

Here, the recombinant ARE-binding region of HuR (residues 18–186) was crystallized in space group P21212, with unit-cell parameters a = 41.2, b = 133.1, c = 31.4 Å.
Keywords: human antigen R, RNA-recognition motif domains, poly(U) binding, post-transcriptional regulation
Abstract
Human antigen R (HuR), a ubiquitously expressed member of the Hu protein family, is an important post-transcriptional regulator which has three RNA-recognition motif (RRM) domains. The two tandem N-terminal RRM domains can selectively bind to the AU-rich element (ARE), while the third one interacts with the poly(A) tail and other proteins. Here, the recombinant ARE-binding region of HuR (residues 18–186) was crystallized in space group P21212, with unit-cell parameters a = 41.2, b = 133.1, c = 31.4 Å. X-ray diffraction data were collected to a resolution of 2.8 Å. Mutagenesis analysis and SPR assays revealed its poly(U)-binding properties.
1. Introduction
Post-transcriptional regulation plays a critical role in the modulation of gene expression, which includes splicing, editing, polyadenylation, nuclear export, mRNA localization, mRNA decay and translation (Hinman & Lou, 2008 ▶). mRNA decay is an important control point of post-transcriptional regulation that controls the level of cellular mRNAs. The stability of a given mRNA transcript is controlled by the interactions between the cis-elements within the transcript and trans-acting RNA-binding proteins. The AU-rich element (ARE), which is located in the 3′-end untranslated region (UTR) of mRNAs, is the most studied cis-element and mediates the degradation of selective cytoplasmic mRNA (Chen & Shyu, 1995 ▶).
Human antigen R (HuR) is a ubiquitously expressed member of the Hu protein family, which also includes three neuronal members (HuB, HuC and HuD; Ma et al., 1996 ▶). HuR contains three classic RNA-recognition motif (RRM) domains that are highly conserved among the family members. The first and second RRM domains (RRM1 and RRM2, respectively) are found to bind and stabilize the AU-rich and U-rich elements (AREs) contained in mRNAs encoding various proteins such as cell-cycle regulators, cytokines and growth factors. The third RRM domain (RRM3) is believed to bind to the poly(A) tail and to contribute to the stabilization of RNA–protein interactions (Yuan et al., 2010 ▶). Between RRM2 and RRM3 is a basic hinge region called HNS which mediates the nucleocytoplasmic shuttling of HuR (Fan & Steitz, 1998 ▶). In order to understand how HuR binds and stabilizes ARE-containing mRNAs, structural investigation of the first two tandem RRM domains (RRM12) of HuR is of great importance. Recently, the crystallization and preliminary X-ray crystallographic study of the HuR RRM12 complexed with RNA has been reported (Iyaguchi et al., 2009 ▶). However, the three-dimensional structure of this complex has not yet been solved. Previous studies showed that RNA binding can change the relative arrangement of two tandem RRM domains and create interdomain interactions (Maris et al., 2005 ▶). For example, in the free forms of sex-lethal protein and nucleolin (for which the structures of two tandem RRMs in complex with RNA have been solved), the two consecutive RRM domains tumble independently and the linkers are disordered. However, upon RNA binding the two RRM domains adopt a fixed orientation and contact each other (Handa et al., 1999 ▶). Therefore, the structure of free-form HuR RRM12 is also of great value. It will not only lead to the structure determination of RRM12 in complex with RNA but also provide information about whether RNA binding changes the relative arrangement of the two tandem RRM domains and creates interdomain interactions and, if so, how. To date, only the first RRM domain (RRM1) of HuR has been structurally determined (Benoit et al., 2010 ▶). Here, we report the crystallization and preliminary X-ray crystallographic study and site-directed mutagenesis results of HuR RRM12.
2. Materials and methods
2.1. Cloning, overexpression and purification
The gene encoding the HuR RNA-binding region RRM12 (residues 18–186) was amplified from a human brain cDNA library by PCR using the primers 5′-AGCATCGCCATATGGGGAGAACGAATTTGA-3′ (forward) and 5′-ATACTCTGCTCGAGGTTGGCTGCAAACTTC-3′ (reverse) (Invitrogen). The PCR product was digested using restriction endonucleases NdeI and XhoI and was then ligated into pET-22b vector (Novagen) with a His6 tag at the C-terminus. The recombinant plasmid was transformed into Rosetta2 (DE3) (Novagen). The cells were grown in LB at 310 K with 100 mg l−1 ampicillin until the OD600 reached 0.8 and were then induced with 1 mM IPTG at 289 K for 20 h. The cells were harvested by centrifugation, resuspended in buffer A (20 mM Tris–HCl pH 8.0, 200 mM NaCl, 20 mM imidazole) and disrupted by sonication. The soluble portion was obtained after centrifugation at 14 000g for 30 min and was applied onto an Ni–NTA column pre-equilibrated with buffer A. The protein was eluted using 20 mM Tris–HCl pH 8.0, 200 mM NaCl, 250 mM imidazole and was then dialyzed against buffer B (20 mM Tris–HCl pH 8.0, 200 mM NaCl). After concentration, the protein was loaded onto HiLoad 16/60 Superdex 200 (GE Healthcare) pre-equilibrated with buffer B. HuR RRM12 mutants were obtained using the PCR protocol in MutanBEST kit (TaKaRa). The mutants were purified using the same method as used for the wild-type protein.
2.2. Lysine methylation
Lysine methylation was basically performed as described by Walter et al. (2006 ▶). The RRM12 protein was diluted to 1 mg ml−1 in 50 mM HEPES pH 7.5, 250 mM NaCl. 20 µl freshly prepared 1 M dimethylamine–borane complex (ABC; Fluka) and 40 µl 1 M formaldehyde (Fluka) were added per millilitre of protein solution. The reaction took place at 277 K. After 2 h, a further 20 µl 1 M ABC and 40 µl 1 M formaldehyde per millilitre of solution were added and incubated for a further 2 h. 10 µl 1 M ABC per millilitre of solution was finally added and the mixture was then incubated at 277 K overnight. Finally, the reaction solution was applied onto Superdex 200 pre-equilibrated with 20 mM Tris–HCl pH 7.5, 200 mM NaCl. The protein solution was collected and concentrated to 9 mg ml−1 by centrifugal ultrafiltration (Millipore, 10 kDa cutoff). The target protein was analyzed using SDS–PAGE (Fig. 1 ▶).
Figure 1.
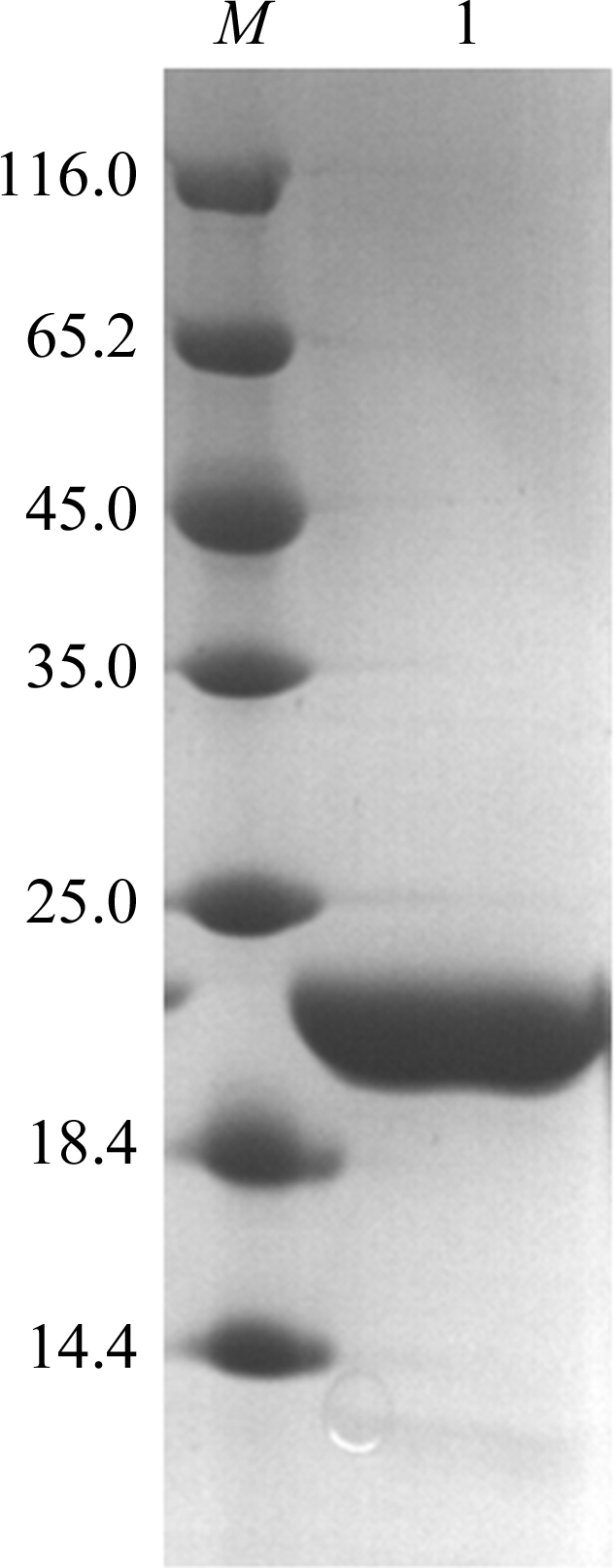
SDS–PAGE of RRM12. Lane M, protein markers (labelled in kDa); lane 1, purified HuR RRM12.
2.3. Crystallization
Methylated RRM12 protein was crystallized using the hanging-drop vapour-diffusion method by mixing 1 µl protein solution and 1 µl reservoir solution at 287 K. Microcrystals were observed using precipitant solution consisting of 1.5 M lithium sulfate, 0.1 M Tris pH 8.5. After optimization, crystals of high quality were obtained for data collection.
2.4. Data collection and processing
For data collection, crystals were transferred to cryoprotectant solution consisting of 1.3 M lithium sulfate, 0.1 M Tris pH 8.1 and 25%(v/v) glycerol and were then frozen in liquid nitrogen. A set of diffraction data was collected at 100 K using synchrotron radiation on beamline BL17U of Shanghai Synchrotron Radiation Facility (SSRF) and was subsequently processed using HKL-2000 (Otwinowski & Minor, 1997 ▶). The data-processing statistics are shown in Table 1 ▶.
Table 1. Data-collection and refinement statistics.
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 41.2, b = 133.1, c = 31.4 |
| Wavelength (Å) | 1.000 |
| Resolution (Å) | 30.0–2.80 (2.85–2.80) |
| No. of observed reflections | 23286 |
| Unique reflections | 4566 |
| Rmerge† (%) | 9.0 (19.1) |
| 〈I/σ(I)〉 | 12.1 (8.6) |
| Completeness (%) | 95.6 (85.0) |
| Multiplicity | 5.1 (4.8) |
R
merge = 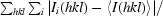
 , where I
i(hkl) is the intensity of an individual reflection and 〈I(hkl)〉 is the average intensity of that reflection.
, where I
i(hkl) is the intensity of an individual reflection and 〈I(hkl)〉 is the average intensity of that reflection.
2.5. Surface plasmon resonance spectroscopy
Surface plasmon resonance (SPR) spectroscopy was carried out using a BIAcore 3000 (GE Healthcare). A 15-base poly(U) segment with the 3′-end biotinylated was immobilized on an SA chip. Wild-type and mutant proteins were passed over the chip at different concentrations in running buffer [20 mM Tris–HCl pH 8.0, 200 mM NaCl, 5%(v/v) glycerol] and the chip was then washed with 100 mM NaOH. SPR assays were conducted at 298 K. The binding curves were fitted according to a one-site binding model using Origin software (http://www.originlab.com).
3. Results and discussion
Full-length RRM12 (residues 18–186) and RRM3 (residues 242–322) from HuR were cloned, but only RRM12 (residues 18–186) could be purified for crystallization. X-ray diffraction was difficult to carry out owing to the poor quality of the microcrystals that were obtained from the initial crystallization screen. In order to obtain high-quality crystals, several parameters (such as temperature, pH and buffer) were changed, but only lysine methylation of the protein was effective (Fig. 2 ▶). A diffraction data set was collected to 2.8 Å resolution. The RRM12 crystal belonged to space group P21212, with unit-cell parameters a = 41.2, b = 133.1, c = 31.4 Å. The Matthews coefficient of 2.05 Å3 Da−1 indicated that there was only one molecule in the asymmetric unit, with a solvent content of 39.98%.
Figure 2.

(a) Crystals of native RRM12; (b) crystal of RRM12 after lysine methylation.
In order to probe the critical residues of RRM12 that are employed in ARE binding, ten residues (Asn21, Lys50, Phe65, Lys89, Thr90, Arg97, Ser99, Tyr109, Arg136 and Arg147) were selected for replacement with alanine based on the known crystal structure of HuD complexed with RNA (Wang & Tanaka Hall, 2001 ▶). Owing to the instability of the mutant proteins, only five mutants (N21A, T90A, Y109A, R136A, R147A) were purified and used in kinetic analyses. An SPR assay was carried out to determine their RNA-binding affinities. Since all of the HuR target mRNAs possess the U-rich motif (López de Silanes et al., 2004 ▶), a 15-base poly(U) segment was used as the RNA substrate. The SPR results demonstrated that RRM12 of HuR bound the poly(U) segment with a K d value of 671 nM, while the K d values for the five mutants N21A, T90A, Y109A, R136A and R147A were 3.61, 1.63, 5.70, 2.43 and 3.52 µM, respectively, indicating that these residues are involved in RNA binding (Fig. 3 ▶). Tyr135 in HuD (equivalent to Tyr109 in HuR) is a residue that makes both stacking and side chain–RNA contacts, while the other four mutant residues make either main-chain or side-chain contacts only. Our SPR results revealed that the Y109A mutant showed the most reduced affinity for RNA binding, which is consistent with a previous study on HuD (Wang & Tanaka Hall, 2001 ▶). All of these mutant residues are located in the β-sheets of the RRM domains and are conserved in Hu proteins (Fig. 4 ▶). Consequently, we concluded that like other RRM domain-containing proteins Hu proteins bind to RNA using the β-sheets of the RRM domains and adopt a similar protein–RNA interaction mode.
Figure 3.

(a) Binding constants of wild-type HuR RRM12 and five mutants for the 15-base poly(U) segment; K d values were calculated based on the steady-state affinity model using Origin software. (b) Histogram showing the reduced affinity of poly(U) binding for all five mutants. Error bars were derived from the calculated K d values.
Figure 4.

Sequence alignment of RRM12 domains of Hu proteins. Residues shown in red were selected for single-amino-acid mutagenesis in HuR.
Acknowledgments
We are grateful to the staff members at SSRF for the collection of diffraction data. Financial support for this project was provided by the Fundamental Research Funds for the Central Universities, the Chinese National Natural Science Foundation (grant Nos. 30900224 and 10979039), the Chinese Ministry of Science and Technology (grant Nos. 2006AA02A318 and 2009CB825500), the Science and Technological Fund of Anhui Province for Outstanding Youth (grant No. 10040606Y11), the Anhui Provincial Natural Science Foundation (grant No. 090413081) and the Education Department of Anhui Province (grant No. 2009SQRZ007ZD).
References
- Benoit, R. M., Meisner, N. C., Kallen, J., Graff, P., Hemmig, R., Cèbe, R., Ostermeier, C., Widmer, H. & Auer, M. (2010). J. Mol. Biol. 397, 1231–1244. [DOI] [PubMed]
- Chen, C.-Y. & Shyu, A.-B. (1995). Trends Biochem. Sci. 20, 465–470. [DOI] [PubMed]
- Fan, X. C. & Steitz, J. A. (1998). Proc. Natl Acad. Sci. USA, 95, 15293–15298. [DOI] [PMC free article] [PubMed]
- Handa, N., Nureki, O., Kurimoto, K., Kim, I., Sakamoto, H., Shimura, Y., Muto, Y. & Yokoyama, S. (1999). Nature (London), 398, 579–585. [DOI] [PubMed]
- Hinman, M. N. & Lou, H. (2008). Cell Mol. Life Sci. 65, 3168–3181. [DOI] [PMC free article] [PubMed]
- Iyaguchi, D., Yao, M., Tanaka, I. & Toyota, E. (2009). Acta Cryst. F65, 285–287. [DOI] [PMC free article] [PubMed]
- López de Silanes, I., Zhan, M., Lal, A., Yang, X. & Gorospe, M. (2004). Proc. Natl Acad. Sci. USA, 101, 2987–2992. [DOI] [PMC free article] [PubMed]
- Ma, W.-J., Cheng, S., Campbell, C., Wright, A. & Furneaux, H. (1996). J. Biol. Chem. 271, 8144–8151. [DOI] [PubMed]
- Maris, C., Dominguez, C. & Allain, F. H. (2005). FEBS J. 272, 2118–2131. [DOI] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Walter, T. S., Meier, C., Assenberg, R., Au, K. F., Ren, J., Verma, A., Nettleship, J. E., Owens, R. J., Stuart, D. I. & Grimes, J. M. (2006). Structure, 14, 1617–1622. [DOI] [PMC free article] [PubMed]
- Wang, X. & Tanaka Hall, T. M. (2001). Nature Struct. Biol. 8, 141–145. [DOI] [PubMed]
- Yuan, Z., Sanders, A. J., Ye, L. & Jiang, W. G. (2010). Histol. Histopathol. 25, 1331–1340. [DOI] [PubMed]