (c) A fluorescence lifetime histogram showing two clear populations corresponding to GFP and autofluorescence. Scale bar 500 μm.
We note that the data sets presented here represent “single time-point” acquisitions and so the time-lapse resolution for this FLIM-OPT acquisition is therefore ~20 minutes, the total data acquisition time. As a result we would not accurately reconstruct the position and fluorescence lifetime of features that move a significant distance during this acquisition time. Furthermore, the acquisition was not synchronized with the heartbeat of the zebrafish and so the reconstructed image in close proximity to the heart will be compromised by the time-averaged motion. The total FLIM-OPT acquisition time can be decreased by optimizing the excitation powers and labeling density, by sampling the fluorescence decays with fewer time gates and by acquiring images at fewer angles of projection although this will, of course, compromise the achievable resolution and accuracy of the reconstructed images. We note, however, that (FLIM) OPT, can potentially provide information with much higher time-lapse resolution than the total data acquisition time by tracking the movement of sample features. For example, by acquiring sequential frames at 90° with respect to each other, it should be possible to track the movement of features such as myeloid cells with a time-lapse resolution movement determined by the time needed to acquire the (~2-4) orthogonal images required to calculate the 3-D coordinates of the moving features.
The tomographic image reconstruction approach used in this demonstration of in vivo FLIM-OPT was based on filtered back-projection, calculated using in-house software implemented in MatLab (Mathworks Inc). The FLIM reconstruction shown in Fig. 5 is comprised of a back-projection reconstruction corresponding to each of the 5 time-gates sampling the fluorescence decays, followed by iterative fitting to determine the fluorescence lifetime in every voxel. This post-processing took ~20 minutes running on a personal computer (2.66 GHz, 3.25 GB RAM). The required processing time could be significantly reduced by parallel processing on multicore computers [44] or by taking advantage of the speed of graphics processor units [45]. In addition, if the fluorescence lifetimes are known in advance, an optimized gating strategy can be used to minimize the required number of time-gates (e.g. 2 gates for a single exponential [46,47]) and therefore reduce the number of 3-D reconstructions required. We note that, since we are imaging with ballistic light, the fluorescence lifetime resolution is determined by the wide-field FLIM system and should be similar to that obtainable in FLIM microscopy, being a function of the instrument response function and the signal to noise ratio. The performance of our time-gated FLIM system has previously been discussed in e.g [21,48].
With respect to sample viability, we note that the zebrafish embryos were alive (with beating heart) at the end of our experiments. While we have not yet been able to determine their long term viability, we have performed multiple fluorescence intensity OPT acquisitions of zebrafish (data not shown), including two single time-point acquisitions of the same zebrafish separated by 48 hours, during which time the zebrafish embryo appeared to develop normally, and a first time lapse study involving 20 intensity OPT acquisitions (each of ~20 s at 2 minute intervals. In both cases the fish were still alive at the end of the experiments.
4. Conclusions
Here we report in vivo OPT and FLIM-OPT of intact viable zebrafish embryos. In order to achieve this result, we have constructed a refractive index-matched chamber designed for in vivo wide-field imaging [41] with computer controlled stable rotation of the sample, as is required for OPT. This technique was implemented on a standard wide-field fluorescence microscope and, in general, OPT can be implemented in almost any wide-field imaging instrument, essentially only requiring the addition of a means to achieve the sample rotation. In principle, this can be extended to FLIM-OPT by incorporating any wide-field FLIM instrumentation. Here we have implemented time-gated imaging, utilizing a tunable supercontinuum excitation source to provide spectral versatility. We note, however, that FLIM could also be realized in the frequency domain using a sinusoidally modulated image intensifier. Looking forward, it may be possible to develop a lower cost implementation using, for example, a directly modulated CMOS camera for detection [49] and sinusoidally modulated LED sources for excitation [50]. The OPT reconstruction method used here was based on filtered back-projection, but it could be improved by using more sophisticated approaches that incorporate the characteristics of the imaging system as a function of defocus [51]. Further advances in the reconstruction process could incorporate optical properties of the specimen including light scattering properties for particular regions (e.g. yolk-sac, eyes) [52].
We have also shown the ability of FLIM to distinguish between an expressed fluorescent protein signal and background autofluorescence in 3-D reconstructions, noting that autofluorescence from the yolk-sac is a particular issue for zebrafish embryos. Thus this work demonstrates the feasibility of using fluorescence lifetime contrast with OPT to provide functional imaging for the investigation of 3-D biological processes in vivo. We believe that it may be feasible to adapt this approach to automated imaging to achieve a higher throughput, e.g. following [53]. It may also be possible to devise a scheme to image multiple zebrafish in a single acquisition to increase imaging throughput, although this would compromise the achievable resolution.
Fluorescence lifetime contrast in OPT could be exploited to distinguish between multiple labels with similar spectral properties but different lifetime values and to map 3-D variations in local fluorophore environment, such as membrane lipid order [54], analyte concentration (e.g. calcium [21]) and FRET. By utilizing specific FRET sensors for probing particular interactions in a signaling pathway or indicating the concentration of specific molecules, FLIM-OPT can provide both 3-D spatial localization and molecular information in transfected/transgenic zebrafish. Combining this with time-lapse acquisition in live samples will allow investigators to correlate 3-D motion with molecular signaling events, for example in elucidating the control and signaling mechanisms behind cell migration during the innate immune response.
Acknowledgments
The authors gratefully acknowledge funding from the UK Biotechnology and Biological Sciences Research Council, the UK Engineering and Physical Sciences Research Council, the Wellcome Trust and GlaxoSmithKline. Paul French acknowledges a Royal Society Wolfson Research Merit Award. We thank Paul Martin (University of Bristol, UK) for provision of the LysC:GFP transgenic zebrafish line and Philip S. Crosier (University of Auckland 1142, NZ) for original production of this line.
References and links
- 1.Tsien R. Y., “The green fluorescent protein,” Annu. Rev. Biochem. 67(1), 509–544 (1998). 10.1146/annurev.biochem.67.1.509 [DOI] [PubMed] [Google Scholar]
- 2.Bastiaens P. I. H., Squire A., “Fluorescence lifetime imaging microscopy: spatial resolution of biochemical processes in the cell,” Trends Cell Biol. 9(2), 48–52 (1999). 10.1016/S0962-8924(98)01410-X [DOI] [PubMed] [Google Scholar]
- 3.Jares-Erijman E. A., Jovin T. M., “FRET imaging,” Nat. Biotechnol. 21(11), 1387–1395 (2003). 10.1038/nbt896 [DOI] [PubMed] [Google Scholar]
- 4.Mank M., Reiff D. F., Heim N., Friedrich M. W., Borst A., Griesbeck O., “A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change,” Biophys. J. 90(5), 1790–1796 (2006). 10.1529/biophysj.105.073536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ueyama H., Takagi M., Takenaka S., “A novel potassium sensing in aqueous media with a synthetic oligonucleotide derivative. Fluorescence resonance energy transfer associated with Guanine quartet-potassium ion complex formation,” J. Am. Chem. Soc. 124(48), 14286–14287 (2002). 10.1021/ja026892f [DOI] [PubMed] [Google Scholar]
- 6.Kuner T., Augustine G. J., “A genetically encoded ratiometric indicator for chloride: capturing chloride transients in cultured hippocampal neurons,” Neuron 27(3), 447–459 (2000). 10.1016/S0896-6273(00)00056-8 [DOI] [PubMed] [Google Scholar]
- 7.Mochizuki N., Yamashita S., Kurokawa K., Ohba Y., Nagai T., Miyawaki A., Matsuda M., “Spatio-temporal images of growth-factor-induced activation of Ras and Rap1,” Nature 411(6841), 1065–1068 (2001). 10.1038/35082594 [DOI] [PubMed] [Google Scholar]
- 8.Nezu A., Tanimura A., Morita T., Shitara A., Tojyo Y., “A novel fluorescent method employing the FRET-based biosensor “LIBRA” for the identification of ligands of the inositol 1,4,5-trisphosphate receptors,” Biochim. Biophys. Acta 1760(8), 1274–1280 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Nishioka T., Aoki K., Hikake K., Yoshizaki H., Kiyokawa E., Matsuda M., “Rapid turnover rate of phosphoinositides at the front of migrating MDCK cells,” Mol. Biol. Cell 19(10), 4213–4223 (2008). 10.1091/mbc.E08-03-0315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li I. T., Pham E., Truong K., “Protein biosensors based on the principle of fluorescence resonance energy transfer for monitoring cellular dynamics,” Biotechnol. Lett. 28(24), 1971–1982 (2006). 10.1007/s10529-006-9193-5 [DOI] [PubMed] [Google Scholar]
- 11.Buurman E. P., Sanders R., Draaijer A., Gerritsen H. C., Vanveen J. J. F., Houpt P. M., Levine Y. K., “Fluorescence lifetime imaging using a confocal laser scanning microscope,” Scanning 14, 155–159 (1992). [Google Scholar]
- 12.Wang X. F., Periasamy A., Herman B., Coleman D. M., “Fluorescence lifetime imaging microscopy (FLIM): instrumentation and applications,” Crit. Rev. Anal. Chem. 23(5), 369–395 (1992). 10.1080/10408349208051651 [DOI] [Google Scholar]
- 13.Dowling K., Hyde S. C. W., Dainty J. C., French P. W. W., Hares J. D., “2-D fluorescence lifetime imaging using a time-gated image intensifier,” Opt. Commun. 135(1-3), 27–31 (1997). 10.1016/S0030-4018(96)00618-9 [DOI] [Google Scholar]
- 14.Talbot C. B., McGinty J., Grant D. M., McGhee E. J., Owen D. M., Zhang W., Bunney T. D., Munro I., Isherwood B., Eagle R., Hargreaves A., Dunsby C., Neil M. A., French P. M. W., “High speed unsupervised fluorescence lifetime imaging confocal multiwell plate reader for high content analysis,” J Biophotonics 1(6), 514–521 (2008). 10.1002/jbio.200810054 [DOI] [PubMed] [Google Scholar]
- 15.Abbott A., “Cell culture: biology’s new dimension,” Nature 424(6951), 870–872 (2003). 10.1038/424870a [DOI] [PubMed] [Google Scholar]
- 16.Keller P. J., Pampaloni F., Stelzer E. H. K., “Life sciences require the third dimension,” Curr. Opin. Cell Biol. 18(1), 117–124 (2006). 10.1016/j.ceb.2005.12.012 [DOI] [PubMed] [Google Scholar]
- 17.Weissleder R., Ntziachristos V., “Shedding light onto live molecular targets,” Nat. Med. 9(1), 123–128 (2003). 10.1038/nm0103-123 [DOI] [PubMed] [Google Scholar]
- 18.Stockholm D., Bartoli M., Sillon G., Bourg N., Davoust J., Richard I., “Imaging calpain protease activity by multiphoton FRET in living mice,” J. Mol. Biol. 346(1), 215–222 (2005). 10.1016/j.jmb.2004.11.039 [DOI] [PubMed] [Google Scholar]
- 19.Skala M. C., Riching K. M., Gendron-Fitzpatrick A., Eickhoff J., Eliceiri K. W., White J. G., Ramanujam N., “In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia,” Proc. Natl. Acad. Sci. U.S.A. 104(49), 19494–19499 (2007). 10.1073/pnas.0708425104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kennedy G. T., Manning H. B., Elson D. S., Neil M. A. A., Stamp G. W., Viellerobe B., Lacombe F., Dunsby C., French P. M. W., “A fluorescence lifetime imaging scanning confocal endomicroscope,” J Biophotonics 3(1-2), 103–107 (2010). 10.1002/jbio.200910065 [DOI] [PubMed] [Google Scholar]
- 21.Kumar S., Alibhai D., Margineanu A., Laine R., Kennedy G., McGinty J., Warren S., Kelly D., Alexandrov Y., Munro I., Talbot C., Stuckey D. W., Kimberly C., Viellerobe B., Lacombe F., Lam E. W.-F., Taylor H., Dallman M. J., Stamp G., Murray E. J., Stuhmeier F., Sardini A., Katan M., Elson D. S., Neil M. A. A., Dunsby C., French P. M. W., “FLIM FRET technology for drug discovery: automated multiwell-plate high-content analysis, multiplexed readouts and application in situ,” ChemPhysChem 12(3), 609–626 (2011). 10.1002/cphc.201000874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huisken J., Swoger J., Del Bene F., Wittbrodt J., Stelzer E. H. K., “Optical sectioning deep inside live embryos by selective plane illumination microscopy,” Science 305(5686), 1007–1009 (2004). 10.1126/science.1100035 [DOI] [PubMed] [Google Scholar]
- 23.Dodt H. U., Leischner U., Schierloh A., Jährling N., Mauch C. P., Deininger K., Deussing J. M., Eder M., Zieglgänsberger W., Becker K., “Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain,” Nat. Methods 4(4), 331–336 (2007). 10.1038/nmeth1036 [DOI] [PubMed] [Google Scholar]
- 24.Sharpe J., Ahlgren U., Perry P., Hill B., Ross A., Hecksher-Sørensen J., Baldock R., Davidson D., “Optical projection tomography as a tool for 3D microscopy and gene expression studies,” Science 296(5567), 541–545 (2002). 10.1126/science.1068206 [DOI] [PubMed] [Google Scholar]
- 25.Dunsby C., “Optically sectioned imaging by oblique plane microscopy,” Opt. Express 16(25), 20306–20316 (2008). 10.1364/OE.16.020306 [DOI] [PubMed] [Google Scholar]
- 26.Sakhalkar H. S., Dewhirst M., Oliver T., Cao Y., Oldham M., “Functional imaging in bulk tissue specimens using optical emission tomography: fluorescence preservation during optical clearing,” Phys. Med. Biol. 52(8), 2035–2054 (2007). 10.1088/0031-9155/52/8/001 [DOI] [PubMed] [Google Scholar]
- 27.Oldham M., Sakhalkar H. S., Oliver T., Allan Johnson G., Dewhirst M., “Optical clearing of unsectioned specimens for three-dimensional imaging via optical transmission and emission tomography,” J. Biomed. Opt. 13(2), 021113 (2008). 10.1117/1.2907968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huisken J., Stainier D. Y. R., “Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM),” Opt. Lett. 32(17), 2608–2610 (2007). 10.1364/OL.32.002608 [DOI] [PubMed] [Google Scholar]
- 29.Vinegoni C., Pitsouli C., Razansky D., Perrimon N., Ntziachristos V., “In vivo imaging of Drosophila melanogaster pupae with mesoscopic fluorescence tomography,” Nat. Methods 5(1), 45–47 (2008). 10.1038/nmeth1149 [DOI] [PubMed] [Google Scholar]
- 30.Birk U. J., Rieckher M., Konstantinides N., Darrell A., Sarasa-Renedo A., Meyer H., Tavernarakis N., Ripoll J., “Correction for specimen movement and rotation errors for in-vivo Optical Projection Tomography,” Biomed. Opt. Express 1(1), 87–96 (2010). 10.1364/BOE.1.000087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beis D., Stainier D. Y. R., “In vivo cell biology: following the zebrafish trend,” Trends Cell Biol. 16(2), 105–112 (2006). 10.1016/j.tcb.2005.12.001 [DOI] [PubMed] [Google Scholar]
- 32.Parng C., Seng W. L., Semino C., McGrath P., “Zebrafish: a preclinical model for drug screening,” Assay Drug Dev. Technol. 1(1), 41–48 (2002). 10.1089/154065802761001293 [DOI] [PubMed] [Google Scholar]
- 33.Hill A. J., Teraoka H., Heideman W., Peterson R. E., “Zebrafish as a model vertebrate for investigating chemical toxicity,” Toxicol. Sci. 86(1), 6–19 (2005). 10.1093/toxsci/kfi110 [DOI] [PubMed] [Google Scholar]
- 34.Barros T. P., Alderton W. K., Reynolds H. M., Roach A. G., Berghmans S., “Zebrafish: an emerging technology for in vivo pharmacological assessment to identify potential safety liabilities in early drug discovery,” Br. J. Pharmacol. 154(7), 1400–1413 (2008). 10.1038/bjp.2008.249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burns C. G., Milan D. J., Grande E. J., Rottbauer W., MacRae C. A., Fishman M. C., “High-throughput assay for small molecules that modulate zebrafish embryonic heart rate,” Nat. Chem. Biol. 1(5), 263–264 (2005). 10.1038/nchembio732 [DOI] [PubMed] [Google Scholar]
- 36.Kari G., Rodeck U., Dicker A. P., “Zebrafish: an emerging model system for human disease and drug discovery,” Clin. Pharmacol. Ther. 82(1), 70–80 (2007). 10.1038/sj.clpt.6100223 [DOI] [PubMed] [Google Scholar]
- 37.Boot M. J., Westerberg C. H., Sanz-Ezquerro J., Cotterell J., Schweitzer R., Torres M., Sharpe J., “In vitro whole-organ imaging: 4D quantification of growing mouse limb buds,” Nat. Methods 5(7), 609–612 (2008). 10.1038/nmeth.1219 [DOI] [PubMed] [Google Scholar]
- 38.McGinty J., Tahir K. B., Laine R., Talbot C. B., Dunsby C., Neil M. A. A., Quintana L., Swoger J., Sharpe J., French P. M. W., “Fluorescence lifetime optical projection tomography,” J Biophotonics 1(5), 390–394 (2008). 10.1002/jbio.200810044 [DOI] [PubMed] [Google Scholar]
- 39.Hall C., Flores M. V., Storm T., Crosier K., Crosier P., “The zebrafish lysozyme C promoter drives myeloid-specific expression in transgenic fish,” BMC Dev. Biol. 7(1), 42 (2007). 10.1186/1471-213X-7-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mathias J. R., Dodd M. E., Walters K. B., Yoo S. K., Ranheim E. A., Huttenlocher A., “Characterization of zebrafish larval inflammatory macrophages,” Dev. Comp. Immunol. 33(11), 1212–1217 (2009). 10.1016/j.dci.2009.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petzold A. M., Bedell V. M., Boczek N. J., Essner J. J., Balciunas D., Clark K. J., Ekker S. C., “SCORE imaging: specimen in a corrected optical rotational enclosure,” Zebrafish 7(2), 149–154 (2010). 10.1089/zeb.2010.0660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dunsby C., Lanigan P. M. P., McGinty J., Elson D. S., Requejo-Isidro J., Munro I., Galletly N., McCann F., Treanor B., Onfelt B., Davis D. M., Neil M. A. A., French P. M. W., “An electronically tunable ultrafast laser source applied to fluorescence imaging and fluorescence lifetime imaging microscopy,” J. Phys. D Appl. Phys. 37(23), 3296–3303 (2004). 10.1088/0022-3727/37/23/011 [DOI] [Google Scholar]
- 43.A. C. Kak and M. Slaney, “Principles of computerized tomographic imaging,” R. E. O’Malley ed. (SIAM, IEEE Press, New York, 1988). [Google Scholar]
- 44.Agulleiro J. I., Fernandez J. J., “Fast tomographic reconstruction on multicore computers,” Bioinformatics 27(4), 582–583 (2011). 10.1093/bioinformatics/btq692 [DOI] [PubMed] [Google Scholar]
- 45.Castaño Díez D., Mueller H., Frangakis A. S., “Implementation and performance evaluation of reconstruction algorithms on graphics processors,” J. Struct. Biol. 157(1), 288–295 (2007). 10.1016/j.jsb.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 46.Ballew R. M., Demas J. N., “An error analysis of the rapid lifetime determination method for the evaluation of single exponential decay,” Anal. Chem. 61(1), 30–33 (1989). 10.1021/ac00176a007 [DOI] [Google Scholar]
- 47.McGinty J., Requejo-Isidro J., Munro I., Talbot C. B., Kellett P. A., Hares J. D., Dunsby C., Neil M A A., French P. M. W., “Signal-to-noise characterization of time-gated intensifiers used for wide-field time-domain FLIM,” J. Phys. D Appl. Phys. 42(13), 135103 (2009). 10.1088/0022-3727/42/13/135103 [DOI] [Google Scholar]
- 48.Dowling K., Dayel M. J., Lever M. J., French P. M. W., Hares J. D., Dymoke-Bradshaw A. K. L., “Fluorescence lifetime imaging with picosecond resolution for biomedical applications,” Opt. Lett. 23(10), 810–812 (1998). 10.1364/OL.23.000810 [DOI] [PubMed] [Google Scholar]
- 49.Esposito A., Gerritsen H. C., Oggier T., Lustenberger F., Wouters F. S., “Innovating lifetime microscopy: a compact and simple tool for life sciences, screening, and diagnostics,” J. Biomed. Opt. 11(3), 034016 (2006). 10.1117/1.2208999 [DOI] [PubMed] [Google Scholar]
- 50.van Geest L. K., Stoop K. W. J., “FLIM on a wide field fluorescence microscope,” Lett. Pept. Sci. 10(5-6), 501–510 (2003). 10.1007/BF02442582 [DOI] [Google Scholar]
- 51.Walls J. R., Sled J. G., Sharpe J., Henkelman R. M., “Resolution improvement in emission optical projection tomography,” Phys. Med. Biol. 52(10), 2775–2790 (2007). 10.1088/0031-9155/52/10/010 [DOI] [PubMed] [Google Scholar]
- 52.Soloviev V. Y., Arridge S. R., “Optical Tomography in weakly scattering media in the presence of highly scattering inclusions,” Biomed. Opt. Express 2(3), 440–451 (2011). 10.1364/BOE.2.000440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pardo-Martin C., Chang T. Y., Koo B. K., Gilleland C. L., Wasserman S. C., Yanik M. F., “High-throughput in vivo vertebrate screening,” Nat. Methods 7(8), 634–636 (2010). 10.1038/nmeth.1481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Owen D. M., Lanigan P. M. P., Dunsby C., Munro I., Grant D., Neil M. A. A., French P. M. W., Magee A. I., “Fluorescence lifetime imaging provides enhanced contrast when imaging the phase-sensitive dye di-4-ANEPPDHQ in model membranes and live cells,” Biophys. J. 90(11), L80–L82 (2006). 10.1529/biophysj.106.084673 [DOI] [PMC free article] [PubMed] [Google Scholar]