

The X-ray crystal structure of human dual-specificity phosphatase 27 (DUSP27) is reported at 2.38 Å resolution.
Keywords: dual-specificity phosphatases, DUSP27, protein tyrosine phosphatases
Abstract
There are over 100 genes in the human genome that encode protein tyrosine phosphatases (PTPs) and approximately 60 of these are classified as dual-specificity phosphatases (DUSPs). Although many dual-specificity phosphatases are still not well characterized, novel functions have been discovered for some of them that have led to new insights into a variety of biological processes and the molecular basis for certain diseases. Indeed, as the functions of DUSPs continue to be elucidated, a growing number of them are emerging as potential therapeutic targets for diseases such as cancer, diabetes and inflammatory disorders. Here, the overexpression, purification and structure determination of DUSP27 at 2.38 Å resolution are presented.
1. Introduction
A wide variety of signaling pathways and other physiological processes are mediated by reversible phosphorylation of proteins by numerous protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs) (Hunter, 1995 ▶; Pawson, 1995 ▶). For example, reversible phosphorylation plays an important role in cell development, differentiation and transformation in higher eukaryotes (Fauman & Saper, 1996 ▶). The precise coordination and tight control of the activities of kinases and phosphatases is pivotal in many signal transduction pathways; disruption of the balance between opposing kinase and phosphatase activities has been linked to many diseases (Levitzki, 2004 ▶; Cohen, 2002a ▶; Tonks, 2006 ▶). PTKs have been studied more extensively than PTPs, in part because the first PTKs were cloned and purified much earlier than the first PTPs (Charbonneau et al., 1989 ▶; Guan et al., 1990 ▶). Consequently, the development of kinase inhibitors into successful drugs has progressed more rapidly than has the development of phosphatase inhibitors (Cohen, 2002b ▶; Hooft van Huijsduijnen et al., 2002 ▶; Zhang et al., 2009 ▶).
The human genome encodes more than 100 PTPs and many of them remain poorly characterized (Alonso et al., 2004 ▶). In fact, the physiological substrates of many PTPs have yet to be identified (Tiganis & Bennett, 2007 ▶; Arnoldussen & Saatcioglu, 2009 ▶). As a result, novel and unexpected functions for PTPs are continuing to be uncovered (Tonks, 2006 ▶; Clark et al., 2008 ▶; Patterson et al., 2009 ▶; Shang et al., 2010 ▶). It seems likely that a more thorough understanding of how reversible phosphorylation is regulated and mediated will lead to the development of novel diagnostic and therapeutic strategies (Yi & Lindner, 2008 ▶; Pulido & Hooft van Huijsduijnen, 2008 ▶; Hooft van Huijsduijnen et al., 2002 ▶). The determination of three-dimensional structures of PTPs can contribute to this goal by providing structural insights into function and mechanism and may possibly also aid in the structure-based design of inhibitors (Almo et al., 2007 ▶; Hoffman et al., 2004 ▶; Zhang, 2002 ▶; Zhang et al., 2002 ▶).
PTPs can be divided into four subfamilies based on the amino-acid sequences of their catalytic domains (Alonso et al., 2004 ▶; Bhaduri & Sowdhamini, 2003 ▶). The largest family are the type I cysteine-based PTPs, which can be further subdivided into two distinct groups based on substrate specificity (Andersen et al., 2004 ▶): the classical tyrosine-specific PTPs and the DUSPs, which are capable of removing phosphates from tyrosine and serine/threonine residues. Additional classes of PTPs include the type II variety, which are low-molecular-weight tyrosine-specific cysteine-based enzymes, the rhodanese-related type III cysteine-based phosphatases with dual specificity for tyrosine and threonine, and the type IV aspartic acid-based PTPs.
DUSPs share a similar catalytic mechanism with the classical PTPs (Denu & Dixon, 1995 ▶). The highly conserved catalytic domains of DUSPs and PTPs contain the consensus sequence HCXXGXXR and the catalytic cysteine is located at the base of the catalytic pocket (Denu & Dixon, 1998 ▶). The hydrolysis of the phosphorylated substrate occurs through the formation of a stable phosphoryl intermediate and a conserved aspartic acid acts as a general acid/base which helps to stabilize the phosphoryl intermediate (Fauman & Saper, 1996 ▶; Camps et al., 2000 ▶; Denu et al., 1995 ▶). The shape, size and depth of the catalytic pocket constitute the key structural determinants of substrate specificity (Denu et al., 1996 ▶). Classical PTPs have deep catalytic pockets that are typically unable to accommodate the shorter phosphothreonine/phosphoserine residue and position it properly for efficient catalysis to occur (Zhang, 2002 ▶; Schumacher et al., 2002 ▶). The catalytic pockets of DUSPs, on the other hand, are typically more shallow and broad than those of the classical PTPs, enabling them to accommodate phosphorylated tyrosine, threonine or serine residues.
Initial characterization of human DUSP27 was performed by Friedberg et al. (2007 ▶). Originally, the computationally predicted open reading frame, designated DUPD1, appeared to encode 446 amino acids that included two catalytic domains: a proline isomerase and a DUSP. However, it was subsequently shown that the true open reading frame only encompassed the DUSP domain and the gene product was therefore renamed DUSP27. Recombinant DUSP27 was shown to have catalytic activity in vitro as demonstrated by the dephosphorylation of para-nitrophenol phosphate and 6,8-difluoro-4-methylumbelliferyl phosphate. Furthermore, DUSP27 exhibited a marked preference for the hydrolysis of phosphotyrosine over phosphothreonine and phosphoserine. It was demonstrated that DUSP27 is a cytosolic enzyme that is highly expressed in skeletal muscle, adipose tissue and liver and the central tissues for energy metabolism. While additional studies of DUSP27 are scarce in the literature, a recent report identified DUSP27 as an enzyme that may contribute to the propensity of individuals to develop heroin addiction (Nielsen et al., 2010 ▶). In this report, we contribute to the characterization of DUSP27 by solving its X-ray crystal structure at 2.38 Å resolution.
2. Materials and methods
2.1. Cloning, expression and purification
The open reading frame encoding human DUSP27 (Thr2–Leu220) was amplified from cDNA (IMAGE clone ID 9021318, American Type Culture Collection, Manassas, Virginia, USA) by the polymerase chain reaction (PCR) using the following oligodeoxyribonucleotide primers: 5′-GAG AAC CTG TAC TTC CAG ACC TCT GGT GAA GTG AAG ACA AGC CTC AAG AAT GC-3′ and 5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT ACA GCT CCC TGC CAT CCT CCT CCT C-3′ (primer R). The resulting PCR amplicon was subsequently used as the template for a second PCR with the following primers: 5′-GGG GAC AAG TTT GTA CAA AAA AGC AGG CTC GGA GAA CCT GTA CTT CCA G-3′ and primer R. The amplicon from the second PCR was inserted by recombinational cloning into the entry vector pDONR221 (Invitrogen, Carlsbad, California, USA) and the nucleotide sequence was confirmed experimentally. The open reading frame of DUSP27, now with a recognition site (ENLYFQ/T) for tobacco etch virus (TEV) protease added to its N-terminus, was moved by recombinational cloning into the destination vector pDEST527 (Protein Expression Laboratory, SAIC-Frederick, Frederick, Maryland, USA) to construct pJT154. This plasmid directs the expression of human DUSP27 with a hexahistidine tag preceding the TEV protease recognition site. The fusion protein was expressed in Escherichia coli strain Rosetta 2 (DE3) (Novagen, Madison, Wisconsin, USA). Cells containing pJT154 were grown to mid-log phase (OD600 ≃ 0.5) at 310 K in Luria broth containing 100 µg ml−1 ampicillin, 30 µg ml−1 chloramphenicol and 0.2% glucose. Overproduction of the fusion protein was induced with isopropyl β-d-1-thiogalactopyranoside at a final concentration of 1 mM for 4 h at 303 K. The cells were pelleted by centrifugation and stored at 193 K.
All procedures were performed at 277–281 K. 10–15 g E. coli cell paste was suspended in 150 ml ice-cold 50 mM MES pH 6.5, 200 mM NaCl, 25 mM imidazole, 10%(v/v) glycerol buffer (buffer A) containing 1 mM benzamidine–HCl (Sigma Chemical Company, St Louis, Missouri, USA) and Complete EDTA-free protease-inhibitor cocktail tablets (Roche Molecular Biochemicals, Indianapolis, Indiana, USA). The cells were lysed using an APV-1000 homogenizer (Invensys APV Products, Albertslund, Denmark) at 69 MPa and centrifuged at 30 000g for 30 min. The supernatant was filtered through a 0.22 µm polyethersulfone membrane and applied onto a 12 ml Ni–NTA Superflow column (Qiagen, Valencia, California, USA) equilibrated in buffer A. The column was washed to baseline with buffer A and eluted with a linear gradient of imidazole to 500 mM. Fractions containing recombinant His6-DUSP27 were pooled, concentrated using an Amicon YM10 membrane (Millipore Corporation, Bedford, Massachusetts, USA), diluted with 50 mM MES pH 6.5, 200 mM NaCl, 10%(v/v) glycerol buffer to reduce the imidazole concentration to about 25 mM and digested overnight at 277 K with His6-tagged S219V TEV protease (Kapust et al., 2001 ▶). The digest was applied onto a 12 ml Ni–NTA Superflow column equilibrated in buffer A and recombinant DUSP27 emerged in the column effluent. The effluent was incubated overnight with 10 mM dithiothreitol, concentrated as above and applied onto a HiPrep 26/60 Sephacryl S-200 HR column (GE Healthcare Bio-Sciences Corp., Piscataway, New Jersey, USA) equilibrated in 25 mM MES pH 6.5, 150 mM NaCl, 2 mM Tris(2-carboxyethyl)phosphine (TCEP), 10%(v/v) glycerol buffer. The peak fractions containing DUSP27 were pooled and concentrated to 10–15 mg ml−1 (estimated at 280 nm using a molar extinction coefficient of 31 400 M −1 cm−1). Aliquots were flash-frozen in liquid nitrogen and stored at 193 K. The final product was judged to be >95% pure by SDS–PAGE. The molecular weight was confirmed by electrospray ionization mass spectrometry.
2.2. Crystallization and data collection
DUSP27 [10 mg ml−1 in 25 mM MES pH 6.5, 150 mM NaCl, 2 mM TCEP and 10%(v/v) glycerol buffer] was screened for crystals using a Phoenix crystallization robot (Art Robbins Instruments, Sunnyvale, California, USA) and commercially available sparse-matrix screens from Qiagen (Valencia, California, USA), Hampton Research (Aliso Viejo, California, USA) and Molecular Dimensions (Apopeka, Florida, USA). Crystals were only obtained under one condition from the Hampton Research PEG/Ion screen [0.2 M ammonium sulfate, 25%(w/v) polyethylene glycol 3350] at 293 K. These initial crystals usually diffracted X-rays to between 3.1 and 2.7 Å resolution using the rotating copper-anode home source. Optimization efforts focused on grid screens around the initial conditions and the use of additive screens from Hampton Research. However, the diffraction limit using X-rays from the home source could not be significantly improved. The crystals ultimately used for data collection and structure solution were grown by mixing 2 µl DUSP27 [10 mg ml−1 in 25 mM MES pH 6.5, 150 mM NaCl, 2 mM TCEP and 10%(v/v) glycerol buffer] with 2 µl well solution [0.2 M ammonium sulfate and 25%(w/v) polyethylene glycol 3350]. The drops were sealed over 1 ml crystallization well solution and incubated at 293 K. Crystals grew within two weeks and were cryoprotected by supplementing the mother liquor with 20%(v/v) glycerol and flash-freezing the crystals in liquid nitrogen. Native X-ray diffraction data were collected from a single crystal at 100 K using a MAR 225 detector on beamline 22-BM of the SER-CAT facilities at the Advanced Photon Source, Argonne National Laboratory. 180 frames of data were collected using an oscillation angle of 1.0°, a crystal-to-detector distance of 200 mm and a 10 s exposure time. The data were integrated and scaled with HKL-3000 (Minor et al., 2006 ▶). The crystals belonged to space group P6422, with unit-cell parameters a = b = 126.0, c = 125.8 Å, α = β = 90.0, γ = 120.0°. The Matthews coefficient of 2.86 Å3 Da−1 and the solvent content of 57.0% suggested that there were two molecules in the asymmetric unit (Matthews, 1968 ▶; Kantardjieff & Rupp, 2003 ▶).
2.3. Structure solution and refinement
The structure of DUSP27 was solved by molecular replacement using the coordinates of the testis-specific dual-specificity phosphatase DUSP13 as a search model (PDB entry 2pq5; 50% sequence identity; E. Ugochukwu et al., unpublished work) after removing all solvent atoms and mutating non-identical residues to alanine. Molecular replacement was performed using Phaser from the CCP4 suite of programs (McCoy et al., 2007 ▶; Winn et al., 2011 ▶). Cross-rotation and translational searches for two molecules in the asymmetric unit were performed with data from 15 to 3.0 Å resolution, followed by rigid-body refinement with REFMAC5 (Murshudov et al., 2011 ▶). The model was rebuilt manually using σA-weighted 2mF o − DF c and mF o − DF c electron-density maps with the Coot molecular-graphics program while gradually introducing higher resolution reflections up to the limit of 2.38 Å (Read, 1997 ▶; Emsley & Cowtan, 2004 ▶). Water molecules were located with Coot and were refined with REFMAC5. TLS refinement was performed in the final stages by identifying TLS parameters using the TLS motion determination server (Painter & Merritt, 2006a ▶,b ▶). For TLS refinement, each protein chain was divided into two TLS groups (residues 32–103 and residues 104–205). The refinement was monitored by setting aside 5% of the reflections for calculation of the R free value (Brünger, 1992 ▶). Model validation was performed with MolProbity (Chen et al., 2010 ▶). The all-atom contacts score was 6.41 (99th percentile) and the MolProbity protein geometry score was 1.95 (94th percentile). The Ramachandran plots were prepared with PROCHECK (Laskowski et al., 1993 ▶). X-ray diffraction data-collection and refinement statistics are presented in Table 1 ▶. All structural alignments were performed with PyMOL (DeLano Scientific LLC, Palo Alto, California, USA) and sequence alignments were produced with ClustalW (Larkin et al., 2007 ▶). The coordinates and structure factors were submitted to the Protein Data Bank under accession code 2y96.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray source | 22-BM, SER-CAT |
| Wavelength () | 1.0 |
| Resolution () | 50.02.38 (2.432.38) |
| Space group | P6422 |
| Unit-cell parameters () | a = b = 126.0, c = 125.8 |
| Total reflections/unique reflections | 401585/24047 |
| Completeness (%) | 100 (100) |
| R merge † (%) | 7.4 (72.5) |
| I/(I) | 48.2 (4.2) |
| Multiplicity | 16.1 (15.5) |
| Refinement statistics | |
| Resolution () | 50.02.38 |
| No. of reflections (working set/test set) | 22618/1220 |
| R work ‡ (%) | 20.1 |
| R free (%) | 22.5 |
| No. of atoms/mean B factor§ (2) | |
| Protein chain A | 1438/46.7 |
| Protein chain B | 1387/48.8 |
| Water | 137/22.0 |
| Sulfate | 35/74.8 |
| R.m.s. deviations from ideal geometry | |
| Bond lengths () | 0.015 |
| Bond angles () | 1.5 |
| Ramachandran plot | |
| Most favored (%) | 88.3 |
| Additionally allowed (%) | 11.4 |
| Generously allowed (%) | 0.3 |
| Disallowed (%) | 0 |
| MolProbity clash score | 6.4 (99th percentile) |
| MolProbity protein geometry score | 1.9 (94th percentile) |
| PDB code | 2y96 |
R
merge = 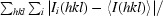
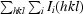 , where I(hkl) is the mean intensity of multiply recorded reflections.
, where I(hkl) is the mean intensity of multiply recorded reflections.
R = 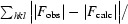
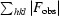 . R
free is the R value calculated for 5% of the data set not included in refinement.
. R
free is the R value calculated for 5% of the data set not included in refinement.
The B factor reported for protein atoms is the residual B factor after TLS refinement.
3. Results and discussion
3.1. Overall structure of DUSP27
The DUSP27 construct consisting of residues 2–220 was successfully crystallized using the hanging-drop vapor-diffusion method and its structure was solved by molecular replacement at 2.38 Å resolution. The Matthews coefficient of 2.86 Å3 Da−1 and the solvent content of 57.0% suggested that there were two molecules in the asymmetric unit. Indeed, the structure reveals that DUSP27 crystallized as a domain-swapped dimer (Fig. 1 ▶), with the domain swapping mediated by interactions mainly between the N-terminal α1 helix of one monomer (Fig. 2 ▶) and the region from helices α7–α8 near the C-terminus of the other monomer to form a four-helix bundle. The elution profile of DUSP27 from size-exclusion chromatography also indicated that DUSP27 exists primarily as a dimer in solution (data not shown). Domain swapping in dual-specificity phosphatases has previously been observed in the crystal structures of the variola H1 phosphatase (Phan et al., 2007 ▶), the vaccinia virus H1 phosphatase (Koksal et al., 2009 ▶) and DUSP26 (PDB entry 2e0t; Y. Xie, S. Kishishita, K. Murayama, C. Hori-Takemoto, L. Chen, Z.-J. Liu, B.-C. Wang, M. Shirozu & S. Yokoyama, unpublished work). In these structures, domain swapping is also mediated by the N-terminal α1 helix interacting with the C-terminal α-helices of the other monomer in the dimer.
Figure 1.
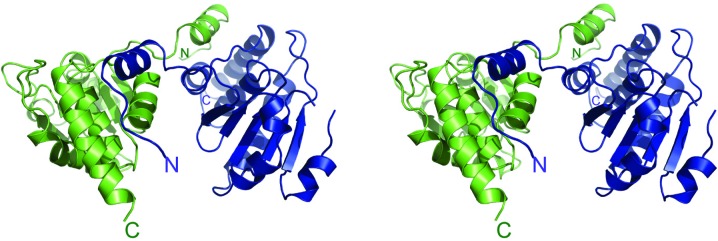
Stereo image of a ribbon model of the three-dimensional structure of DUSP27 with chain A colored green and chain B blue. The enzyme crystallized as a domain-swapped dimer. The N- and C-termini are labeled.
Figure 2.

Stereo image of monomeric DUSP27 illustrated in ribbon format. The bound sulfate molecule is depicted as a blue sphere.
The catalytic core domain of DUSP27 is formed by a central five-stranded β-sheet that is surrounded by helices α2, α3 and α4 on one side and α5–α8 on the opposite side (Fig. 2 ▶). The N-terminal tail including helix α1 is tilted away from the core domain. Residues 2–31 in the N-terminal region and residues 207–220 in the C-terminal tail were not visible in the electron-density maps and thus were not included in the model. Additionally, while chain A is well ordered throughout the structure, the electron density for residues 90–94 in chain B is poorly defined, indicating disorder in this region. These residues are part of a loop between strand β3 and helix α4 in chain A. Chain A was therefore used for structural analysis of the monomer. The electron-density maps also clearly indicated the presence of a bound sulfate molecule in the active site of DUSP27 and five additional bound sulfates throughout the structure. Many of these additional bound sulfate ions appear to mediate crystal contacts between symmetry mates in the crystal lattice.
3.2. Comparison with structural homologs
Using PDBeFold to perform a three-dimensional alignment of protein structures in the PDB, several DUSPs were identified as structural homologs (Krissinel & Henrick, 2004 ▶). The closest three-dimensional structural homologs for DUSP27 were identified as DUSP13 (Kim et al., 2007 ▶) and vaccinia H1-related phosphatase (VHR), also known as DUSP3 (Yuvaniyama et al., 1996 ▶). As VHR has been one of the most extensively studied DUSPs at the biochemical, kinetic and structural levels, it is often considered to be the prototypical member of the DUSP family (Schumacher et al., 2002 ▶). The r.m.s.d. values after structural alignment of DUSP27 with DUSP13 and VHR are 0.65 Å (over 153 residues) and 0.78 Å (over 153 residues), respectively. The core catalytic structure of DUSP27 superimposes well with that of VHR (Fig. 3 ▶), the only significant difference being the conformation of the loop between strand β3 and helix α4 (highlighted by the black arrow). However, the most significant difference between the two molecules is found in the orientation of the α1 helix in the N-terminal tail. While in VHR this helix packs against the core catalytic domain to form a four-helix bundle (Yuvaniyama et al., 1996 ▶), the analogous helix in DUSP27 is flipped 180° in the opposite direction and therefore projects away from the catalytic core. This extended conformation of the α1 helix in DUSP27 is likely to mediate domain swapping between the two molecules in the dimer structure. A similar conformation of the α1 helix projecting away from the catalytic core was observed in the structure of DUSP26, which was solved by a structural genomics consortium (PDB entry 2e0t) and also crystallized as a domain-swapped dimer.
Figure 3.
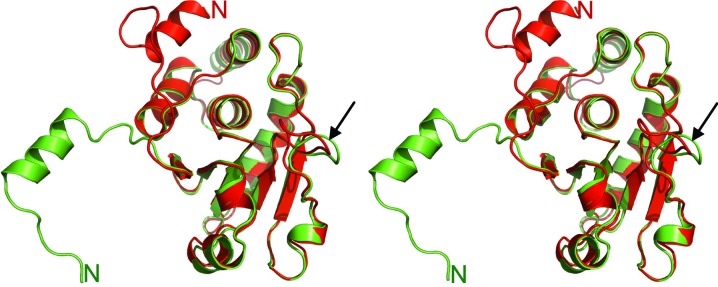
A stereoview of the superimposed coordinates of DUSP27 (green ribbons) with those of VHR (red ribbons; PDB code 1vhr). The N-terminal regions are labeled and the black arrow highlights the structural deviation of the loop between strand β3 and helix α4.
3.3. The active site
The presence of a bound sulfate molecule (a phosphate mimic) from the crystallization solution in the active site of DUSP27 provides an opportunity for structural examination of its interactions with the catalytic residues. An example of the quality of the electron-density maps for the active-site residues and bound sulfate ion is shown in Fig. 4 ▶. PTPs mediate the removal of phosphate groups from substrates via a two-step mechanism that proceeds via a cysteinyl-phosphate intermediate (Denu & Dixon, 1995 ▶, 1998 ▶). The phosphate-binding pocket is formed by the HCXXGXXR motif, where X is occupied by a variable residue (Denu & Dixon, 1998 ▶). Removal of the tyrosine (as well as serine or threonine) phosphate group is catalyzed by a conserved cysteine thiolate anion through a nucleophilic attack of the P atom to form a cysteinyl-phosphate intermediate (Zhou et al., 1994 ▶). The unphosphorylated tyrosine is then released from the active site by the donation of a proton from an invariant aspartic acid that serves as a general acid and then also acts as a general base that abstracts a proton from a water molecule to allow the removal of phosphate and regenerate the active enzyme by a nucleophilic reaction (Denu et al., 1995 ▶).
Figure 4.
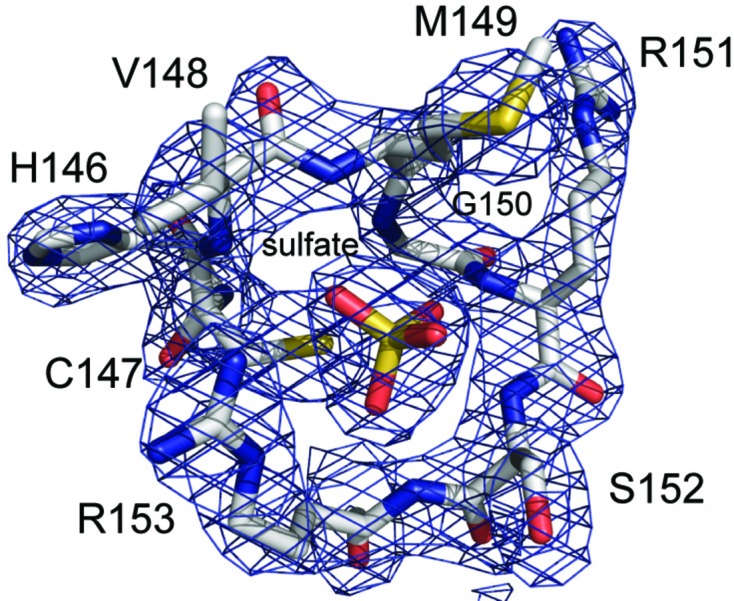
The σA-weighted 2mF o − DF c electron-density map (2.38 Å resolution, contoured at the 1.5σ level) for the residues of the active-site phosphate-binding loop and bound sulfate ion. The residues are depicted in stick format with carbon in gray, nitrogen in blue, oxygen in red and sulfur in yellow.
The HCXXGXXR motif of DUSP27 consists of residues 146–154 (HCVMGRSR) that form the phosphate-binding pocket (Fig. 5 ▶ a). These residues are part of the loop between strand β5 and helix α6. The α6 helix projects toward the sulfate ion and is capped by Arg153, which creates a dipole moment that contributes to the binding of the sulfate ion (Zhang, 2002 ▶; Denu & Dixon, 1998 ▶). The positively charged Arg153 residue also creates a stabilizing interaction with the negatively charged sulfate and contributes a hydrogen bond between the side-chain guanidinium N atom and one of the sulfate O atoms (2.8 Å). The sulfate ion is also held in the active site by an extensive hydrogen-bonding network between the sulfate O atoms and the backbone amide NH atoms of Val148 (3.1 Å), Met149 (2.9 Å), Arg151 (2.9 Å), Ser152 (2.9 Å) and Arg153 (2.8 Å). Cys147, which corresponds to the conserved catalytic nucleophile, is located approximately 3.4 Å from the S atom (Zhou et al., 1994 ▶). Asp115, which is located on a loop between helix α5 and strand β4, corresponds to the conserved aspartic acid residue that acts a general acid/base in the reaction mechanism and is located 3.1 Å away from one of the O atoms of the sulfate (Denu et al., 1995 ▶). That the proper orientation of this aspartic acid in the substrate-bound form of the enzyme is important for efficient catalysis can be construed from other PTP crystal structures that exhibit a large conformational change of the loop containing the aspartic acid upon substrate binding (Denu & Dixon, 1998 ▶; Zhang, 2002 ▶).
Figure 5.

(a) A view of the active site of DUSP27 (green ribbons) with bound sulfate ion. The residues are depicted in stick format with carbon in gray, nitrogen in blue, oxygen in red and sulfur in yellow. Hydrogen bonds are illustrated by red dashed lines. (b) Sequence alignment of the active site HCXXGXXR motif of DUSP27 and selected homologs. The alignment was performed with ClustalW and conserved residues are highlighted in yellow.
As illustrated in Fig. 5 ▶(b), there are some significant differences between the HCXXGXXR motif of DUSP27 and those of other dual-specificity phosphates. Residues Met149 and Arg151 are of particular interest. Schumacher and coworkers were the first to obtain a crystal structure of a dual-specificity phosphatase in complex with a phosphorylated substrate (Schumacher et al., 2002 ▶). Composed of the inactive Cys124Ser mutant of human VHR and a bisphosphorylated peptide corresponding to the MAP kinase p38 activation lip, the structure provided insights into the structural determinants for substrate binding. We superimposed our DUSP27 crystal structure onto the coordinates of the VHR–peptide crystal structure (PDB code 1j4x; Schumacher et al., 2002 ▶) to make comparisons (Figs. 6 ▶ a and 6 ▶ b). The depth of the active-site pocket is a key structural determinant of phosphotyrosine or dual-specificity substrate specificity. Typically, tyrosine-specific PTPs have deep pockets that are unable to accommodate the shorter phosphothreonine/phosphoserine residue and position it appropriately for efficient catalysis (Denu et al., 1996 ▶, Zhang, 2002 ▶). VHR differs from most other DUSPs in that its preference for phosphotyrosine appears to be dictated by the narrow entrance to the active site created by the side chains of Glu126 (Met149 in DUSP27) and Tyr128 (Arg151 in DUSP27) (Schumacher et al., 2002 ▶). Most of the other dual-specificity phosphatases contain smaller hydrophobic residues in the same positions. These smaller residues form a shallower pocket that would allow phosphothreonine or phosphoserine binding. In DUSP27, a bulky Met149 residue is located in the analogous Glu126 position of VHR and contributes to the formation of a deeper active-site pocket. This structural feature of DUSP27 is likely to explain why it prefers phosphotyrosine.
Figure 6.

(a) Surface representation of the crystal structure of DUSP27 with the computed electrostatic surface potential. Positively charged regions are depicted in blue and negatively charged regions in red. The position of the of the DDE(Nle)pTGpYVATR peptide (yellow sticks), a VHR substrate, was modeled based on superposition of DUSP27 with the VHR–peptide complex (PDB code 1j4x). (b) The same view as in (a) illustrated in green ribbon format. (c) Electrostatic surface representation of the crystal structure of the VHR–DDE(Nle)pTGpYVATR peptide complex (PDB code 1j4x). The peptide is illustrated in yellow stick format. (d) The same view as in (c) illustrated in red ribbon format.
3.4. Environment of Arg151
Another unusual feature of DUSP27 that distinguishes it from most other DUSPs is the presence of Arg151 after the conserved glycine residue in the HCXXGXXRS motif (Fig. 5 ▶ b). A sequence alignment with selected DUSPs shows that this position is usually occupied by a hydrophobic residue. We therefore examined the environment of Arg151 in the crystal structure of DUSP27 (Fig. 7 ▶). The side-chain guanidinium of Arg151 is held in place by a 3.0 Å hydrogen bond to the backbone carbonyl O atom of Thr53, which is located on the loop between helix α1 and strand β1, and a 2.9 Å hydrogen bond to the backbone carbonyl O atom of Cys182. Additionally, the side chain of Arg151 stacks against the Tyr52 side chain and may potentially be involved in a π–cation interaction. Tyr52 is not conserved in either VHR or DUSP13, where this position is instead occupied by a proline residue (Yuvaniyama et al., 1996 ▶; Kim et al., 2007 ▶). Interestingly, the region immediately before Tyr52 is where the orientation of the N-terminal tail differs significantly from that of VHR. Consequently, the environment around the Tyr52–Arg151 interaction is very different from that of VHR. Based on the superimposition with the VHR–peptide crystal structure, the Arg151 side chain in DUSP27 is located approximately 3.6 Å from the peptide backbone and projects away from the phosphotyrosine-binding pocket. The backbone amide NH atom, however, does make a hydrogen bond to one of the sulfate O atoms. As Arg151 is part of the phosphate-binding loop, it appears that the hydrogen-bonding interaction of the side chain may serve a structural role in stabilizing the conformation of the loop. While the phosphate-binding loop backbone atoms overlay well with those of VHR, close inspection shows that there is a slight offset of the backbone atoms at the position equivalent to Arg151 in the two PTPs. Additionally, the guanidinium side chain of Arg151 is located at the base of a shallow groove that is separated from the active-site pocket by a ridge formed by Met149. Together with Arg181 (which is conserved as Arg158 in VHR), these residues impart a positively charged nature to this groove. In VHR, this analogous region forms the phosphothreonine recognition site or second phosphate-binding site (Schumacher et al., 2002 ▶). The overlay of DUSP27 with the VHR–peptide crystal structure reveals that the Arg151 side chain is located approximately 3.6 Å from one of the O atoms in the second phosphorylated residue of the VHR peptide in the model. Although we observed five additional sulfate ions that were bound throughout the DUSP27 structure, we did not observe any bound in this groove. Rather, it is occupied by two water molecules in DUSP27. Therefore, although it appears that the Arg151 side chain has potential to interact with the incoming substrate, the extensive hydrogen-bonding network of the side chain with other residues suggests it is most likely to play a stabilizing structural role for the phosphate-binding loop.
Figure 7.
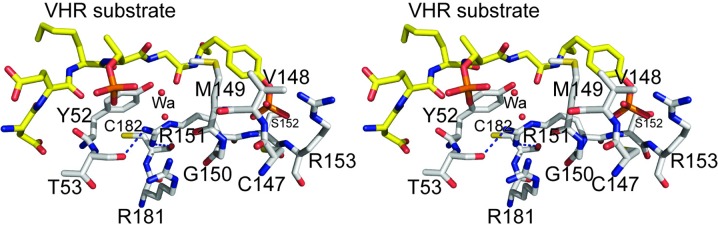
Stereoview of a hypothetical model of DUSP27 with the VHR substrate DDE(Nle)pTGpYVATR peptide (stick format with C atoms in yellow) obtained after the superposition of DUSP27 with the VHR–peptide complex crystal structure.
3.5. Comparison with the peptide-bound VHR structure
The electrostatic surface potentials of DUSP27 and VHR are compared in Fig. 6 ▶. A striking feature is that the surface-charge patterns near the substrate-binding region are significantly different in the two enzymes. The surface below and to the right of the active-site pocket in DUSP27 is negatively charged, whereas the region above and to the left consists of positively charged patches (Fig. 6 ▶ a). The charge distribution in VHR is the opposite of that of DUSP27 in the analogous regions (Fig. 6 ▶ c; Schumacher et al., 2002 ▶). Another significant difference between DUSP27 and VHR is the location of the N-terminal α1 helix (Figs. 6 ▶ b and 6 ▶ d). In the VHR–peptide complex the peptide binds in an extended conformation through a groove that includes the N-terminal α1 helix, which packs against the α6 helix in the catalytic core. Therefore, the VHR α1 helix is involved in the formation of the substrate-binding groove. However, in DUSP27 the α1 helix projects away from the core catalytic domain and therefore cannot engage in similar interactions with the substrate as are observed in monomeric VHR. The different orientations also open up the possibility of new substrate-binding interactions. Interestingly, structural alignment of the domain-swapped DUSP27 dimer with VHR reveals that the α1 helix of one molecule in the DUSP27 dimer aligns well with the VHR α1 helix (Fig. 8 ▶). Hence, domain swapping in DUSP27 creates a substrate-binding groove not unlike that of VHR, although structural differences still exist in the loop region between helix α1 and strand β1. Similar domain swapping involving the α1 helix has also been observed in the crystal structures of DUSP26, variola H1 phosphatase and vaccinia VH1 phosphatase (Phan et al., 2007 ▶; Koksal et al., 2009 ▶). Although the physiological substrates of DUSP27 are unknown (Camps et al., 2000 ▶; Friedberg et al., 2007 ▶), the differences in surface-charge distribution and the orientation of the α1 helix suggests that DUSP27 and VHR have different physiological substrates.
Figure 8.
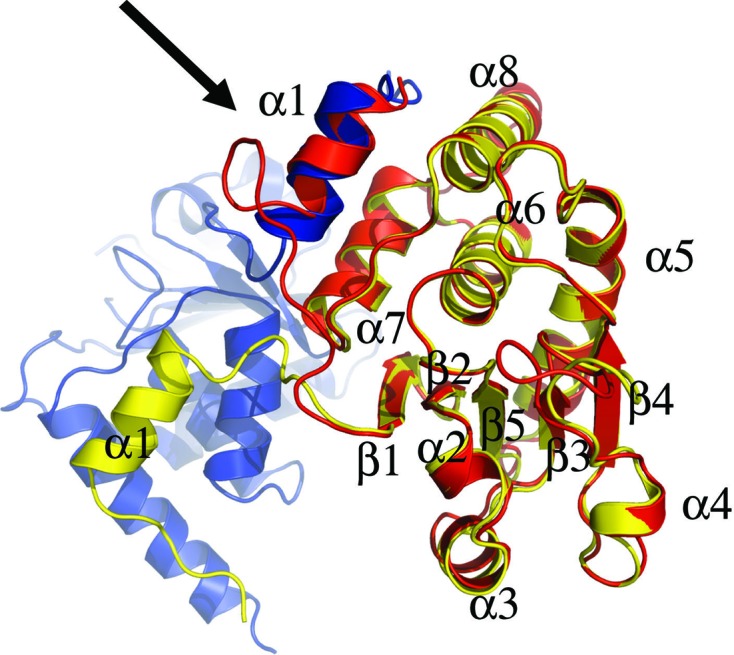
Superposition of monomeric VHR (red) onto chain B (yellow) of the DUSP27 dimer. Chain A of DUSP27 is colored blue. The black arrow highlights the superposition of the α1 helix of DUSP27 chain A onto the α1 helix of VH1 as a result of domain swapping.
4. Conclusions
In summary, we have determined the crystal structure of human dual-specificity phosphatase 27. DUSP27 crystallized as a domain-swapped dimer and the overall fold of the core catalytic domain is highly similar to those of many other DUSPs. However, the orientation of the N-terminal α1 helix differs from that of many DUSPs such as VHR. Comparison of the structure of DUSP27 with that of the VHR–peptide complex reveals differences in the electrostatic surface potentials of the two homologs that may govern their substrate specificities in vivo. The crystal structure of DUSP27 should provide valuable data for structure-guided characterization of the enzyme and the discovery of inhibitors.
Supplementary Material
PDB reference: DUSP27, 2y96
Acknowledgments
We thank Scott Cherry for technical assistance. This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. Electrospray mass-spectrometry experiments were conducted on the LC/ES-MS instrument maintained by the Biophysics Resource in the Structural Biophysics Laboratory, Center for Cancer Research, National Cancer Institute at Frederick. X-ray diffraction data were collected on the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-BM at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract No. W-31-109-Eng-38.
References
- Almo, S. C. et al. (2007). J. Struct. Funct. Genomics, 8, 121–140. [DOI] [PMC free article] [PubMed]
- Alonso, A., Sasin, J., Bottini, N., Friedberg, I., Friedberg, I., Osterman, A., Godzik, A., Hunter, T., Dixon, J. & Mustelin, T. (2004). Cell, 117, 699–711. [DOI] [PubMed]
- Andersen, J. N., Jansen, P. G., Echwald, S. M., Mortensen, O. H., Fukada, T., Del Vecchio, R., Tonks, N. K. & Møller, N. P. (2004). FASEB J. 18, 8–30. [DOI] [PubMed]
- Arnoldussen, Y. J. & Saatcioglu, F. (2009). Mol. Cell. Endocrinol. 309, 1–7. [DOI] [PubMed]
- Bhaduri, A. & Sowdhamini, R. (2003). Protein Eng. 16, 881–888. [DOI] [PubMed]
- Brünger, A. T. (1992). Nature (London), 355, 472–475. [DOI] [PubMed]
- Camps, M., Nichols, A. & Arkinstall, S. (2000). FASEB J. 14, 6–16. [PubMed]
- Charbonneau, H., Tonks, N. K., Kumar, S., Diltz, C. D., Harrylock, M., Cool, D. E., Krebs, E. G., Fischer, E. H. & Walsh, K. A. (1989). Proc. Natl Acad. Sci. USA, 86, 5252–5256. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Clark, A. R., Martins, J. R. & Tchen, C. R. (2008). J. Biol. Chem. 283, 25765–25769. [DOI] [PMC free article] [PubMed]
- Cohen, P. (2002a). Nature Cell Biol. 4, E127–E130. [DOI] [PubMed]
- Cohen, P. (2002b). Nature Rev. Drug Discov. 1, 309–315. [DOI] [PubMed]
- Denu, J. M. & Dixon, J. E. (1995). Proc. Natl Acad. Sci. USA, 92, 5910–5914. [DOI] [PMC free article] [PubMed]
- Denu, J. M. & Dixon, J. E. (1998). Curr. Opin. Chem. Biol. 2, 633–641. [DOI] [PubMed]
- Denu, J. M., Stuckey, J. A., Saper, M. A. & Dixon, J. E. (1996). Cell, 87, 361–364. [DOI] [PubMed]
- Denu, J. M., Zhou, G., Guo, Y. & Dixon, J. E. (1995). Biochemistry, 34, 3396–3403. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fauman, E. B. & Saper, M. A. (1996). Trends Biochem. Sci. 21, 413–417. [DOI] [PubMed]
- Friedberg, I., Nika, K., Tautz, L., Saito, K., Cerignoli, F., Friedberg, I., Godzik, A. & Mustelin, T. (2007). FEBS Lett. 581, 2527–2533. [DOI] [PubMed]
- Guan, K. L., Haun, R. S., Watson, S. J., Geahlen, R. L. & Dixon, J. E. (1990). Proc. Natl Acad. Sci. USA, 87, 1501–1505. [DOI] [PMC free article] [PubMed]
- Hoffman, B. T., Nelson, M. R., Burdick, K. & Baxter, S. M. (2004). Curr. Pharm. Des. 10, 1161–1181. [DOI] [PubMed]
- Hooft van Huijsduijnen, R., Wälchli, S., Ibberson, M. & Harrenga, A. (2002). Expert Opin. Ther. Targets, 6, 637–647. [DOI] [PubMed]
- Hunter, T. (1995). Cell, 80, 225–236. [DOI] [PubMed]
- Kantardjieff, K. A. & Rupp, B. (2003). Protein Sci. 12, 1865–1871. [DOI] [PMC free article] [PubMed]
- Kapust, R. B., Tözsér, J., Fox, J. D., Anderson, D. E., Cherry, S., Copeland, T. D. & Waugh, D. S. (2001). Protein Eng. 14, 993–1000. [DOI] [PubMed]
- Kim, S. J., Jeong, D.-G., Yoon, T.-S., Son, J.-H., Cho, S. K., Ryu, S. E. & Kim, J.-H. (2007). Proteins, 66, 239–245. [DOI] [PubMed]
- Koksal, A. C., Nardozzi, J. D. & Cingolani, G. (2009). J. Biol. Chem. 284, 10129–10137. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R., Thompson, J. D., Gibson, T. J. & Higgins, D. G. (2007). Bioinformatics, 23, 2947–2948. [DOI] [PubMed]
- Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. (1993). J. Appl. Cryst. 26, 283–291.
- Levitzki, A. (2004). Semin. Cancer Biol. 14, 219–221. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Nielsen, D. A., Ji, F., Yuferov, V., Ho, A., He, C., Ott, J. & Kreek, M. J. (2010). Psychiatr. Genet. 20, 207–214. [DOI] [PMC free article] [PubMed]
- Painter, J. & Merritt, E. A. (2006a). Acta Cryst. D62, 439–450. [DOI] [PubMed]
- Painter, J. & Merritt, E. A. (2006b). J. Appl. Cryst. 39, 109–111.
- Patterson, K. I., Brummer, T., O’Brien, P. M. & Daly, R. J. (2009). Biochem. J. 418, 475–489. [DOI] [PubMed]
- Pawson, T. (1995). Nature (London), 373, 573–580. [DOI] [PubMed]
- Phan, J., Tropea, J. E. & Waugh, D. S. (2007). Acta Cryst. D63, 698–704. [DOI] [PubMed]
- Pulido, R. & Hooft van Huijsduijnen, R. (2008). FEBS J. 275, 848–866. [DOI] [PubMed]
- Read, R. J. (1997). Methods Enzymol. 277, 110–128. [DOI] [PubMed]
- Schumacher, M. A., Todd, J. L., Rice, A. E., Tanner, K. G. & Denu, J. M. (2002). Biochemistry, 41, 3009–3017. [DOI] [PubMed]
- Shang, X. et al. (2010). Oncogene, 29, 4938–4946. [DOI] [PMC free article] [PubMed]
- Tiganis, T. & Bennett, A. M. (2007). Biochem. J. 402, 1–15. [DOI] [PMC free article] [PubMed]
- Tonks, N. K. (2006). Nature Rev. Mol. Cell Biol. 7, 833–846. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yi, T. & Lindner, D. (2008). Curr. Oncol. Rep. 10, 114–121. [DOI] [PubMed]
- Yuvaniyama, J., Denu, J. M., Dixon, J. E. & Saper, M. A. (1996). Science, 272, 1328–1331. [DOI] [PubMed]
- Zhang, J., Yang, P. L. & Gray, N. S. (2009). Nature Rev. Cancer, 9, 28–39. [DOI] [PubMed]
- Zhang, Z.-Y. (2002). Annu. Rev. Pharmacol. Toxicol. 42, 209–234. [DOI] [PubMed]
- Zhang, Z.-Y., Zhou, B. & Xie, L. (2002). Pharmacol. Ther. 93, 307–317. [DOI] [PubMed]
- Zhou, G., Denu, J. M., Wu, L. & Dixon, J. E. (1994). J. Biol. Chem. 269, 28084–28090. [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: DUSP27, 2y96