

An engineered deletion of a nonconserved loop in the RUN domain of Rab6-interacting protein 1 enhances the solubility of the recombinant protein when expressed in E. coli. Crystals of the engineered effector in complex with Rab6a diffracted X-rays to 3.25 Å resolution.
Keywords: Rab6-interacting protein 1, DENN domain-containing protein 5, RUN domain
Abstract
Effectors of the Rab small GTPases are large multi-domain proteins which have proved difficult to express in soluble form in Escherichia coli. Generally, effectors are recruited to a distinct subcellular compartment by active (GTP-bound) Rabs, which are linked to membranes by one or two prenylated Cys residues at their C-termini. Following recruitment via their Rab-binding domain (RBD), effectors carry out various aspects of vesicle formation, transport, tethering and fusion through their other domains. Previously, successful purification of the RUN–PLAT tandem domains (residues 683–1061) of the 1263-residue Rab6-interacting protein 1 (R6IP1) required co-expression with Rab6, as attempts to solubly express the effector alone were unsuccessful. R6IP1 is also known as DENN domain-containing protein 5 (DENND5) and is expressed as two isoforms, R6IP1A/B (DENND5A/B), which differ by 24 amino acids at the N-terminus. Here, a deletion in R6IP1 was engineered to enable soluble expression and to improve the quality of the crystals grown in complex with Rab6. A large 23-residue loop linking two α-helices in the RUN1 domain was removed and replaced with a short linker. This loop resides on the opposite face to the Rab6-binding site and is not conserved in the RUN-domain family. In contrast to wild-type R6IP1–Rab6 crystals, which took several weeks to grow to full size, the engineered R6IP1 (RPdel)–Rab6 crystals could be grown in a matter of days.
1. Introduction
Rab GTPases are the main regulators of the endocytic and secretory trafficking pathways (Zerial & McBride, 2001 ▶). The thermodynamic characterization of Rab–effector interactions is an important step in understanding the molecular basis of cellular transport. Each Rab typically binds to several effectors; conversely, several effectors are known to be promiscuous and to interact with more than one Rab protein (Fukuda et al., 2008 ▶; Kanno et al., 2010 ▶). Rab6 in particular regulates the transport pathway into and out of the Golgi apparatus and numerous Rab6 effectors have been identified to date. Recently, the minimal Rab6-binding domains (RBDs) of three effectors have been identified and their affinities (K d) for Rab6 have been measured and shown to be in the low micromolar range (Bergbrede et al., 2009 ▶). These Rab6–effector complexes display rapid on/off rates, thus providing a rationale for Rab6 promiscuity which may be critical for cellular trafficking (Bergbrede et al., 2009 ▶).
The interaction of Rab6 with the effector R6IP1 was initially detected using a two-hybrid system screen (Janoueix-Lerosey et al., 1995 ▶). The crystal structure of the largest Rab–effector complex determined to date (Rab6 with two domains of R6IP1 protein) was recently solved in our laboratory (PDB entry 3cwz; Recacha et al., 2009 ▶). The crystallized fragment of R6IP1 (residues 683–1061) includes a RUN domain (702–920) and a PLAT domain (927–1057). All of the interactions with Rab6 emanate from two noncontiguous α-helices, α1 and α8, from the RUN domain. However, biophysical studies have been hampered by the limited availability of purified soluble R6IP1. Bioinformatic analyses revealed that a loop bridging α3 and α4 of the RUN1 domain is not conserved in structurally related proteins, including the second RUN domain (RUN2) of R6IP1 at its C-terminus. Moreover, the 3.2 Å crystal structure of R6IP1–Rab6 revealed that this loop is disordered (Recacha et al., 2009 ▶).
In order to enhance solubility and possibly improve crystal quality, we designed a new construct spanning the RUN1 and PLAT domains (residues 702–1057) with a deletion of the loop connecting α3 and α4, hereafter referred to as RPdel (Fig. 1 ▶). This involved the deletion of residues between positions 813–835 inclusive; these were replaced by a glycine residue to enable sufficient flexibility in bridging the two helices. Molecular modelling of the short loop (not shown) supported the idea that the loop was sufficiently long and flexible to bridge the two α-helices. This manuscript describes the crystallization of the engineered effector in complex with Rab6, revealing that its crystallization conditions and morphology are similar to those of wild-type complexes.
Figure 1.

The domain arrangement of R6IP1, with the deletion between α3 and α4 indicated. The acronym DENN refers to differentially expressed in neoplastic versus normal cells, while uDENN refers to the ‘upstream’ DENN domain and dDENN refers to the ‘downstream’ DENN domain. Also shown is a sequence alignment of the RUN1 and RUN2 domains of R6IP1, the RUN domains of human Rap2IPx (RPIPX; NP_001032519.1 residues 65–236), murine RabIP4 (NP_766145.1 residues 113–286) and the engineered RUN1 domain of RPdel. The asterisks in the R6IP1del sequence represent residues which have been deleted from the loop spanning α-helices 3 and 4 of murine R6IP1. The residue numbering above is in accordance with that adopted in the structure deposited as PDB entry 3cwz; however, it is worth noting that residue 719 of Rab6IP1 above corresponds to residue 743 of the full-length Denn5a gene sequence (Gene ID 19347).
2. Materials and methods
2.1. Cloning and purification of RPdel
A full-length version of the R6IP1 gene (Gene ID 19347) containing a corresponding deletion of the loop between α3 and α4 was generated by Geneart AG (Regensburg, Germany). The residues deleted were 813-LSTSGILLDSERRKSDASAVMSP-835 and the synthetic gene was cloned into the expression vector pNIC28-BSA4 (GenBank Accession No. EF198106; Stols et al., 2002 ▶). The deleted residues were replaced by a single glycine in order to enhance flexibility within this region. The segment from 702 to 1057 was PCR-amplified using the forward primer 5′-CAGGATCCATGGGCAGTACCATCCGTG-3′ and the reverse primer 5′-CGGAATTCTCAGGACTGCTGTAGTGGCGGAGT-3′. The BamHI/EcoRI-cut fragment generated from this PCR reaction was cloned into a pHIS-parallel vector (pHis-1). The expressed RPdel protein thus contains a tobacco etch virus (TEV) cleavable N-terminal His6 tag. The construct was transformed into Escherichia coli BL21 (DE3) cells and all subsequent cultures were grown in 2×YT medium supplemented with 34 mg l−1 kanamycin. For large-scale cultures, 1 l 2×YT was inoculated with 10 ml of an overnight culture. The cultures were incubated at 310 K with shaking (120 rev min−1) until the A 600 reached ∼0.6, at which point expression was induced by addition of 0.5 mM IPTG for approximately 6 h. The cells were harvested by centrifugation at 2700g for 10 min. The bacterial pellets were washed once with ice-cold phosphate-buffered saline buffer and stored as frozen pellets at 253 K.
The pellets were resuspended in 300 mM NaCl, 5 mM MgCl2, 10 mM imidazole, 10 mM β-mercaptoethanol, 0.5 mM PMSF in 10 mM Tris–HCl pH 8.0. The cells were disrupted by sonication and the bacterial lysate was centrifuged at 20 000g for 30 min to eliminate cellular debris. The cleared lysate was passed through a 0.2 µm filter and loaded onto Ni2+ agarose (Chromatrin). The column was washed with ten volumes of 300 mM NaCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, 10 mM imidazole in 10 mM Tris–HCl pH 8.0 and then with sequential washes with this buffer supplemented with 20, 30 and 40 mM imidazole. Finally, the protein was eluted with a step gradient to 200 mM imidazole. The His6 tag was cleaved by overnight dialysis in 150 mM NaCl, 5 mM MgCl2, 10 mM β-mercaptoethanol, 10 mM Tris–HCl pH 8.0 with 10 µg rTEV per milligram of eluted protein. Following dialysis, the protein solution was supplemented with NaCl and imidazole to final concentrations of 300 and 10 mM, respectively. The rTEV protease, the cleaved tag and any uncleaved RPdel were removed by passage through a second Ni2+ agarose column. The unbound fraction containing cleaved RPdel was pooled, concentrated and loaded onto a Superdex-200 size-exclusion column (mounted on AKTAbasic FPLC, GE Healthcare) equilibrated with 10 mM Tris–HCl, 100 mM NaCl, 5 mM MgCl2 and 1 mM DTT pH 8.0. The cleaved protein has a serine residue preceding the start methionine at the N-terminus.
2.2. The Rab6–RPdel complex
A truncated version of human Rab6a (Gene ID 5870) between residues 8 and 195 and with a Q72L mutation to lock the GTPase in the GTP-bound state was cloned into the NdeI/XhoI restriction sites of pET28 with ampicillin resistance (Recacha et al., 2009 ▶). The expressed Rab6aQ72L contains an N-terminal His6 tag with a thrombin-cleavable site between the tag and the Rab6aQ72L protein. After cleavage, the resulting protein had the sequence GSHM at the N-terminus.
To generate large amounts of the complex for crystallization, RPdel and Rab6aQ72L constructs were co-transformed into E. coli BL21 (DE3) cells and all subsequent cultures were supplemented with 100 mg l−1 ampicillin and 34 mg l−1 kanamycin. Protein expression in large-scale cultures was induced by addition of 0.5 mM IPTG for approximately 6 h at 298 K. The cells were harvested and the complex was purified as described above for the RPdel protein but with the addition of 10 units ml−1 thrombin (GE Healthcare) during dialysis.
2.3. Rab6–RPdel crystallization
Prior to crystallization, all proteins (including the wild-type complex) were stored in the buffer from the gel-filtration chromatography step: 10 mM Tris–HCl, 100 mM NaCl, 5 mM MgCl2 and 1 mM DTT pH 8.0. Initial crystallization screening was carried out on a submicrolitre scale by the sitting-drop vapour-diffusion method (100 nl protein solution was mixed with 100 nl reservoir solution and equilibrated against 70 µl reservoir solution) using a Mosquito automated pipetting system (TTP LabTech) and screening kits from Hampton Research, Molecular Dimensions and a PEG 4000/MPD custom screen. Larger crystals were grown by hanging-drop vapour diffusion at 291 K. 1 µl 7 mg ml−1 Rab6aQ72L–RPdel was mixed with 1 µl mother liquor consisting of 100 mM HEPES pH 7.1, 2%(w/v) PEG 4000 and 4% 2,4-methylpentanediol (MPD) on a siliconized cover slip. Hexagonal-shaped crystals appeared overnight and grew to full size (0.2 × 0.2 × 0.6 mm) in 2–4 d (Fig. 2 ▶). Crystals were cryoprotected in 100 mM HEPES pH 7.1, 2%(w/v) PEG 4000, 4% MPD and 25% glycerol and then flash-cooled in liquid nitrogen.
Figure 2.

Single crystals of the Rab6–RPdel complex. The average dimensions of the crystals are 0.2 × 0.2 × 0.6 mm.
2.4. X-ray diffraction analysis and phase-problem solution
X-ray diffraction data extending to 3.25 Å resolution were collected on the 24-ID-E beamline at the APS synchrotron (Argonne National Laboratory, Illinois, USA). A data set consisting of 120 frames was collected with an oscillation width of 1° per frame and a crystal-to-detector distance of 450 mm (Fig. 3 ▶). Diffraction images were integrated with MOSFLM (Leslie, 1999 ▶) and scaled using SCALA (Evans, 2006 ▶) from the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶). Data-collection statistics are summarized in Table 1 ▶. An estimation of the number of molecules in the asymmetric unit (Matthews, 1968 ▶) indicated the presence of one RPdel–Rab6 complex in the asymmetric unit with a solvent content of 69.6%. A molecular-replacement search was performed with Phaser (McCoy et al., 2007 ▶) using the crystal structure of R6IP1 from the wild-type complex (PDB entry 3cwz; Recacha et al., 2009 ▶). The rotation/translation search led to a Z score of 52.7 and a final log-likelihood gain (LLG) of 3251.
Figure 3.

An X-ray diffraction image recorded from a single crystal of RPdel–Rab6 using synchrotron radiation (1° oscillation).
Table 1. Data-collection and processing statistics.
Values in parentheses are for the highest resolution shell.
| No. of crystals | 1 |
| Beamline | 24-ID-E, APS |
| Wavelength (Å) | 0.97916 |
| Detector | ADSC Quantum 315 |
| Crystal-to-detector distance (mm) | 450 |
| Rotation range per image (°) | 1 |
| Total rotation range (°) | 120 |
| Exposure time per image (s) | 1.5 |
| Resolution range (Å) | 50.0–3.25 (3.43–3.25) |
| Space group | P6122 |
| Unit-cell parameters (Å, °) | a = b = 100.1, c = 301.8, α = β = 90, γ = 120 |
| Mosaicity (°) | 0.26 |
| Total No. of measured intensities | 203233 |
| Unique reflections | 14971 |
| Multiplicity | 13.6 (14.2) |
| Mean I/σ(I) | 18.5 (5.7) |
| Completeness (%) | 99.9 (100) |
| Rmerge† (%) | 8.4 (48.2) |
| Rmeas‡ (%) | 8.7 (50.1) |
| Overall B factor from Wilson plot (Å2) | 117.9 |
R
merge = 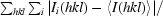
 , where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all i observations of reflection hkl.
, where Ii(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all i observations of reflection hkl.
R meas is the redundancy-independent (multiplicity-weighted) R merge.
3. Results and discussion
The engineered construct RPdel was transformed into E. coli cells and successfully expressed as a soluble protein. Moreover, gel-filtration chromatography confirmed that Rab6aQ72L (GTP-locked) and RPdel form a complex when co-expressed in cells or when purified individually and mixed together (not shown). We performed crystallization trials of the Rab6Q72L–RPdel complex to confirm the structural integrity of the engineered protein with respect to the previously reported structure of the wild-type complex (PDB entry 3cwz). The initial crystallization screens did not produce any new hits, apart from the PEG 4000/MPD custom screen. After optimization, hexagonal crystals appeared within a day and grew to full size (0.1–0.6 mm) in a few days. The crystal dimensions and morphology are very similar to those of the wild-type crystals. However, the speed of crystal growth contrasts sharply with the wild-type crystals, which took a week to appear and 4–6 weeks to reach full size. We eliminated the possibility that the longer N-terminus of the published structure (683–1061) is responsible for the alteration in the kinetics of crystal growth by generating a complex of wild-type R6IP1 (702–1057) with Rab6. Crystals of this complex also took several weeks to grow to full size (data not shown). A complete data set from a single Rab6–RPdel crystal was collected to 3.25 Å resolution on APS beamline 24-ID-E.
Initially, the expression of full-length R6IP1 (1263 residues) and a series of truncations encompassing the RUN1 and PLAT domains failed to produce soluble protein. To overcome this problem, the wild-type R6IP1–Rab6 complex was purified by co-expression of the two proteins. One of our key objectives was to generate a soluble R6IP1 construct to enable biophysical analyses of its interactions with Rab6. Our initial assessment of RPdel suggests that it is fully functional in its ability to bind Rab6. The lack of electron density in the loop bridging α3 and α4 in the structure reported previously (Recacha et al., 2009 ▶) and information from sequence alignments led us to engineer the truncation, which successfully enhances solubility (data not shown) while maintaining Rab6-binding properties. Biophysical characterization of the complex of Rab6 and the engineered effector RPdel is now feasible and we are performing kinetic and thermodynamic studies of the association. Furthermore, the PLAT domain is predicted to interact with lipid membranes (Aleem et al., 2008 ▶) and the soluble RPdel protein described here will allow biochemical characterization of effector function. Whether the truncation compromises any other functional aspect of the effector remains to be determined through cellular studies.
An additional objective was to improve the diffraction quality of the crystals through protein engineering. Unfortunately, the crystals diffracted to the same resolution as the wild-type crystals, although crystal growth proceeded much more rapidly. It is widely accepted that pruning unstructured or disordered protein elements improves the chances of crystallization (Derewenda, 2004 ▶, 2010 ▶). Indeed, in the Rab field it is common practice to delete the unstructured C-terminal region of the proteins before attempting crystallization (Derewenda, 2004 ▶). Given the lack of electron density in the segment connecting α3 and α4 (Recacha et al., 2009 ▶) and also because of its complete absence in other RUN-domain-containing homologues (Fig. 1 ▶), it was predicted that a corresponding loop truncation would retain structural integrity and possibly enhance solubility. The fact that this loop is also located in the vicinity of crystal contacts yet does not appear to contribute directly to lattice formation provided an incentive to engineer a modification of this unstructured region. This disordered loop is found to make twofold-symmetric interactions with itself along the c axis in the hexagonal space group, such that the neighbour is related by the transformation (x, x − y, −z + 1/6). By deleting the loop, we endeavoured to reduce the entropic contribution to crystal lattice formation (Price et al., 2009 ▶) and to minimize the protein conformational heterogeneity and thereby enhance the quality of the crystals (Derewenda, 2010 ▶). Additionally, a more stable loop could potentially mediate more lattice contacts either directly or possibly by extending a network of hydrogen bonds via water/solvent molecules (Salemme et al., 1988 ▶). Interestingly, the loop deletion accelerated the rate of crystal lattice formation but it is apparent that it did not have an impact on the quality of the lattice, as shown by the identical diffracting power of the crystals. These observations suggest that loops can have complicated effects on the kinetics of crystalline growth and lattice order. An understanding of the principles in order to guide loop engineering of protein crystal lattices will require further examples and detailed analyses of other systems.
Acknowledgments
We would like to thank the staff of NE-CAT at the Advanced Photon Source, Argonne, Illinois, USA for their help in the collection of X-ray diffraction data. This work was supported by the Science Foundation Ireland (grant No. 03/IN.3/B371 to AK).
References
- Aleem, A. M., Jankun, J., Dignam, J. D., Walther, M., Kühn, H., Svergun, D. I. & Skrzypczak-Jankun, E. (2008). J. Mol. Biol. 376, 193–209. [DOI] [PubMed]
- Bergbrede, T., Chuky, N., Schoebel, S., Blankenfeldt, W., Geyer, M., Fuchs, E., Goody, R. S., Barr, F. & Alexandrov, K. (2009). J. Biol. Chem. 284, 2628–2635. [DOI] [PubMed]
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Derewenda, Z. S. (2004). Methods, 34, 354–363. [DOI] [PubMed]
- Derewenda, Z. S. (2010). Acta Cryst. D66, 604–615. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fukuda, M., Kanno, E., Ishibashi, K. & Itoh, T. (2008). Mol. Cell. Proteomics, 7, 1031–1042. [DOI] [PubMed]
- Janoueix-Lerosey, I., Jollivet, F., Camonis, J., Marche, P. N. & Goud, B. (1995). J. Biol. Chem. 270, 14801–14808. [DOI] [PubMed]
- Kanno, E., Ishibashi, K., Kobayashi, H., Matsui, T., Ohbayashi, N. & Fukuda, M. (2010). Traffic, 11, 491–507. [DOI] [PubMed]
- Leslie, A. G. W. (1999). Acta Cryst. D55, 1696–1702. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Price, W. N. II et al. (2009). Nature Biotechnol. 27, 51–57.
- Recacha, R., Boulet, A., Jollivet, F., Monier, S., Houdusse, A., Goud, B. & Khan, A. R. (2009). Structure, 17, 21–30. [DOI] [PubMed]
- Salemme, F. R., Genieser, L., Finzel, B. C., Hilmer, R. M. & Wendoloski, J. J. (1988). J. Cryst. Growth, 90, 273–282.
- Stols, L., Gu, M., Dieckman, L., Raffen, R., Collart, F. R. & Donnelly, M. I. (2002). Protein Expr. Purif. 25, 8–15. [DOI] [PubMed]
- Zerial, M. & McBride, H. (2001). Nature Rev. Mol. Cell Biol. 2, 107–117. [DOI] [PubMed]