

Abstract
Ulcerative colitis (UC) is a chronic, relapsing inflammatory condition of the gastrointestinal tract with a complex genetic and environmental etiology. We performed two distinct UC genome-wide association (GWA) studies, and analyzed these jointly with a previously published scan1, comprising, in aggregate, 2,693 patients with UC and 6,791 controls. A total of 59 SNPs from 14 independent loci attained P < 10−5. Seven of these loci exceeded genome-wide significance (P < 5 × 10−8). After testing an independent cohort of 2009 patients with UC and 1580 controls, 14 loci were significantly associated, including novel UC associations with FCGR2A, 5p15, 2p16, CARD9 and ORMDL3. In our study we confirmed association with 14 previously identified UC susceptibility loci, while an analysis of acknowledged Crohn's disease (CD) loci showed that roughly half of known CD associations are shared with UC. These data implicate approximately 30 loci for UC, providing novel insights into disease pathogenesis.
Epidemiological studies suggest that UC and CD share some, but not all, susceptibility genes, a hypothesis supported by recent GWA studies. Meta-analysis of three CD GWA studies increased the number of susceptibility loci known for CD to over 302. UC is a condition of more modest heritability (λs 10–15) compared to CD (λs 20–35), and perhaps as a result, fewer loci have been identified for UC to date1,3–6. Previous experience with CD implies that the currently identified UC susceptibility loci explain only a fraction of the genetic contribution to disease susceptibility. Considerations of statistical power also suggest that additional loci could be found by enlarging the number of cases and controls used in genome-wide discovery. Here, we combine data from two new UC GWA studies, and perform a meta-analysis with a recently published study1. This brings together a discovery set of 2,693 patients with UC and 6,791 controls, all of European descent (Supplementary Table 1). Independent replication of top results from this meta-analysis was then performed in 2009 cases and 1580 controls, also of European origin.
All primary studies used similar Illumina BeadChips, allowing us to directly examine 266,047 (258,137 autosomal and 7,910 X-chromosomal) SNPs that passed QC in each study. The three studies were analyzed and corrected for population structure separately. P-values from each study were converted to Z-scores summarizing the direction and magnitude of association evidence, and combined (weighted by the relative size of each study) using standard methods7. A Q-Q plot (Supplementary Fig. 1) shows a significant excess of likely true positives in the tail of the distribution, against only modest overall inflation (λGC=1.036).
We sought to replicate 14 independent loci represented by one or more of the 59 SNPs with P < 10−5 in the meta-analysis (Table 1 and Supplementary Table 2). Several previously identified UC loci were among these identified in the meta-analysis, although not all had previously attained genome-wide significance. Loci with prior UC association included IL23R3, the HLA region (including the BTNL2 association)1,3,8, MST19, CARD96, 1q32 (near IL10)5, 1p36 (RNF186/OTUD3/PLA2G2E)1, DLD/LAMB11 (recently confirmed in a British UC GWA study10) 12q15 (neighboring to IFNG/IL26)1, and 21q2211. A second, back-up, SNP was chosen for the six regions with P < 10−6 in the discovery set. Additional SNPs (N=4) from the 1p36, 2p16 and 12q15 regions were chosen, because these SNPs appeared to be independently associated (r2 < 0.2 in CEU HapMap). We also included 10 SNPs with P values between 10−4 and 10−5. Finally, previously published associations with CD and UC were included. These SNPs were typed in 2009 UC cases and 1580 controls of Dutch and Italian descent (complete data in Supplementary Table 1).
Table 1.
Loci with association with UC in GWA STUDY meta-analysis (p > 10−8) with replication cohort results
| GWA STUDIES | REPLICATION | GWA + REPLIC | @ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||
| SNP | Chr | Position | Genes | variant (major/ minor) | CEDARS | SWEDEN | NIDDK | Combined | ITALIAN | DUTCH | Combined | ||
| rs3806308 | 1 | 19612341 | RNF186 OTUD3 PLA2G2E |
G/A | 0.034 | 0.047 | 4.70E-08 | 3.28E-08 | - | - | N/A | N/A | 1 |
| rs1317209 | 1 | 20012623 | RNF186 OTUD3 PLA2G2E |
C/T | 2.43E-04 | 6.26E-03 | 0.034 | 8.60E-07 | 0.046 | 1.56E-04 | 4.41E-05 | 1.60E-10 | |
| rs6426833 | 1 | 20044447 | RNF186 OTUD3 PLA2G2E |
G/A | 1.02E-04 | 2.79E-04 | 6.8E-10 | 2.66E-15 | 4.89E-04 | 3.87E-05 | 7.65E-08 | 1.70E-21 | |
| rs2201841 | 1 | 67466790 | IL23R | T/C | 0.023 | 4.90E-03 | 1.10E-06 | 8.91E-09 | 6.72E-03 | 9.85E-05 | 3.01E-06 | 1.28E-13 | 2 |
| rs11209026 | 1 | 67478546 | IL23R | G/A | 7.28E-04 | 1.47E-03 | 3.10E-05 | 5.91E-10 | 0.012 | 1.32E-03 | 5.18E-05 | 1.89E-13 | |
| rs10800309 | 1 | 159738782 | FCGR2A FCGR2C |
G/A | 2.64E-03 | 0.083 | 7.30E-04 | 2.78E-06 | 2.15E-04 | 0.14 | 2.52E-04 | 2.76E-09 | |
| rs3024505 | 1 | 205006527 | IL10 IL19 | C/T | 3.06E-03 | 0.012 | 9.30E-03 | 3.24E-06 | 2.09E-03 | 0.12 | 1.06E-03 | 1.37E-08 | |
| rs6706689 | 2 | 61024549 | REL CCDC139 PUS10 |
G/A | 0.038 | 1.96E-04 | 0.031 | 4.40E-06 | 1.11E-03 | 0.15 | 8.88E-04 | 1.53E-08 | |
| rs13003464 | 2 | 61040333 | REL CCDC139 PUS10 |
A/G | 0.012 | 2.41E-03 | 9.50E-05 | 4.67E-08 | 0.21 | 0.027 | 0.014 | 7.40E-09 | 2 |
| rs3197999 | 3 | 49696536 | MST1 | C/T | 4.55E-03 | 2.00E-04 | 0.07 | 1.36E-06 | 1.90E-04 | 0.28 | 6.67E-04 | 3.76E-09 | |
| rs4957048 | 5 | 636180 | CEP72 TPPP | C/T | 0.19 | 5.28E-03 | 1.20E-03 | 2.28E-05 | 3.52E-04 | 7.09E-03 | 9.38E-06 | 1.18E-09 | |
| rs2395185 | 6 | 32541145 | C6orf10 BTNL2 |
G/T | 1.36E-07 | 4.42E-12 | 1.40E-06 | 8.75E-23 | - | - | N/A | N/A | 1 |
| rs4598195 | 7 | 107290677 | DLD LAMB1 | A/C | 0.098 | 4.94E-04 | 1.30E-05 | 4.18E-08 | 0.46 | 0.08 | 0.075 | 7.70E-08 | |
| rs4077515 | 9 | 138386317 | CARD9 | A/G | 2.02E-03 | 0.012 | 4.90E-03 | 1.17E-06 | 6.34E-04 | 0.75 | 8.26E-03 | 5.48E-08 | 2 |
| rs11190140 | 10 | 101281583 | NKX2-3 | C/T | 8.84E-04 | 0.028 | 0.058 | 1.85E-05 | 7.61E-06 | 0.37 | 1.45E-04 | 1.07E-08 | |
| rs1558744 | 12 | 66790859 | IFNG IL26 | G/A | 4.24E-03 | 0.028 | 5.50E-10 | 8.14E-11 | 2.38E-04 | 0.57 | 2.70E-03 | 4.18E-12 | |
| rs971545 | 12 | 66877952 | IFNG IL26 | A/G | 2.40E-04 | 0.097 | 3.10E-04 | 2.44E-07 | 0.012 | 0.055 | 1.73E-03 | 2.23E-09 | |
| rs2305480 | 17 | 35315722 | ORMDL3 region | C/T | 3.93E-04 | 8.45E-04 | 0.18 | 2.06E-06 | 0.19 | 4.29E-03 | 3.22E-03 | 3.01E-08 | 2, 4 |
| rs2836878 | 21 | 39387404 | near PSMG1 | G/A | 1.92E-03 | 2.67E-04 | 2.10E-03 | 1.42E-08 | - | - | N/A | N/A | 1 |
@ Notes:
SNP assays failed to design or failed genotyping in Replication Stage
Highly correlated proxies (r2>0.9) were typed in the Replication Stage (see Supplementary Table 2 for further details
Region contains SULT6B1 CEBPZ C2orf56 PRKD3 QPCT
Region contains STAC2 FBXL20 MED1 CRKRS NEUROD2 PPP1R1B STARD3 TCAP PNMT PERLD1 ERBB2 C17orf37 GRB7 IKZF3 ZPBP2 GSDML ORMDL3
Overall 13 loci achieved association with genome-wide statistical significance (P < 5 × 10−8) (Table 1, Supplementary Table 2). A fourteenth locus, containing the CARD9 gene, did not reach genome-wide significance but was significant after Bonferroni correction (Pnominal = 5.48 × 10−8; Pcorrected = 0.014). We achieved genome-wide significance for at least 4 novel loci in populations of European origins, including 1q21 (FCGR2A/FCGR2C)(this locus was recently shown to be associated with UC in a Japanese population12), 2p16 (REL/PUS10), 17q12 (ORMDL3) and 5p15 (rs4957048; approximately 30 kilobases from CEP72) (Table 1). Fourteen loci either suggestively or conclusively associated with UC in prior studies, including TNFSF15, NKX2-3, IL12B, MST1, IL18RAP, HLA, IBD5 locus (5q31), RNF186/OTUD3/PLA2G2E, DLD/LAMB1, IL10, CARD9, 12q15 (IFNG/IL26), JAK2, and IL23R were replicated in our study. Finally, we demonstrated association with 10 additional loci previously associated with CD or IBD, including the IRGM, KIF21B, IKZF1, ICOSLG, CCL2/CCL7, 5p13 (near to PTGER4), 21q21, CUL2/CREM, PSMG1 and STAT3 (Table 2). Replication of these previously identified UC, CD or IBD loci was defined as an association at a level of P < 0.05, with the same risk allele as identified in the index studies.
Table 2.
Association between UC and known CD loci.
| GWA STUDIES | REPLICATION | GWA + REPLICATION | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Gene of interest | SNP from Barrett et al. | Chr# | CD meta p-value (Barrett et al.) | CEDARS | SWEDEN | NIDDK | Combined | ITALIAN | DUTCH | Combined | P-value | Notes |
| IL23R | rs11465804 | 1p31 | 6.38E-34 | 7.28E-04 | 1.47E-03 | 3.10E-05 | 5.91E-10 | 0.012 | 1.32E-03 | 5.18E-05 | 1.89E-13 | 1,2,4,5 |
| ATG16L1 | rs3828309 | 2q37 | 2.57E-21 | 0.24 | 0.18 | 0.25 | 0.57 | 0.28 | 0.63 | 0.67 | 0.48 | 1 |
| MST1 | rs3197999 | 3p21 | 2.17E-07 | 4.55E-03 | 2.00E-04 | 0.07 | 1.36E-06 | 1.90E-04 | 0.28 | 6.67E-04 | 3.76E-09 | 4 |
| PTGER4 | rs4613763 | 5p13 | 5.02E-22 | 0.57 | 0.62 | 0.0049 | 0.11 | 0.048 | 1.58E-03 | 2.81E-04 | 4.21E-04 | |
| IBD5 | rs2188962 | 5q31 | 4.58E-09 | 0.14 | 0.40 | 0.32 | 0.35 | 0.044 | 2.50E-03 | 3.68E-04 | 2.90E-03 | 2 |
| IRGM | rs13361189 | 5q33 | 8.17E-11 | - | - | - | N/A | 0.015 | 4.78E-05 | 4.33E-06 | N/A | |
| TNFSF15 | rs4263839 | 9q32 | 2.61E-07 | 0.024 | 0.42 | 0.56 | 0.035 | 0.2996 | 2.85E-04 | 9.72E-04 | 2.01E-04 | 1,2 |
| ZNF365 | rs10995271 | 10q21 | 1.56E-07 | 0.024 | 0.73 | 0.89 | 0.24 | 0.1852 | 0.96 | 0.34 | 0.127 | 1 |
| NKX2-3 | rs11190140 | 10q24 | 1.71E-10 | 8.84E-04 | 0.028 | 0.058 | 1.85E-05 | 7.61E-06 | 0.37 | 1.45E-04 | 1.07E-08 | |
| NOD2 (R702W) | rs2066844 | 16q12 | N/A | - | - | - | N/A | 0.25 | 0.64 | 0.25 | N/A | |
| NOD2 (G908R) | rs2066845 | 16q12 | N/A | - | - | - | N/A | 0.69 | 0.017 | 0.049 | N/A | |
| NOD2 (insC) | rs2066847 | 16q12 | N/A | - | - | - | N/A | 0.90 | 0.077 | 0.18 | N/A | |
| NOD2 (R459R) | rs2066843 | chr16 | 1.20E-27 | 0.36 | 0.99 | 0.62 | 0.41 | - | - | N/A | N/A | 3 |
| PTPN2 | rs2542151 | 18p11 | 6.54E-11 | 0.14 | 0.57 | 0.75 | 0.17 | 0.98 | 0.17 | 0.32 | 091 | 1 |
| PTPN22 | rs2476601 | 1p13 | 1.81E-05 | 0.11 | 0.32 | 0.096 | 0.19 | 0.25 | 0.15 | 0.067 | 0.88 | |
| ITLN1 | rs2274910 | 1q23 | 3.51E-07 | 0.11 | 0.15 | 0.24 | 0.45 | 0.51 | 0.52 | 0.36 | 0.24 | 2 |
| - | rs9286879 | 1q24 | 4.01E-07 | 0.90 | 0.75 | 0.85 | 0.71 | 0.19 | 0.063 | 0.70 | 0.60 | |
| KIF21B | rs11584383 | 1q32 | 1.94E-06 | 0.16 | 0.024 | 8.50E-02 | 1.88E-03 | 0.020 | 1.08E-04 | 1.17E-05 | 2.22E-07 | 1 |
| IL12B | rs10045431 | 5q33 | 8.81E-09 | - | - | - | N/A | 0.024 | 0.023 | 1.39E-03 | N/A | |
| CDKAL1 | rs6908425 | 6p22 | 2.53E-07 | 0.49 | 0.61 | 0.17 | 0.14 | 0.29 | 0.97 | 0.44 | 0.51 | |
| ATG5 | rs7746082 | 6q21 | 3.13E-04 | 0.069 | 5.68E-03 | 0.042 | 1.34E-04 | 0.23 | 0.88 | 0.46 | 6.08E-04 | 1 |
| CCR6 | rs2301436 | 6q27 | 3.29E-07 | 0.044 | 0.16 | 0.094 | 3.30E-03 | 0.38 | 0.28 | 0.89 | 0.029 | |
| IKZF1 | rs1456893 | 7p12 | 3.16E-05 | 0.93 | 0.66 | 0.53 | 0.960 | 0.15 | 1.14E-03 | 9.05E-04 | 0.040 | 1 |
| - | rs1551398 | 8q24 | 4.90E-06 | 0.57 | 0.32 | 0.73 | 0.27 | 4.24E-04 | 0.057 | 0.25 | 0.90 | 2 |
| JAK2 | rs10758669 | 9p24 | 6.80E-07 | 0.023 | 0.028 | 0.49 | 2.88E-03 | 8.17E-04 | 0.023 | 7.06E-05 | 1.42E-06 | |
| CUL2/CREM | rs17582416 | 10p11 | 2.21E-05 | 0.69 | 0.028 | 0.92 | 0.15 | 0.032 | 0.45 | 0.040 | 0.016 | 1,2 |
| C11orf30 | rs7927894 | 11q13 | 1.43E-07 | 0.021 | 0.79 | 0.062 | 0.010 | 0.20 | 0.69 | 0.23 | 0.22 | 1 |
| LRRK2, MUC19 | rs11175593 | 12q12 | 1.33E-07 | - | - | - | N/A | 0.071 | 0.77 | 0.28 | N/A | |
| - | rs3764147 | 13q14 | 1.44E-05 | 0.27 | 0.010 | 0.041 | 0.34 | 0.59 | 0.76 | 0.55 | 0.27 | 1 |
| ORMDL3 | rs2872507 | 17q21 | 2.13E-06 | 3.93E-04 | 8.45E-04 | 0.18 | 2.06E-06 | 0.19 | 4.29E-03 | 3.22E-03 | 3.01E-08 | 1,4 |
| STAT3 | rs744166 | 17q21 | 5.94E-06 | 0.35 | 0.77 | 0.012 | 0.031 | 0.40 | 0.084 | 0.069 | 4.83E-03 | |
| - | rs1736135 | 21q21 | 3.28E-05 | 0.36 | 0.053 | 5.90E-04 | 2.88E-04 | 0.60 | 8.21E-07 | 1.15E-04 | 1.54E-07 | 1 |
| ICOSLG | rs762421 | chr21 | 1.08E-05 | - | - | - | N/A | 1.70E-03 | 0.59 | 9.32E-03 | N/A | 2 |
| - | rs4807569 | 19p13 | 1.30E-08 | 0.99 | 0.53 | 0.058 | 0.15 | - | - | N/A | 1,3 | |
| GCKR | rs780094 | 2p23 | 7.24E-05 | 0.70 | 0.95 | 0.18 | 0.30 | 0.29 | 0.47 | 0.81 | 0.81 | 2 |
| BTNL2, SLC26A3, HLA-DRB1, HLA-DQA1 | rs3763313 | 6p21 | 1.45E-08 | 0.015 | 0.20 | 0.25 | 5.05E-03 | 0.020 | 0.71 | 0.17 | 0.19 | |
| PUS10 | rs13003464 | 2p16 | 7.65E-06 | 0.012 | 2.41E-03 | 9.50E-05 | 4.67E-08 | 0.21 | 0.027 | 0.014 | 7.40E-09 | 4 |
| CCL2, CCL7 | rs991804 | 17q12 | 4.01E-06 | 0.52 | 0.060 | 0.46 | 0.25 | 0.013 | 0.028 | 9.33E-04 | 2.88E-03 | 2 |
| LYRM4 | rs12529198 | 6p25 | 7.08E-07 | - | - | - | N/A | 0.23 | 0.78 | 0.51 | N/A | |
| SLC22A23 | rs17309827 | 6p25 | 2.08E-06 | - | - | - | N/A | - | - | N/A | N/A | 3 |
| - | rs7758080 | 6q25 | 7.28E-06 | 0.67 | 0.18 | 0.54 | 0.38 | 0.50 | 0.079 | 0.44 | 0.84 | |
| - | rs8098673 | 18q11 | 3.17E-05 | 0.97 | 0.86 | 0.92 | 0.89 | 0.16 | 0.29 | 0.81 | 0.96 | |
| IL18RAP | rs917997 | 2q11 | 2.17E-05 | 0.58 | 0.58 | 0.012 | 0.15 | 0.35 | 0.026 | 0.025 | 0.011 | |
Notes:
= Highly correlated proxies were evaluated in the UC GWA studies (See Supplementary Table 2 for further details)
= Highly correlated proxies were typed in the Replication Stage (See Supplementary Table 2 for further details)
= SNP assays failed to design or failed genotyping in Replication Stage
= also reported in Table 1
= rs11465804 was captured by the highly correlated proxy rs11209026 in both scan and replication (Table q and Supplementary Table 2)
Two recently published studies performed in populations of European descent identified additional IBD associated loci10,13. We examined these loci within our discovery set (using the exact SNP or a perfect proxy (r2 = 1.0)) and were able to demonstrate association between UC and 22q1213 (an IBD locus)(p = 0.049), 16q22/CDH110 (a UC locus)(p = 0.0061), as well as with the DLD/LAMB110 locus discussed earlier. Furthermore we are able to provide further evidence of association between the 1p36 locus (rs7524102, p = 0.0015) which demonstrated strong (but not genome-wide significant) association in the WTCCC study10. We were not able to replicate the UC associations seen at 13q1310 (p = 0.15), 20q13/HNF4A10 and 2q37/GPR35 (SNPs not in our dataset and no proxy SNPs available) or the IBD associations seen at 16p11 (p = 0.17), 10q22/ZMIZ1 (p = 0.22), and 19q13 (SNP not in our dataset and no proxy SNP available). Therefore, adding these additional associations we provide evidence for at least 30 distinct risk factors for UC.
In three regions (1p36 [RNF186/OTUD3/PLA2G2E], 2p16 [PUS10/REL] and 12q15 [IFNG/IL26]), our scan identified multiple SNPs with P < 10−5 that showed low LD with each other (r2 < 0.2) in HapMap CEU samples. To determine if multiple independent alleles were contributing at these loci, we performed conditional analyses in all scan and replication samples (Table 3). While the signals for each SNP conditional on the other are greatly diminished in the 2p16 region (suggesting these may both be in LD with a single, yet to be discovered, causal variant), we have extended prior evidence for multiple independent associations at 1p36 and 12q151. Particularly striking are three SNPs at 1p36, each of which is genome-wide significant even while conditional on the other two.
Table 3.
Conditional analyses for three regions with multiple associated SNPs in linkage equilibrium.
| SNP | Location | Conditional on | CEDARS | NIDDK | SWEDEN | ITALIAN | DUTCH | Combined-P |
|---|---|---|---|---|---|---|---|---|
| rs1317209 | 1p36 | rs3806308 | 7.95E-05 | 0.043 | 0.00445 | 2.75E-06 | ||
| rs6426833 | 0.0002674 | 0.044 | 0.01 | 0.04976 | 0.0001907 | 3.10E-08 | ||
| rs3806308 | 1p36 | rs1317209 | 0.02201 | 3.40E-08 | 0.06 | 1.39E-08 | ||
| rs6426833 | 0.0348 | 2.50E-07 | 0.13 | 2.64E-07 | ||||
| rs6426833 | 1p36 | rs3806308 | 6.37E-05 | 3.10E-09 | 0.00035 | 5.05E-14 | ||
| rs1317209 | 0.0001061 | 5.70E-10 | 0.00022 | 0.00055 | 7.48E-05 | <1e-16 | ||
| rs6706689 | 2p16 | rs13003464 | 0.3137 | 8.30E-01 | 0.01663 | 0.00256 | 0.7145 | 0.0091 |
| rs13003464 | 2p16 | rs6706689 | 0.08581 | 7.00E-04 | 0.12 | 0.8752 | 0.0681 | 0.0011 |
| rs1558744 | 12q15 | rs971545 | 0.04439 | 5.00E-08 | 0.2 | 0.0018 | 0.9499 | 2.20E-08 |
| rs971545 | 12q15 | rs1558744 | 0.002071 | 4.60E-02 | 0.2 | 0.09206 | 0.06118 | 0.000386 |
We also performed interaction analysis between all pairs of SNPs listed in table 1. Among the 496 pairs of SNPs examined (Supplementary Table 3) one pair (an interaction between genome-wide significant hits at CARD9 and REL/PUS10) was significant after correction for the number of tests performed and a second interaction at this locus approached significance after replication. Few such interactions have been documented in complex disease14 and further replication is warranted before this is considered confirmed but the known functional interaction between CARD9 and REL should not be overlooked.
It is clear from these (Table 2) and other data that UC and CD share some mechanistic pathways and susceptibility genes but that some are particular to each condition. We sought to estimate the proportion of alleles with an influence on both CD and UC by calculating the likelihood of the observed UC genotype data at each CD locus, under the alternate hypotheses that the UC sample has the same allele frequency as either a CD sample or of a control sample. The maximum data set likelihood was achieved when 15 or 16 of the CD loci – essentially half of the 31 - were presumed to affect UC risk. This is consistent with the summary results in table 2, where 14 of the 31 confirmed previous findings are significant at p<.01, three have 0.01<p<.05, while the remainder fit the null distribution. The lack of complete overlap is unlikely to result entirely from limited power because our likelihood analysis limits the possibility that more than 20 of the Crohn's loci are shared, and some of the strongest CD variants (e.g., NOD2, ATG16L1) are among the loci not associated with UC, despite having increased power to detect these variants of larger effect size.
All of the genes implicated by our work could plausibly play a functional role in UC (see Supplementary Table 4 for genes located nearby to replicated UC-risk SNPs not shared with CD). A key next step in translating genetic loci to function requires that we understand gene function in the context of cell and tissue type relevant to human disease. Expression studies may help identify relevant cell types for functional studies, the nature of which are likely to differ for genes expressed in epithelial cells, macrophages or lymphocytes. We focused on five UC susceptibility loci containing genes whose expression and function are not well established. We performed quantitative real-time PCR in human intestinal and immune cDNA panels (Supplementary Fig. 2a). The RNF186/PLA2G2E/OTUD3 genes are located at 1p36 which harbors distinct UC-risk SNPs (rs1317209, rs6426833). While RNF186 and OTUD3 are proteins with unknown function, both contain protein domains that have been associated with protein ubiquitination. Ubiquitin modifications are known to regulate immune responses, since the OTU protein TNFAIP3 was identified as an important negative regulator of NFkB15. Interestingly, the TNFAIP3 locus, which has been associated with multiple autoimmune conditions16,17, is in the vicinity of one the UC-risk regions (rs2327832) although this locus achieved association only at a suggestive level of genome-wide significance (P = 3.92 × 10−05). The expression of RNF186 is higher in intestinal tissues than immune tissues. Immuno-staining indicated that RNF185 is expressed at the basal pole of epithelial cells and lamina propria within colonic tissues (Supplementary Fig. 2a and 2b). In contrast, OTUD3 transcripts had higher levels in immune tissues (spleen, lymph nodes and PBMCs) and in lymphocytes compared to CD14 positive cells (not shown). Phospholipase A2 group IIE (PLA2G2E) is a secretory PLA2 involved in the production of various types of proinflammatory lipid mediators18. PLA2G2E was undetectable in most tissues but showed weak expression in bone marrow, lymph node and thymus suggesting a role in immunity. Surprisingly, in the intestinal panel, PLA2G2E was detected very specifically in the small intestine, but not colon. RNF186, OTUD3 and PLA2G2E showed very different expression patterns suggesting that investigations of the biological functions of these candidate genes will require disparate strategies.
CEP72/TPPP genes are located close to the UC-risk SNP rs4957048. CEP72 and TPPP are both involved in microtubule organization and are expressed ubiquitously (Supplementary Fig. 2a and 2b). LAMB1 and DLD are located near the UC risk SNPs rs4598195 and rs2237686 at 7q31 and were both expressed ubiquitously (Supplementary Fig. 2a). LAMB1 is an extracellular matrix glycoprotein constituent of basement membranes. These data together with recently reported associations between UC and CHD1 and CHD310 further implicate defects in barrier integrity in the development of colonic inflammation. Importantly future studies will need to integrate disease associations and the consequences of risk allele specific expression to uncover functional role for genes in diseases such as UC.
GSDMB and ORMDL3 genes are located nearby to the UC risk SNPs rs2305480 and rs8067378. Both ORMDL3 and GSDMB showed higher expression in immune tissues in comparison to intestinal tissues (Supplementary Fig. 2a). We selected the ORMDL3 region for functional analysis not only because of the novel association with UC presented herein but also because ORMDL3 has been implicated in many diseases involving dysregulated immune responses, although the underlying mechanisms of this association remain unclear2,19–21. ORMDL3 is suggested to be involved in protein folding and growing evidence demonstrate interactions between the Unfolded Protein Response (UPR) and immune responses22,23. The ORMDL3-GFP protein is localized in the ER (Fig. 1a), confirming previous published data24. We next investigated whether ORMDL3 expression is involved in the UPR in epithelial cells. Over-expression of ORMDL3 decreased both the basal and ER-stress induced UPR (Fig. 1b). Knockdown of ORMDL3 expression induced a higher UPR following tunicamycin or thapsigargin stimulation (Fig. 1c), indicating that ORMDL3 expression levels can regulate UPR and that ORMDL3 maybe an important factor to ensure ER homeostasis.
Figure 1.
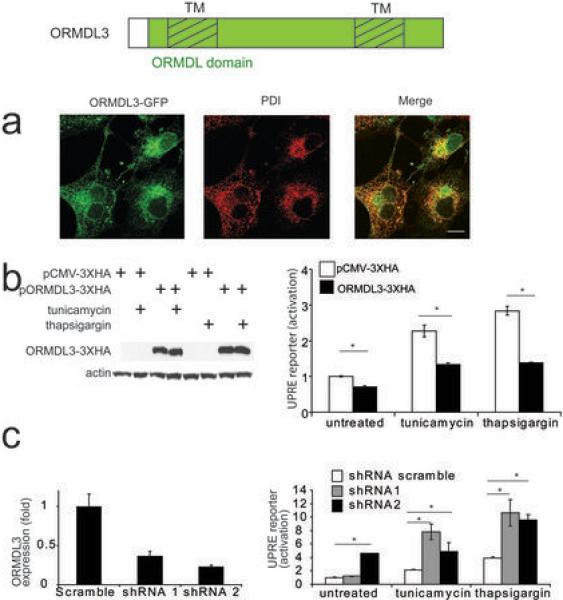
(a) Cos-7 cells were transfected with pCMV-ORMDL3-GFP. After 24h, cells were fixed using 4% paraformaldehyde and subjected to immunostaining using anti-PDI antibodies as an ER marker. Fluorescence signals were acquired with a confocal microscope. Displayed images represent a single confocal section.
(b) HEK293T cells were co-transfected with a firefly luciferase reporter p5xUPRE-GL3, renilla luciferase as transfection control and pCMV-3XHA or pCMV-ORMDL3-3XHA. After 24h, cells were left untreated or treated using 5μg/ml of tunicamycin or 10μM of thapsigargin for 6h. ORMDL3-3XHA was detected using an anti-HA antibody. Luciferase activities were measured to detect UPRE transcription activation.
(c). HEK293T cells were co-transfected with plasmids encoding shRNA scramble or directed against ORMDL3, firefly luciferase reporter p5xUPRE-GL3 and renilla luciferase as transfection control. After 48h, cells were left untreated or treated using 5μg/ml of tunicamycin or 10μM of thasigargin. ORMDL3 expression was determined by quantitative RT-PCR (left). Luciferase activities were measured to detect UPRE transcription activation (right)
The data presented herein significantly increase our understanding of the pathogenesis of UC and its relationship to CD, as well as providing novel expression and functional data for some of the implicated genes. The genetic associations described in this report taken together with recently published papers10,12,13 highlight the importance of barrier function, cell specific innate responses (ER stress, microbe elicited responses ROS production, NF-kB activation), gene sets that coordinately regulate key functional programs in adaptive immunity and resolution of inflammation in UC pathogenesis. Taken together we have explained less than 10% of the variance of UC and the challenge now is to both identify additional genetic factors and to translate these advances into real benefits for patients.
Methods
Two unpublished UC GWAS were combined with the published NIDDK UC GWAS1 (Supplementary Table 1).
Cedars-Sinai (CS) UC GWAS
The CS UC scan consisted of 852 cases and 3271 controls. Samples with more than 1% missing genotype data (1 case, 278 controls) were removed. Standard searches for relatedness (using PLINK –genome) and population structure (using PLINK –mds)25 were performed. Identity-by descent of >20% (catching half-sibs and above) was identified in 81 pairs (primarily controls) and one member from each pair was removed. The first ten principal components: PC1 represented the expected N. Europe – S. Europe – Ashkenazi axis were detected. The second PC identified outliers that were removed as they were not readily matched in a case-control sense. For the final analysis of 723 cases and 2880 controls a logistic regression analysis correcting for MDS covariates 1 and 3–10 was performed. This QC process reduced the λGC from ~2.5 to 1.074. This residual inflation was genomic-control corrected before inclusion in the meta-analysis.
Swedish UC GWAS
Cases were enrolled at three sites in Sweden. All genotyping was performed on the Illumina Hap550 array and Illumina Quad-610 at the Genome Institute of Singapore. Genotype data from 640 previously QC'd controls, all free of inflammatory diseases, was combined with common controls from the same Epidemiological Investigation of Rheumatoid Arthritis genotyped on the Illumina Hap550 (N=460) and Quad-610 (N=378) BeadChips (total of 1,478 controls). Alleles were called in the Illumina BeadStudio software by reclustering with cases and controls included for each chip type. Sample call-rates exceeded 97.29%, and none were removed for this reason. Four samples with low heterozygosity were removed from further analysis. Fifteen samples (10 cases and 5 controls) were excluded on the basis of sex discrepancies between database records and X chromosome zygosity. 112 (106 cases and 6 controls) sample duplicates and first-degree relatives were identified by excess allele sharing as calculated in PLINK, and subsequently excluded. 138 population outliers (82 cases and 56 controls) were identified by a principal components analysis implemented in Eigenstrat.
SNPs with call-rates <95%; minor allele frequency < 0.005, and a Hardy-Weinberg equilibrium P < 10−7 in controls together with non-autosomal SNPs were removed. Finally, the common set of directly genotyped SNPs across all three chip types were used, resulting in a post-QC dataset of 948 cases and 1,408 controls for 297,031 SNPs. Trend tests of association were calculated in PLINK. λGC for the entire dataset was 1.04, uncorrected. Eigenstrat correction based on the top ten PC's (bringing λGC to 1.03), followed by genomic control correction was used to remove the small residual stratification.
Meta-analysis
As all studies utilized compatible Illumina platforms, we combined 266,047 (258,137 autosomal and 7,910 X chromosome) SNPs passing QC in all three studies. P-values from the population structure corrected analyses were converted to Z-scores, consistently oriented to the combined minor allele. These consistently oriented Z-scores were then combined, and the square of those scores evaluated as a chi-square. The Q-Q plot is shown in Supplementary Figure 2 – while overall there is only very modest inflation (λGC=1.036), there is a significant excess of true positive results in the tail of the distribution. There were 59, 126 and 511 SNPs from the 266,047 that exceed .00001, .0001 and .001 respectively (~20, 5 and 2-fold in excess of null expectation).
Replication cohorts
Two independent case-control cohorts were examined for the replication phase. An Italian study population composed of 1094 UC patients and 908 controls collected at the S. Giovanni Rotondo “CSS” (SGRC) Hospital in Italy and secondly a Dutch study population composed of 1090 UC patients of Caucasian ethnicity recruited through the Inflammatory Bowel Disease unit of the University Medical Center Groningen, Groningen; the Academic Medical Center, Amsterdam; the Leiden University Medical Center, Leiden and the Radboud University Medical Center, Nijmegen. Healthy controls (N=804) of self-declared European ancestry were drawn from volunteers at the University Medical Center, Utrecht. Patients were diagnosed according to accepted clinical, endoscopic, radiological and histological findings. All patients and controls gave informed consent and the study was approved by the ethics review committees of each participating hospital. All DNA samples and data in this study were handled anonymously.
Replication Genotyping and quality control
Selected SNPs were designed into multiplex assays, and genotyped using primer extension chemistry and mass spectrometric analysis (iPlex assay, Sequenom, San Diego, California, USA) on the Sequenom MassArray at the Laboratory for Genetics and Genomic Medicine of Inflammation (www.inflammgen.org) of the Universite de Montreal. In cases where SNPs did not design into multiplex assays or failed the quality control thresholds, a proxy was selected. Samples showing >10% missing data, as well as SNPs with >10% missing data or significantly out of Hardy-Weinberg equilibrium (P < 0.001), were excluded from the analyses. The overall genotyping call rate in the replication dataset following QC was >99%, and consisted of 993 UC cases and 826 controls (Italian), and 1016 UC cases and 754 controls (Dutch).
Association analysis of the replication phase
Association testing of single SNPs in the replication cohorts was performed by standard χ2 test carried out on a 2 × 2 contingency table (PLINK). Combination of the results from the two replication cohorts, as well as combination of the results from the screen and replication, was achieved by the calculation of a combined weighted z -score. The threshold for significant independent replication was set at P-values of < 0.05 in the combined Italian and Dutch datasets.
Antibodies
Experiments were performed using the following antibodies: anti-RNF186 (Abnova), anti-CEP72 (Novus Biological NB100-60661), anti-Troma-1 (developmental Studies Hybridoma Bank, Iowa City) and anti-PDI (Stressgen).
Reverse transcription and real time polymerase chain reaction
For expression maps, immune and intestinal cDNA panels were obtained from Clontech (two independent panels of cDNA). For ORMDL3 functional studies, RNA extraction was performed using RNeasy kit (Qiagen) according to the manufacturer's instructions. One μg of total RNA was reverse-transcribed using an iScriptTM cDNA synthesis kit (Bio-Rad). The displayed gene expression is representative of 3 independent experiments. Real time quantitative PCR was performed in duplicate in a Bio-Rad iCycler thermal cycler equipped of an iQ5 optical module using the iQTMSYBR®Green super mix (Bio-Rad). Briefly, 100 ng of reverse transcribed cDNA were used for each PCR with 250 nM forward and reverse primers. The thermal cycling conditions were 4 min at 95°C, followed by 40 cycles at 94°C for 15 s, 59°C for 1 minute. Values were normalized to GAPDH and the condition containing the lowest mRNA content was defined as 1 arbitrary unit. All the PCR products were analyzed with a 2% agarose gel to check that the size of the amplicons were as predicted.
Patient intestinal biopsies
Following informed consent and the approval of the Human Research Committee of the Massachusetts General Hospital, colonic biopsies from non-inflamed regions were obtained from IBD patients undergoing colonoscopy. Tissues were embedded and frozen in OCT compound (Fisher).
Immunostaining and fluorescence microscopy
Cells or frozen tissue sections were fixed using 4% paraformaldehyde and permeabilized PBS-Triton X-100 0.1%. After washing with PBS, the sections were incubated 30 minutes in PBS containing 3M glycine to block the reactive groups of paraformaldehyde. The sections were incubated (1 hour) with a blocking solution containing 10% of donkey serum (Rockland Immunochemicals) and 10% of Human Fc block reagent (Miltenyi Biotec). The preparations were then incubated with the primary antibodies (1 hour), washed using PBS, incubated with fluorescent secondary antibodies (Jackson Immunoresearch)(1 hour), washed using PBS and incubated with PBS containing 100μg/ml of DABCO (Sigma) as antifading reagent before mounting in Glycergel medium (Dako). Fluorescence signals were captured using a laser confocal microscope (model Radiance 2000 Bio-Rad). Image acquisition was performed with LaserSharpScanning software (Bio-Rad).
Plasmids
ORMDL3 constructs for mammalian expression: a clone of ORMDL3 in pCMV-SPORT6 was obtained from Open Biosystems. ORMDL3 coding sequence was amplified by PCR using forward and reverse primers (supplementary table 7) that contain the enzyme restriction sites EcoRI and NotI, respectively. Using EcoRI and NotI digestion, the ORMDL3 coding sequence was sub-cloned into C-terminal tagged pCMV-3xHA vector derived from the Clontech pCMV-Myc vector (Cat. # 631604) and into a pcDNA4/TO-GFP-C vector derived from Invitrogen's pcDNATM4/TO vector by inserting EGFP sequence from XhoI and ApaI sites in pcDNATM4/TO vector.
ShRNA constructs. ShRNA hairpins directed against the human transcript of ORMDL3 were designed using the tools of the RNAi consortium from the Broad Institute (http://www.broadinstitute.org/rnai/trc/lib). The pairs of oligonucleotides purchased to construct the hairpins are listed in supplementary table 7.
After annealing and according to the RNAi consortium instructions, the paired oligonculeotides were inserted using AgeI EcoRI enzyme restriction sites in TRC22 vector derived from pLKO1.
ER-Stress Reporter
HEK293T cells were plated on 24-well plates at a density of 2 × 105 cells per well. After 24 hours, cells of each well were transfected with 2 ng of the ER-stress firefly luciferase reporters p5xUPRE-GL326 and 0.025ng of renilla luciferase (Promega) using TransFectin Lipid Reagent (Bio Rad). Luciferase activities were measured using the Dual-Luciferase Reporter Assay System (Promega) and were normalized to the internal transfection control of renilla luciferase activity.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported in part by NCRR grant M01-RR00425 to the Cedars-Sinai General Research Center Genotyping core; NIH/NIDDK grant P01-DK046763; the Diabetes Endocrinology Research Center grant, DK 063491; Cedars-Sinai Medical Center Inflammatory Bowel Disease Research Funds; The Feintech Family Chair in IBD (S.R.T.); The Abe and Claire Levine Chair in Pediatric IBD (M.D.) and The Cedars-Sinai Board of Governors' Chair in Medical Genetics (J.I.R.). Additional funding through grants DK76984 (MD) and DK084554 MD and DPBM) CHS research reported in this article was supported by contract numbers N01-HC-85079 through N01-HC-85086, N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133; grant numbers U01 HL080295 and R01 HL087652 from the NHLBI, with additional contribution from the National Institute of Neurological Disorders and Stroke. A full list of principal CHS investigators and institutions can be found at http://www.chs-nhlbi.org/pi.htm. AG is supported by the Crohn's and colitis foundation of America. RJX and MJD are supported by the following grants DK83756, DK086502, and DK043351.
The NIDDK IBD Genetics Consortium is funded by the following grants: DK062431 (S.R.B.), DK062422 (J.H.C.), DK062420 (R.H.D.), DK062432 (J.D.R.), DK062423 (M.S.S.), DK062413 (D.P.B.M.) and DK062429 (J.H.C.). JHC is also funded by Bohmfalk Funds for Medical Research, Burroughs Wellcome Medical Foundation, and the Crohn's and Colitis Foundation of America. JDR is also funded by grants from the National Institutes of Allergy and Infectious Diseases (AI065687; AI067152) and from the National Institute of Diabetes and Digestive and Kidney Diseases (DK064869).
Activities in Sweden were supported by the Swedish Society of Medicine; the Bengt Ihre Foundation; the Karolinska Institutet; the Swedish National Program for IBD genetics and SOIBD; the Swedish Medical Research Council; the Soderbergh foundation; and the Swedish Cancer foundation. Support for genotyping and genetic data analysis was provided by the National Cancer Centre, Singapore General Hospital; and the Singapore Millenium Foundation (to S.P.); and the Agency for Science Technology and Research (A*STAR), Singapore to (M.L.H. and M.S.). Genotyping and DNA handling at the Genome Institute of Singapore was performed by Wee Yang Meah, Khai Koon Heng, Hong Boon Toh, Xiaoyin Lin, Sigeeta Rajaram, Dennis Tan and Chang Hua Wong. We are grateful to the funders and investigators of the Epidemiological Investigation of Rheumatoid Arthritis, for the provision of genotype data from healthy Swedish controls.
Footnotes
Author contributions: DPBM, MJD, RJX, JDR, JhC, PG, RhD and MS participated in the study design and conception. DPBM, AG, MJD, RJX, JDR and MS wrote the manuscript with contributions from RHD and JIR. DPBM, LT, KT, CL, CB, PRF, MC, MD'A, JH, MLH, ML, LP, AA, EC, AL, OP, E-J B, CD, DWH, DJdeJ, PCS, RKW, YS, MS, JHC, SRB, LPS, RHD, MCD, NLG, TH, AI, GYM, DSS, EV, SRT, VA, CW and SP performed patient diagnosis, enrollment, and collection of clinical data. Replication genotyping was performed by CL and CB in the laboratory of JDR. Expression analysis, immunohistochemistry and shRNA studies were designed by AG and RJX and performed by CL and AG. MJD, JE, BN, KR, JW, JDR, PG and RTHO provided statistical analyses. All authors contributed to the final paper.
The authors report no competing financial interests.
Note: Supplementary information is available on the Nature Genetics website.
References
- 1.Silverberg MS, et al. Ulcerative colitis-risk loci on chromosomes 1p36 and 12q15 found by genome-wide association study. Nat Genet. 2009;41:216–20. doi: 10.1038/ng.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barrett JC, et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn's disease. Nat Genet. 2008;40:955–62. doi: 10.1038/NG.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fisher SA, et al. Genetic determinants of ulcerative colitis include the ECM1 locus and five loci implicated in Crohn's disease. Nat Genet. 2008;40:710–2. doi: 10.1038/ng.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Franke A, et al. Replication of signals from recent studies of Crohn's disease identifies previously unknown disease loci for ulcerative colitis. Nat Genet. 2008;40:713–5. doi: 10.1038/ng.148. [DOI] [PubMed] [Google Scholar]
- 5.Franke A, et al. Sequence variants in IL10, ARPC2 and multiple other loci contribute to ulcerative colitis susceptibility. Nat Genet. 2008;40:1319–23. doi: 10.1038/ng.221. [DOI] [PubMed] [Google Scholar]
- 6.Zhernakova A, et al. Genetic analysis of innate immunity in Crohn's disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am J Hum Genet. 2008;82:1202–10. doi: 10.1016/j.ajhg.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bakker PI, et al. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–8. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Franke A, et al. Systematic association mapping identifies NELL1 as a novel IBD disease gene. PLoS ONE. 2007;2:e691. doi: 10.1371/journal.pone.0000691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Goyette P, et al. Gene-centric association mapping of chromosome 3p implicates MST1 in IBD pathogenesis. Mucosal Immunol. 2008;1:131–8. doi: 10.1038/mi.2007.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrett JC, et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009 doi: 10.1038/ng.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kugathasan S, et al. Loci on 20q13 and 21q22 are associated with pediatric-onset inflammatory bowel disease. Nat Genet. 2008;40:1211–5. doi: 10.1038/ng.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asano K, et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat Genet. 2009 doi: 10.1038/ng.482. [DOI] [PubMed] [Google Scholar]
- 13.Imielinski M, et al. Common variants at five new loci associated with early-onset inflammatory bowel disease. Nat Genet. 2009 doi: 10.1038/ng.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McGovern DP, et al. Genetic epistasis of IL23/IL17 pathway genes in Crohn's disease. Inflamm Bowel Dis. 2009 doi: 10.1002/ibd.20855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun SC. Deubiquitylation and regulation of the immune response. Nat Rev Immunol. 2008;8:501–11. doi: 10.1038/nri2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graham RR, et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–61. doi: 10.1038/ng.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trynka G, et al. Coeliac disease-associated risk variants in TNFAIP3 and REL implicate altered NF-kappaB signalling. Gut. 2009;58:1078–83. doi: 10.1136/gut.2008.169052. [DOI] [PubMed] [Google Scholar]
- 18.Burke JE, Dennis EA. Phospholipase A2 structure/function, mechanism, and signaling. J Lipid Res. 2009;50(Suppl):S237–42. doi: 10.1194/jlr.R800033-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hirschfield GM, et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N Engl J Med. 2009;360:2544–55. doi: 10.1056/NEJMoa0810440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Verlaan DJ, et al. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet. 2009;85:377–93. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moffatt MF, et al. Genetic variants regulating ORMDL3 expression contribute to the risk of childhood asthma. Nature. 2007;448:470–3. doi: 10.1038/nature06014. [DOI] [PubMed] [Google Scholar]
- 22.Kitamura M. Biphasic, Bidirectional Regulation of NF-ĸB by Endoplasmic Reticulum Stress. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2008.2391. [DOI] [PubMed] [Google Scholar]
- 23.Todd DJ, Lee AH, Glimcher LH. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol. 2008;8:663–74. doi: 10.1038/nri2359. [DOI] [PubMed] [Google Scholar]
- 24.Hjelmqvist L, et al. ORMDL proteins are a conserved new family of endoplasmic reticulum membrane proteins. Genome Biol. 2002;3:RESEARCH0027. doi: 10.1186/gb-2002-3-6-research0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y, et al. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J Biol Chem. 2000;275:27013–20. doi: 10.1074/jbc.M003322200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.