

The crystal structure of C. thermocellum CelK CMB4 confirms unique extended binding pocket features of clostridial CBM4s.
Keywords: carbohydrate-binding modules, CBM, CBM4, CelK
Abstract
Here, a 2.0 Å resolution X-ray structure of Clostridium thermocellum cellulase K family 4 carbohydrate-binding module (CelK CBM4) is reported. The resulting structure was refined to an R factor of 0.212 and an R free of 0.274. Structural analysis shows that this new structure is very similar to the previously solved structure of C. thermocellum CbhA CBM4. Most importantly, these data support the previously proposed notion of an extended binding pocket using a novel tryptophan-containing loop that may be highly conserved in clostridial CBM4 proteins.
1. Introduction
Some bacteria use large multi-protein complexes called cellulosomes to deconstruct lignocellulosic substrates (Bayer et al., 2008 ▶). In the cellulosomal system, multi-domain enzymes are attached to scaffoldin proteins that are bound to the bacterial cell wall. Cellulosomes are known to contain carbohydrate-binding modules (CBMs), glycoside hydrolases and fibronectin type III-like modules.
Here, we report the X-ray structure of Clostridium thermocellum cellulase K family 4 CBM (CelK CBM4). By structural classification, this module was predicted to be a type B CBM that binds to soluble glucan chains with its cleft-like binding pocket (Boraston et al., 2004 ▶). However, actual binding studies with CelK CBM4 and the insoluble substrate bacterial microcrystalline cellulose (BMCC) suggest that it can also bind to this form of cellulose (Kataeva et al., 2001 ▶). Furthermore, the recent three-dimensional structure of C. thermocellum CbhA CBM4 showed a functional peptide loop that is not found in most bacterial CBM4 structures (Alahuhta et al., 2010 ▶). These combined observations suggested that other clostridial CBM4 proteins should be characterized. We have thus solved the unliganded structure of CelK CBM4 to confirm that these unique features found for the CbhA CBM4 are conserved in other C. thermocellum CBM4s even in the absence of ligand.
Our long-term strategy is to better understand the structure–function relationship of and the cellulose degradation accomplished by bacterial cellulosomal proteins. This publication is the third in a series of structural and functional studies on critical cellulosomal domains from C. thermocellum. Although the main goal of this study is to confirm the unique features of clostridial CBM4s, this structure is only the second from the bacterial cellulosome and therefore helps us to better understand how this complex plant cell-wall-degrading machinery works at the molecular level.
2. Materials and methods
2.1. Cloning, expression and purification
The CelK CBM4 is from the C. thermocellum cellulosomal cellulase gene (UniProt ID P0C2S1). The overall architecture of this gene is CBM4-(family 9 glycosyl hydrolase)-(type I dockerin) (EC 3.2.1.91). The expressed protein contained an N-terminal linker including a histidine tag: MGSSHHHHHHSSGLVPRGSHM.
The CelK CBM4 module was amplified by polymerase chain reaction using the primers 5′-GATATACATATGGCTTTGGAAGACAA-3′ and 5′-AGAGAGGAATTCTCACGGAAGTACATATTCAA-3′ with the genomic DNA from C. thermocellum. The PCR fragment of CelK CBM4 was inserted into a pET28b plasmid (Novagen, Madison, Wisconsin, USA) via NdeI and EcoRI to generate the expression plasmids. It was overexpressed in Escherichia coli (BL21) (Stratagene, La Jolla, California, USA) with induction by 0.3 mM isopropyl β-d-1-thiogalactopyranoside. The recombinant CelK CBM4 containing an N-terminal His tag was purified using a QIAexpress Ni–NTA protein-purification system (Qiagene, Valencia, California, USA) following the manufacturer’s recommended protocol.
2.2. Crystallization
Crystals for CelK CBM4 data collection were obtained by sitting-drop vapor diffusion using a 96-well plate with Crystal Screen HT from Hampton Research (Aliso Viejo, California, USA). 50 µl well solution was added to the reservoir and drops were made up of 1 µl well solution and 1 µl protein solution. The crystals were grown at 293 K with 1.5 M ammonium sulfate, 0.1 M Tris pH 8.5 and 12%(v/v) glycerol as the well solution. The protein solution contained 7.3 mg ml−1 protein, 20 mM acetic acid pH 5 and 100 mM NaCl.
2.3. Data collection and processing
The CelK CBM4 crystal was briefly dipped into a 5 µl drop of 50:50 paraffin:Paratone solution to remove excess water before flash-freezing in a nitrogen-gas stream at 100 K. Home-source data collection was performed using a Bruker X8 MicroStar X-ray generator with Helios mirrors and Bruker Platinum 135 CCD detector. Data were indexed and processed with the Bruker Suite of programs v.2008.1-0 (Bruker AXS, Madison, Wisconsin, USA).
2.4. Structure solution and refinement
Intensities were converted into structure factors and 5% of the reflections were flagged for R free calculations using the programs F2MTZ, TRUNCATE, CAD and UNIQUE from the CCP4 package of programs (Collaborative Computational Project, Number 4, 1994 ▶). The program MOLREP v.10.2.23 (Vagin & Teplyakov, 2010 ▶) was used for molecular replacement using the C. thermocellum CbhA CBM4 module (PDB entry 3k4z; Alahuhta et al., 2010 ▶) as the search model. Refinement and manual corrections were performed using REFMAC5 v.5.5.0109 (Murshudov et al., 1997 ▶) and Coot v.0.6 (Emsley & Cowtan, 2004 ▶). The MolProbity method (Chen et al., 2010 ▶) was used to analyze the Ramachandran plot, and the root-mean-square deviations (r.m.s.d.s) of the bond lengths and angles were calculated from the ideal values of stereochemical parameters (Engh & Huber, 1991 ▶). The Wilson B factor was calculated using CTRUNCATE v.1.0.11 (Collaborative Computational Project, Number 4, 1994 ▶). The r.m.s.d. of Cα atoms, average B factors and the final structure analysis were performed using the program ICM v.3.6-1i (Molsoft LLC, La Jolla, California, USA). The figures were created using PyMOL (http://www.pymol.org). Data-collection and refinement statistics are shown in Table 1 ▶.
Table 1. X-ray data-collection and refinement statistics.
Values in parentheses are for the highest resolution bin.
| Data collection | |
| Space group | P41212 |
| Unit-cell parameters (, ) | a = 65.95, b = 65.95, c = 272.44, = = = 90.0 |
| Wavelength () | 1.54178 |
| Temperature (K) | 100 |
| Resolution () | 252.0 (2.12.0) |
| Unique reflections | 41971 (5536) |
| Observed reflections | 273231 (21867) |
| R int † | 0.057 (0.540) |
| Average multiplicity | 6.5 (4.0) |
| I/(I) | 20.84 (2.08) |
| Completeness (%) | 99.8 (99.5) |
| Refinement | |
| R/R free | 0.212 (0.320)/0.274 (0.408) |
| Protein atoms | 1526 |
| Water molecules | 353 |
| Other atoms | 16 |
| R.m.s.d. from ideal bond lengths‡ (%) | 0.02 |
| R.m.s.d. from ideal bond angles‡ () | 1.9 |
| Wilson B factor (2) | 30.2 |
| Average B factor for protein atoms (2) | 31.1 |
| Average B factor for water molecules (2) | 38.0 |
| Ramachandran plot statistics§ (%) | |
| Allowed | 100 |
| Favored | 97.9 |
| Outliers | 0 |
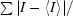
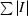
3. Results and discussion
We have solved the X-ray structure of CelK CBM4 from C. thermocellum to 2.0 Å resolution. It has two molecules in the asymmetric unit. Five of the C-terminal residues were found to protrude out of the molecule and are stabilized by the C-terminal residues of the neighboring symmetry-related molecule (Fig. 1 ▶). There are no significant differences between these two molecules (r.m.s.d. of 0.25 Å for all Cα atoms). The typical β-sandwich fold of CBM4s, with two β-sheets containing five antiparallel β-strands each, is shown in Fig. 1 ▶. There is one glycerol molecule located in the binding site of one of the two protein molecules in the asymmetric unit; the other protein molecule has some undefined density, probably another weakly bound glycerol molecule. This binding does not appear to be biologically relevant. This structure has been deposited in the Protein Data Bank with code 3p6b.
Figure 1.

The overall structure of C. thermocellum CelK CBM4 with two molecules in the asymmetric unit. β-Strands are shown in yellow, α-helices in red and loops in green.
Because of weak electron density, the N-terminal peptide was modeled starting from residue 19 and only the main-chain portion of the C-terminal end proline is shown. Also, the side chains of His19, Met20 and Lys25 are not visible in the electron density and have not been modeled. This relatively high proportion of residues without clear electron density and the overall quality of the data (Table 1 ▶) are the reasons for the somewhat high R factors (R factor of 0.212 and R free of 0.274) compared with other structures of similar resolution. The binding cleft and the tryptophan-displaying loop between the anchoring residues Trp134 and Phe141 have well defined electron density (Fig. 2 ▶).
Figure 2.
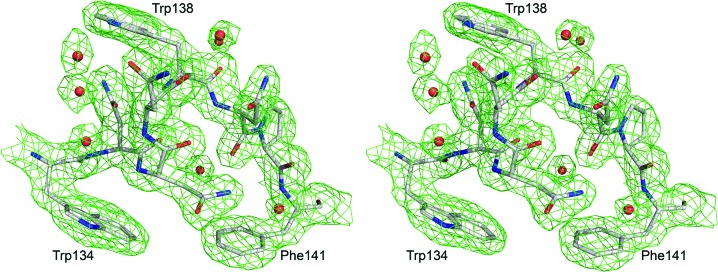
The electron-density map of the Trp138 loop between the anchoring residues Trp134 and Phe141. This 2F o − F c map was calculated at 1.5σ after one cycle of REFMAC5 (Murshudov et al., 1997 ▶). The residues are shown in stick representation, with red O atoms, blue N atoms and gray C atoms.
Structural comparison with the DALI search tool (Holm et al., 2008 ▶) found 667 structures with a Z score of five or higher. Pairwise secondary-structure matching by PDBefold (Krissinel & Henrick, 2004 ▶) found 491 unique structures with at least 50% secondary-structure similarity. By far the most similar structure is the C. thermocellum CbhA CBM4 (PDB entry 3k4z), with a DALI Z score of 32.8, and r.m.s.d. of 0.62 Å from PDBefold, and a sequence identity of 77%. The Cα-atom r.m.s.d. for the CelK and CbhA CBM4 structures calculated by ICM (Molsoft LLC, La Jolla, California, USA) is only 0.84 Å. The binding-cleft residues Trp88 (68 in CbhA CBM4) and Tyr130 (110 in CbhA CBM4) are conserved. The extended binding pocket as well as the Trp138 (118 in CbhA CBM4) loop of CbhA CBM4 (Alahuhta et al., 2010 ▶) are essentially identical (PDBefold residue distances of less than 0.8 Å), with overlapping side-chain conformations even though the CelK CBM4 structure is unliganded (Fig. 3 ▶). All the other hits have DALI Z scores of 18 or lower and sequence identities of below 30%.
Figure 3.
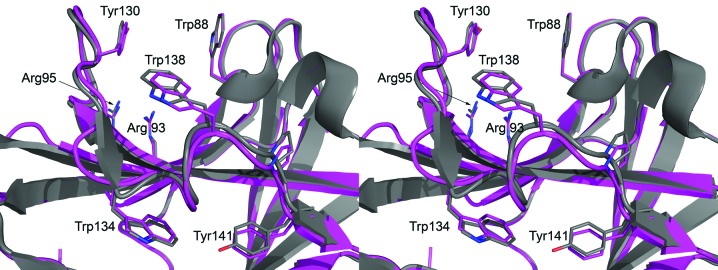
The conserved binding-pocket features of clostridial CBM4 modules. The residues and the cellobiose of CbhA CBM4 are shown in stick representation, with red O atoms, blue N atoms and magenta C atoms for the CelK CBM4 structure and gray C atoms for the CbhA CBM4 module.
The CelK and CbhA CBM4s clearly share the same secondary-structure and binding-pocket features. Our previous mutation and binding studies with CbhA CBM4 showed that the Trp138 (118 in CbhA CBM4) loop interacts with soluble oligodextrins, specifically cellopentaose. However, only the binding-cleft Trp88 (68 in CbhA CBM4) and Tyr130 (110 in CbhA CBM4) are absolutely needed for this interaction (Alahuhta et al., 2010 ▶). This observation, together with the fact that both CelK and CbhA CBM4 bind to bacterial microcrystalline cellulose (known to be about 75% crystalline cellulose; Alahuhta et al., 2010 ▶; Kataeva et al., 2001 ▶), implies that this tryptophan-containing peptide loop has a role in binding to the crystalline cellulose surface as well as to amorphous cellulose. It may also simply guide or stabilize a soluble cellodextrin in the binding cleft. Only CBM4s from clostridia have been shown to bind to crystalline cellulose (Zverlov et al., 2001 ▶) and they are the only CBMs that have been shown to have this tryptophan-containing loop. Furthermore, the cleft aromatic residues Trp88 and Tyr130 are conserved between clostridial and non-clostridial CBM4s; however, Trp138 and the surrounding loop are not (Alahuhta et al., 2010 ▶).
The CbhA CBM4 and CelK CBM4 proteins from C. thermocellum are the only known CBM4 structures that display the tryptophan-containing loop. We have proposed that this structure plays an important role in the binding of the module to free cellulose chain ends accessible from the amorphous regions of the cellulose crystal for CbhA (Alahuhta et al., 2010 ▶). This new structure confirms the existence of the extended binding pocket and tryptophan-containing loop in other clostridial CBM4 proteins as was predicted previously by sequence alignments. The question now arises as to the actual role that is played by bacterial CBMs that display such broad interactive modalities with cellulose.
Supplementary Material
PDB reference: cellulase K CBM4, 3p6b
Acknowledgments
This work was supported by the DOE Office of Science, Office of Biological and Environmental Research through the BioEnergy Science Center (BESC), a DOE Bioenergy Research Center.
References
- Alahuhta, M., Xu, Q., Bomble, Y. J., Brunecky, R., Adney, W. S., Ding, S.-Y., Himmel, M. E. & Lunin, V. V. (2010). J. Mol. Biol. 402, 374–387. [DOI] [PubMed]
- Bayer, E. A., Lamed, R., White, B. A. & Flint, H. J. (2008). Chem. Rec. 8, 364–377. [DOI] [PubMed]
- Boraston, A. B., Bolam, D. N., Gilbert, H. J. & Davies, G. J. (2004). Biochem. J. 382, 769–781. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Engh, R. A. & Huber, R. (1991). Acta Cryst. A47, 392–400.
- Holm, L., Kääriäinen, S., Rosenström, P. & Schenkel, A. (2008). Bioinformatics, 24, 2780–2781. [DOI] [PMC free article] [PubMed]
- Kataeva, I. A., Seidel, R. D., Li, X.-L. & Ljungdahl, L. G. (2001). J. Bacteriol. 183, 1552–1559. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Murshudov, G. N., Vagin, A. A. & Dodson, E. J. (1997). Acta Cryst. D53, 240–255. [DOI] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Zverlov, V. V., Volkov, I. Y., Velikodvorskaya, G. A. & Schwarz, W. H. (2001). Microbiology, 147, 621–629. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: cellulase K CBM4, 3p6b