

The sequence design, purification, crystallization and preliminary X-ray diffraction analysis of the novel cap-independent translation RNA element from the 3′ untranslated region of the barley yellow dwarf virus genome is reported.
Keywords: barley yellow dwarf virus-like translation element, translation initiation, 3′ translation enhancers, RNA
Abstract
Barley yellow dwarf virus (BYDV) RNA lacks a 5′ m7GTP cap, yet it is translated efficiently because it contains a 105-base BYDV-like cap-independent translation element (BTE) in the 3′ untranslated region (UTR). To understand how the BTE outcompetes the host mRNA for protein-synthesis machinery, its three-dimensional structure is being determined at high resolution. The purification using transcription from DNA containing 2′-O-methyl nucleotides and preliminary crystallographic analyses of the BTE RNA are presented here. After varying the BTE sequence and crystallization-condition optimization, crystals were obtained that diffracted to below 5 Å resolution, with a complete data set being collected to 6.9 Å resolution. This crystal form indexes with an R merge of 0.094 in the monoclinic space group C2, with unit-cell parameters a = 316.6, b = 54.2, c = 114.5 Å, α = γ = 90, β = 105.1°.
1. Introduction
Translation initiation is the first and the most regulated step of protein synthesis (Sonenberg & Hinnebusch, 2009 ▶; Jackson et al., 2010 ▶). The 40S ribosomal subunit complexed with tRNA and initiation factors is recruited to the mRNA by the eukaryotic initiation factor 4F (eIF4F) complex (Gingras et al., 1999 ▶; Marintchev et al., 2009 ▶). Plant eIF4F consists of the large scaffolding protein eIF4G and the cap-binding protein eIF4E (Browning, 2004 ▶). Unlike host mRNAs, many viral RNAs lack the 5′ m7GTP cap that is normally required for eIF4F to bind the mRNA and yet these viral RNAs are translated efficiently (Doudna & Sarnow, 2007 ▶). The 3′ UTRs of many plant viral RNAs contain cap-independent translation elements (CITEs) that facilitate efficient translation initiation at the 5′ end of the RNA (Kneller et al., 2006 ▶; Miller et al., 2007 ▶).
Among the best characterized and most efficient 3′ CITEs is that of barley yellow dwarf virus (BYDV) and related viruses (Guo et al., 2001 ▶). BYDV-like translation elements (BTEs) display diverse secondary structures (Wang et al., 2010 ▶), but they share the following features: (i) a long basal helix (S-IV in the BYDV BTE) from which a variable number of helices radiate, (ii) a 17-nucleotide consensus sequence (17 nt CS), GGAUCCUGGGAAACAGG, that forms a stem-loop topped by a GNRNA pentaloop, (iii) a stable stem-loop (SL-III in the BYDV BTE) topped by a loop that base-pairs to the 5′ UTR (Guo et al., 2001 ▶) and (iv) variable numbers of essential but nonconserved non-Watson–Crick paired bases around the junction of the helices (Wang et al., 2010 ▶). To facilitate translation, the BTE binds and requires eIF4G but not eIF4E (Treder et al., 2008 ▶). To understand how the BTE interacts with the translational machinery, we are determining its structure. Here, we describe the purification, crystallization and preliminary X-ray diffraction analysis of BYDV BTE crystals in our attempt to determine the first three-dimensional structure of a 3′ CITE at near-atomic resolution.
2. Materials and methods
2.1. T7 RNA polymerase purification
T7 RNA polymerase (with a His6 tag) was purified by modifying the procedure of Grodberg & Dunn (1988 ▶). Briefly, three BL21/pT7-911 colonies (Ichetovkin et al., 1997 ▶) were suspended in 1 ml 10 mM Tris pH 7.5, 10 mM MgSO4. 100 µl of this suspension was added to 500 ml M9TB expression medium [1%(v/v) tryptone, 100 mM NaCl, 40 mM ammonium sulfate, 44 mM KH2PO4, 100 mM Na2HPO4, 0.1 mM CaCl2, 1 mM MgSO4, 0.2% glycerol per 500 ml]. The culture was grown overnight at 303 K until A 600 reached 0.5–0.6.
To induce T7 RNA polymerase expression, 0.5 ml 1 M IPTG was added to the bacterial culture and the cells were harvested 3 h later. The pelleted cells were resuspended in 25 ml lysis buffer [50 mM Tris pH 8, 100 mM NaCl, 5%(v/v) glycerol, 5 mM β-mercaptoethanol, 1 mM imidazole] supplemented with lysozyme and one protease-inhibitor cocktail minitablet (Roche) per pellet. Each pellet was lysed by sonication and centrifuged (15 500 rev min−1, 30 min, 277 K). The supernatant was applied onto an Ni–NTA column (Qiagen) equilibrated with 15 ml lysis buffer. After washing with 15 ml lysis buffer supplemented with 10 mM imidazole, bound protein was eluted with lysis buffer supplemented with 500 mM imidazole. Eluted T7 RNA polymerase was dialyzed into storage buffer [20 mM KHPO4 pH 7.5, 100 mM NaCl, 50%(v/v) glycerol, 1 mM DTT, 0.1 mM EDTA, 0.2% NaN3].
2.2. RNA purification
Templates to transcribe the BYDV BTE sequence variants used in this study (Fig. 1 ▶ a) were prepared by PCR using an upstream oligonucleotide containing either class III T7 promoter (TAATACGACTCACTATAG) for the BTE RNA transcripts that start with GTP or a class II T7 promoter (Coleman et al., 2004 ▶) (TAATACGACTCACTATTA) for the non-GTP-initiating transcripts. The downstream oligonucleotide contained two 2′-O-methyl RNA bases in place of the deoxyribonucleotides at the 5′ end to prevent the addition of nontemplated 3′ nucleotides by the polymerase (Fig. 1 ▶ b; Kao et al., 1999 ▶). DNA was amplified in a 1 ml PCR reaction [100 ng plasmid DNA p5′UTR-LUC-TE869 (Guo et al., 2000 ▶), 0.3 mM of each dNTP, 2.5 mM MgCl2, 0.4 µM primers, 1× High Fidelity PCR buffer and 10 U Platinum Taq DNA polymerase (Invitrogen)] for 30 cycles of 1 min at 367 K, 40 s at 329 K and 90 s at 343 K and a final extension for 6 min at 343 K.
Figure 1.

(a) The secondary structure of the construct 87c BTE used in crystallization trials is shown with deleted wild-type bases in gray and base substitutions in red. The 17 nt conserved sequence (17 nt CS) is highlighted in italics. Bold bases participate in long-distance base-pairing to the 5′ UTR. (b) The production of homogeneous T7 RNA polymerase transcripts by PCR, fusing a primer with modified 5′-terminal bases, followed by transcription with T7 polymerase. (c) 10% denaturing PAGE of purified BTE transcripts. Lane M, low-range ssRNA ladder (New England Biolabs); lane 1, BTE105 RNA; lane 2, 87c BTE. (d) Folding of 87c BTE assessed by 10% native PAGE containing 10 mM HEPES–KOH pH 7.5 and either 0 mM (lane 1) or 8 mM (lane 2) magnesium chloride.
The gel-purified PCR product (0.7 µM) was incubated in 1× transcription buffer (30 mM Tris–HCl pH 8.1, 2 mM spermidine, 0.01% Triton X-100 and 10 mM DTT), 25 mM MgCl2, 5 mM of each NTP and 50 µg ml−1 T7 RNA polymerase for 2 h at 310 K. Following transcription, RNA was phenol–chloroform extracted and precipitated using a half volume of 7.5 M ammonium acetate and two volumes of ethanol. The dried product of a 1 ml transcription reaction was resuspended in 500 µl nuclease-free water (Ambion) and passed through a Micro Bio-Spin 30 column (Bio-Rad) equilibrated with nuclease-free water. The integrity of the eluted RNA sample was verified by 10% denaturing polyacrylamide gel electrophoresis (Fig. 1 ▶ c).
2.3. Crystallization and data collection
Prior to crystallization screening, BTE RNA was heated in water to 348 K followed by slow cooling to 298 K over 3 h to drive RNA folding to its lowest energy native conformation. The RNA solution was subjected to crystal screening using Natrix and Nucleic Acid Mini (NAM) screens from Hampton Research. 1 µl 0.3 mM BTE RNA was mixed with an equal volume of reservoir solution and equilibrated against 500 µl well solution using hanging-drop vapor diffusion at 298 K.
Crystallization screening and initial imaging were performed at the Iowa State University X-ray Crystallography Facility. BTE RNA crystals were soaked for 30 s in a solution consisting of 25%(w/v) glycerol in Natrix 26 solution and flash-cooled in liquid nitrogen before data collection. Initial diffraction images were collected from a single crystal plate at the Advanced Light Source (beamline 4.2.2) at 100 K and a wavelength of 0.979 Å using a CCD detector. Images were recorded with 1 s exposure at a crystal-to-detector distance of 200 mm using an oscillation range of 1°. According to analysis with Phaser (McCoy et al., 2007 ▶), the most probable number of molecules in the asymmetric unit is six, with a Matthews coefficient of 2.55 Å3 Da−1 and a solvent content of 51.77%. Diffraction data were processed using XDS (Kabsch, 2010 ▶).
3. Results and discussion
3.1. RNA construct design and preparation
To ensure the complete homogeneity of BTE RNA while allowing the rapid purification of multiple constructs, we developed a PCR scheme to generate the transcription template. The upstream primer contained either the common class III T7 RNA polymerase promoter for guanosine-initiating transcripts or a class II T7 promoter for adenosine-initiating transcripts. The class II T7 promoter gives superior 5′-end homogeneity (Coleman et al., 2004 ▶) and produced yields comparable to those obtained with the class III T7 promoter. Using a downstream primer containing 2′-methoxy modifications at its 5′ end prevented 3′-end heterogeneity (Kao et al., 1999 ▶). This approach allowed the rapid production of clean, homogeneous and acrylamide-free RNA samples suitable for structural studies as demonstrated by the 5 Å resolution diffracting crystals.
Crystalline material obtained from wild-type BTE105 RNA did not diffract X-rays. Therefore, we tested variant sequences designed to enhance crystallization while taking care to maintain BTE functionality. The functional construct 87c BTE RNA (Guo et al., 2000 ▶), in which the terminal 24 bases of the basal helix were truncated and a GC-rich ‘clamp’ was appended to the ends (Fig. 1 ▶ a), gave many crystals. Natrix solution 26 (0.05 M sodium cacodylate pH 6.5, 0.2 M potassium chloride, 0.1 M magnesium acetate and 10% PEG 8000) yielded plates nucleating from a common center (‘RNA flowers’) at 298 K two weeks after setup (Fig. 2 ▶ a). These BTE crystals diffracted to >25 Å resolution and were subjected to additive screening (Hampton Research) for optimization. Single crystal plates formed in 4% 1,3-propanediol and 0.025% dichloromethane diffracted to <10 Å resolution 3–5 d after screen setup (Fig. 2 ▶ b). Finer additive screening with 1,3-propanediol and dichloromethane, where the percentages varied in 1% increments from 3 to 10% for 1,3-propanediol and 0.01% increments from 0.005 to 0.065% for dichloromethane, yielded large RNA plates (Fig. 2 ▶ c) that diffracted to <5 Å resolution (Fig. 3 ▶).
Figure 2.

Typical crystals of 87c BTE obtained using Natrix 26 [0.2 M potassium chloride, 0.1 M magnesium acetate tetrahydrate, 0.05 M sodium cacodylate trihydrate pH 6.5, 10%(w/v) polyethylene glycol 8000] and (a) no additive (dimensions of 0.54 × 0.08 × 0.01 mm), (b) 6% 1,3-propanediol (dimensions of 0.2 × 0.02 × <0.01 mm in diameter) and (c) 0.045% dichloromethane (dimensions of 0.4 × 0.06 × 0.01 mm).
Figure 3.

X-ray diffraction pattern for the 87c BTE crystals, showing diffraction extending to approximately 6.9 Å resolution.
3.2. Crystallization screening and diffraction experiments
Crystal screening of BTE RNA sequences using commercial sparse-matrix screens revealed a preference for positively charged ions, especially magnesium and potassium, lower percentages of PEG as precipitant and a wide pH range. It is no surprise that magnesium was a critical ion in all conditions that generated BTE RNA crystals, because magnesium often facilitates ion-dependent RNA folding by neutralizing negatively charged phosphates (Woodson, 2005 ▶). The best diffracting 87c BTE RNA crystals formed large thin plates (>0.4 mm in length and 0.01 mm in diameter) in the presence of small organic molecules within 3 d that consistently diffracted to below 5 Å resolution.
The large 87c BTE crystal plates were fragile and difficult to cryoprotect, resulting in radiation damage that weakened diffraction below 7 Å resolution. Despite the crystal damage, we processed the data using the XDS program package to obtain the statistics summarized in Table 1 ▶. To generate better diffracting crystals, we will vary the sequence of the BTE RNA. The promise of this approach is shown by the significant improvement obtained by altering BTE105 to 87c BTE. Also, native PAGE analysis revealed that 8 mM magnesium chloride relieves the conformational heterogeneity of 87c BTE and makes the BTE adopt a more compact conformation, as indicated by its faster mobility (Fig. 1 ▶ d, lane 2). Hence, to ensure structural homogeneity of the RNA we modified the folding step by dialyzing purifed RNA into buffer containing 8 mM magnesium chloride and 10 mM HEPES–KOH pH 7.5 prior to dilution with reservoir solution.
Table 1. Data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Beamline | MBC 4.2.2 |
| Wavelength (Å) | 0.979 |
| Space group | C2 |
| Unit-cell parameters (Å, °) | a = 316.6, b = 54.2, c = 114.5, α = γ = 90, β = 105.1 |
| Measured reflections | 10409 |
| Unique reflections | 3157 |
| Resolution range (Å) | 21.69–6.86 (7.27–6.86) |
| Completeness (%) | 94.5 (87.3) |
| Multiplicity | 3.3 (3.0) |
| Rmerge† | 0.094 (0.54) |
| Average I/σ(I) | 8.4 (1.9) |
R
merge = 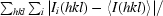
 .
.
In addition to optimizing BTE RNA crystal growth, we are pursuing different conditions of dehydration and crystal annealing in order to extend the diffraction limit of the crystals that we have generated so far. Preliminary cryoscreening using the CryoPro kit (Hampton Research) indicated that 10–30%(v/v) MPD causes no crystal damage even after a prolonged 60 min incubation.
For phasing, we will soak RNA crystals with heavy-atom lanthanide-series compounds which bind to RNA at magnesium sites (Cate & Doudna, 1996 ▶; Feig et al., 1998 ▶) and crystallize similarly to native RNA. Because there is no certainty that the metal will bind at the desired sites, we may position G·U pairs in helices to form a cation-binding pocket lined with negatively charged phosphate groups (Keel et al., 2007 ▶). The BTEs already contain several G·U pairs surrounded by Watson–Crick pairs that could be mutated to fit the consensus phasing sequence (Keel et al., 2007 ▶; Edwards et al., 2009 ▶) while maintaining the structure and function of the BTE prior to heavy-atom screening.
This research may provide fundamental insight into mechanisms of translation factor (and ultimately ribosome) recruitment by eukaryotic mRNAs. We are encouraged by initial incremental success in crystallizing the native RNA and therefore plan to pursue phasing and higher resolution data collection for structure determination.
Acknowledgments
This work was supported by NIH grant R01 GM067104 with an American Recovery and Reinvestment Act supplement. We thank Jay Nix at the Advanced Light Source of Lawrence Berkeley National Laboratory for assistance with data collection, Jennifer Doudna for advice, Mark Hargrove for laboratory support and Jen Kaiser for assistance with data processing.
References
- Browning, K. S. (2004). Biochem. Soc. Trans. 32, 589–591. [DOI] [PubMed]
- Cate, J. H. & Doudna, J. A. (1996). Structure, 4, 1221–1229. [DOI] [PubMed]
- Coleman, T. M., Wang, G. & Huang, F. (2004). Nucleic Acids Res. 32, e14. [DOI] [PMC free article] [PubMed]
- Doudna, J. A. & Sarnow, P. (2007). In Translational Control in Biology and Medicine, edited by M. B. Mathews, N. Sonenberg & J. Hershey. New York: Cold Spring Harbor Laboratory Press.
- Edwards, A. L., Garst, A. D. & Batey, R. T. (2009). Methods Mol. Biol. 535, 135–163. [DOI] [PMC free article] [PubMed]
- Feig, A. L., Scott, W. G. & Uhlenbeck, O. C. (1998). Science, 279, 81–84. [DOI] [PubMed]
- Gingras, A. C., Raught, B. & Sonenberg, N. (1999). Annu. Rev. Biochem. 68, 913–963. [DOI] [PubMed]
- Grodberg, J. & Dunn, J. J. (1988). J. Bacteriol. 170, 1245–1253. [DOI] [PMC free article] [PubMed]
- Guo, L., Allen, E. & Miller, W. A. (2000). RNA, 6, 1808–1820. [DOI] [PMC free article] [PubMed]
- Guo, L., Allen, E. M. & Miller, W. A. (2001). Mol. Cell, 7, 1103–1109. [DOI] [PubMed]
- Ichetovkin, I. E., Abramochkin, G. & Shrader, T. E. (1997). J. Biol. Chem. 272, 33009–33014. [DOI] [PubMed]
- Jackson, R. J., Hellen, C. U. & Pestova, T. V. (2010). Nature Rev. Mol. Cell Biol. 11, 113–127. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kao, C., Zheng, M. & Rüdisser, S. (1999). RNA, 5, 1268–1272. [DOI] [PMC free article] [PubMed]
- Keel, A. Y., Rambo, R. P., Batey, R. T. & Kieft, J. S. (2007). Structure, 15, 761–772. [DOI] [PMC free article] [PubMed]
- Kneller, E. L., Rakotondrafara, A. M. & Miller, W. A. (2006). Virus Res. 119, 63–75. [DOI] [PMC free article] [PubMed]
- Marintchev, A., Edmonds, K. A., Marintcheva, B., Hendrickson, E., Oberer, M., Suzuki, C., Herdy, B., Sonenberg, N. & Wagner, G. (2009). Cell, 136, 447–460. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Miller, W. A., Wang, Z. & Treder, K. (2007). Biochem. Soc. Trans. 35, 1629–1633. [DOI] [PMC free article] [PubMed]
- Sonenberg, N. & Hinnebusch, A. G. (2009). Cell, 136, 731–745. [DOI] [PMC free article] [PubMed]
- Treder, K., Kneller, E. L., Allen, E. M., Wang, Z., Browning, K. S. & Miller, W. A. (2008). RNA, 14, 134–147. [DOI] [PMC free article] [PubMed]
- Wang, Z., Kraft, J. J., Hui, A. Y. & Miller, W. A. (2010). Virology, 402, 177–186. [DOI] [PMC free article] [PubMed]
- Woodson, S. A. (2005). Curr. Opin. Chem. Biol. 9, 104–109.