

Mushroom tyrosinase was crystallized in two different space groups. The crystals diffracted to 3.0 Å resolution (P21) or 2.6 Å resolution (P21212).
Keywords: tyrosinase, Agaricus bisporus
Abstract
Tyrosinase catalyzes the conversion of tyrosine to dihydroxyphenylalanine quinone, which is the main precursor for the biosynthesis of melanin. The enzyme from Agaricus bisporus, the common button mushroom, was purified and crystallized in two different space groups. Crystals belonging to space group P21 (unit-cell parameters a = 104.2, b = 105.0, c = 119.1 Å, β = 110.6°, four molecules per asymmetric unit) diffracted to 3.0 Å resolution. Crystals belonging to space group P21212 (unit-cell parameters a = 104.0, b = 104.5, c = 108.4 Å, two molecules per asymmetric unit) diffracted to 2.6 Å resolution. It was essential to include 5 mM HoCl3 in all crystallization conditions in order to obtain well diffracting crystals.
1. Introduction
Tyrosinase (EC 1.14.18.1) is a copper-containing monooxygenase that is widely distributed in nature, occurring in organisms from all phyla. It catalyzes the o-hydroxylation of monophenols (monophenolase activity) and the subsequent oxidation of the o-diphenols formed to the corresponding o-quinones (diphenolase activity) (Van Gelder et al., 1997 ▶), which are precursors for the biosynthesis of melanins. The precise physiological function of the enzyme is not well understood. In mammals, including humans, the enzyme is mainly found in melanocytes, which are pigment-producing cells. The mammalian enzyme has been implicated in pigmentary diseases such as albinism, vitiligo and other melanin-related syndromes (Cesarini, 1996 ▶; Hearing, 2005 ▶).
In the mushroom Agaricus bisporus, tyrosinase has been proposed to be involved in defence mechanisms against pathogens because of the bacteriostatic properties of o-quinones and melanins and because of the inducibility of at least one tyrosinase isoform upon microbial challenge (Soler-Rivas et al., 1999 ▶). The enzyme is also an important factor in the browning of the mushroom upon aging and microbial infection (Soler-Rivas et al., 1999 ▶) and consequently there is substantial interest in its catalytic properties (Espin, Varon et al., 2000 ▶; Fenoll et al., 2002 ▶; Sánchez-Ferrer et al., 1995 ▶) and its inhibition (Kubo et al., 2000 ▶; Ley & Bertram, 2001 ▶; Zhang et al., 2006 ▶).
To date, four genes coding for A. bisporus tyrosinase have been described. These genes, Abppo1 (UniProt protein-sequence database entry Q00024) and Abppo2 (O42713) (Wichers et al., 2003 ▶), and Abppo3 (C7FF04) and Abppo4 (C7FF05) (Wu et al., 2010 ▶), code for proteins with a molecular mass of 64–67 kDa. However, isolation of the enzyme directly from mushroom fruit bodies yields a heterotetramer composed of two subunits of 43–48 kDa (H chain) and two subunits of 13–14 kDa (L chain) (H2L2 configuration; Strothkamp et al., 1976 ▶). This information led to the hypothesis that tyrosinase is initially formed as a monomeric 64 kDa latent precursor which can be proteolytically activated, resulting in ∼43 kDa active forms (Espin, Soler-Rivas et al., 2000 ▶). The nature of the L chain is not known. Although the L chain may represent the C-terminal fragment resulting from proteolytic cleavage of the latent precursor, it may also be an unrelated protein that is copurified with active tyrosinase and that is difficult to remove (Flurkey & Inlow, 2008 ▶).
X-ray absorption studies have indicated that A. bisporus tyrosinase contains two copper ions in its active site with spectroscopic properties resembling those of the two copper ions found in molluscan haemocyanin, a copper-containing oxygen-carrier protein (Della Longa et al., 1996 ▶). The presence of two copper ions in tyrosinase has also been shown in the crystal structure of Streptomyces castaneoglobisporus tyrosinase (Matoba et al., 2006 ▶), with each of the two copper ions being coordinated by three histidine side chains. However, in A. bisporus tyrosinase the side chain of one of these copper-coordinating histidine residues is covalently linked via a thioether bond to the side chain of the preceding cysteine residue in the C-X-H sequence motif (Van Gelder et al., 1997 ▶). The role of this thioether bond remains elusive. Here, we report the purification and crystallization of commercially available mushroom tyrosinase and preliminary X-ray diffraction data analysis as an initial step towards the elucidation of the three-dimensional structure of the enzyme.
2. Materials and methods
2.1. Sample preparation
Mushroom tyrosinase powder was purchased from Sigma (Sigma–Aldrich, catalogue No. T3824-250KU) and was purified at 277 K essentially as described previously (Schurink et al., 2007 ▶). The mushroom tyrosinase powder was dissolved in 50 mM sodium phosphate buffer pH 6.5 to a concentration of 10 mg ml−1. Insoluble material was removed by centrifugation at 277 K and 12 000g for 15 min. The supernatant was carefully recovered and was loaded onto a 10/30 Superdex S-200 column (Amersham Biosciences) equilibrated with 100 mM HEPES buffer pH 7.5 containing 10 mM calcium chloride. Elution from the gel-filtration column was carried out with the same buffer at a constant flow rate of 0.25 ml min−1. Fractions with tyrosinase activity were pooled and concentrated with concomitant buffer exchange to 10 mM HEPES pH 7.5 over a 10 kDa cutoff Amicon membrane (Millipore) to a final protein concentration of approximately 6 mg ml−1. The concentrated protein was either directly subjected to crystallization experiments or frozen at 253 K for storage.
2.2. Determination of protein concentration and enzyme activity
The protein concentration was estimated from the absorption at 280 nm using the experimentally determined ∊ value of 132 000 M −1 cm−1. Absorbance measurements were performed with a Nanodrop ND-1000 spectrometer (Nanodrop Technologies Inc., Wilmington, USA).
Enzyme activity was measured at 298 K by a colorimetric method essentially as described previously (Espin et al., 1997 ▶) using 3,4-dihydroxyphenylalanine (l-DOPA) as substrate. One unit of tyrosinase activity is defined as the formation of 1 µmol l-DOPA quinone per minute. Briefly, in a 1 ml cuvette, 100 µl of a 5 mM 3-methyl-2-benzothiazolinone hydrazone (MBTH) solution in water was mixed with 800 µl l-DOPA solution (18 mM in 50 mM sodium phosphate buffer pH 6.5). The reaction was started by the addition of 100 µl enzyme solution to the reaction mixture. The increase in the absorbance at 484 nm was followed during the first 3 min. The measurements were carried out in an Ultrospec 4300 Pro spectrometer equipped with a temperature-control unit (Amersham Biosciences) interfaced online to a computer. The spectra were recorded and processed with the time-drive measurement module (SWIFT II program, v.2.01, Amersham Biosciences).
2.3. Characterization of protein samples
The purity of the enzyme was routinely checked by SDS–PAGE using a PhastGel System (Amersham Biosciences) with pre-cast SDS gels and electrode strips. Staining was conducted in the same system using silver nitrate as the staining solution.
The temperature-stability of the protein in solution was analyzed by thermal shift assay (Ericsson et al., 2006 ▶). Briefly, 5 µl 200-fold diluted SYPRO Orange solution (Invitrogen, Breda, The Netherlands) as a molecular probe, 5 µl 4.5 mg ml−1 enzyme solution in water, 2.5 µl 1 M buffer stock solution and 2.5–5 µl additive stock solution were mixed in a 96-well thin-wall PCR plate (Bio-Rad). The final reaction volume was adjusted to 25 µl by adding an appropriate volume of filtered and degassed Milli-Q water. The plate was sealed with optical quality sealing tape (Bio-Rad Laboratories BV, Veenendaal, The Netherlands). The sealed plate was inserted into a real-time PCR machine (iCycler, Bio-Rad Laboratories BV, Veenendaal, The Netherlands) and was heated from 293 to 363 K with a 0.5 K increment per 20 s. The changes in fluorescence of the SYPRO Orange probe were monitored with a fluorescence detector (MyIQ single-colour RT-PCR detection system, Bio-Rad) at excitation and emission wavelengths of 490 and 575 nm, respectively.
The monodispersity of the enzyme was analyzed by dynamic light scattering using a DynaPro dynamic light-scattering instrument (Wyatt Technologies, Santa Barbara, USA). The protein sample (1 mg ml−1) was centrifuged at 12 000g for 15 min at 277 K and the supernatant was carefully transferred into a DLS cuvette. Measurements were performed at both 298 and 277 K.
2.4. Crystallization procedures
Initial screening for crystallization conditions was carried out at 298 K by the sitting-drop vapour-diffusion method employing an Oryx-6 crystallization robot (Douglas Instruments, Hungerford, England) equipped with an anti-evaporation shielding plate. The following commercial screens were used in 96-well CrystalClear strips for sitting-drop setups (Hampton Research): Structure Screens I and II (Molecular Dimensions, Apopka, USA), Cryoscreens I and II (Emerald BioSystems, Bainbridge Island, USA) and the JCSG+ Suite (Qiagen, Venlo, The Netherlands). The starting volume of the drop was 0.3 µl, consisting of 0.12 µl protein solution and 0.18 µl reservoir solution. Optimization of promising crystallization conditions was performed with the hanging-drop vapour-diffusion method, varying the precipitant type and concentration, the temperature and the pH and using additives.
2.5. Data collection and processing
Prior to X-ray diffraction analysis, crystals were transferred into a cryoprotectant solution consisting of 15% PEG 4000, 20% PEG 400, 1 mM HoCl3 and 100 mM sodium acetate buffer pH 4.6 before flash-freezing them in liquid nitrogen. X-ray diffraction experiments were carried out on beamlines BM16, ID23-2 and ID14-4 at the ESRF (Grenoble, France). Diffraction data were recorded at 100 K with the CCD detector available at the beamline with 0.5–1.0° oscillations. The data sets were processed using either XDS (Kabsch, 2010 ▶) or MOSFLM (Leslie, 2006 ▶). Scaling and merging were performed with SCALA (Evans, 2006 ▶), which is part of the CCP4 program suite (Collaborative Computational Project, Number 4, 1994 ▶).
3. Results and discussion
Commercially available mushroom tyrosinase was successfully purified using a single gel-filtration step. In the elution profile, tyrosinase activity was detected in a peak with a retention time corresponding to a molecular mass of ∼120 kDa, as expected for the mass of the H2L2 form of mushroom tyrosinase (Strothkamp et al., 1976 ▶). Furthermore, SDS–PAGE analysis of this peak confirmed the presence of the heavy (H) and light (L) chains, with molecular weights of 45 and 14 kDa, respectively, which is in agreement with previous reports (Schurink et al., 2007 ▶). Analysis of the protein content and specific activity indicated that approximately 50% of the total protein in the mushroom tyrosinase powder consisted of tyrosinase. The purification scheme recovered up to 90% of the enzyme activity.
The thermostability of tyrosinase was investigated by following the unfolding of the protein as a function of temperature under various conditions (Ericsson et al., 2006 ▶). A shift in the melting temperature (T m) of the protein to higher values indicates an increase in thermostability. Fig. 1 ▶(a) shows that the enzyme demonstrated the highest T m value in 100 mM HEPES buffer pH 7.5, 10 mM CaCl2. In the absence of calcium or when the calcium was replaced by sodium the T m value shifted to lower temperatures. Based on this observation, 100 mM HEPES buffer pH 7.5 with 10 mM CaCl2 was chosen for the purification.
Figure 1.

Stability and monodispersity of tyrosinase in solution. (a) Thermal unfolding of tyrosinase in 100 mM HEPES buffer pH 7.5 (black line), in 100 mM HEPES buffer with an additional 10 mM calcium chloride (circles) or in 100 mM HEPES buffer with an additional 10 mM sodium chloride (crosses). (b) Monodispersity of tyrosinase in 100 mM HEPES buffer pH 7.5 containing 10 mM CaCl2 at 298 K.
To assess the aggregation behaviour of tyrosinase, a dynamic light-scattering experiment was performed. Without applying any noise reduction or peak filtering, the fit on mass showed one main peak with a mean R h of 4.4 nm and 6% polydispersity, indicating a monodisperse solution at 298 K (Fig. 1 ▶ b). The calculated mass of this peak was 110 kDa, which is in agreement with the results of size-exclusion chromatography, and confirmed the presence of an H2L2 tetramer in solution (Strothkamp et al., 1976 ▶). Although some noise was observed owing to protein aggregation, it resulted from less than 0.1% of the mass in the sample. Because the monodispersity of the enzyme solution was reduced at 277 K (data not shown), subsequent crystallization experiments were carried out at 298 K.
Initial crystals were obtained with the JCSG+ screen after 48 h at 298 K using a reservoir solution composed of 8% PEG 4000 in 100 mM sodium acetate buffer pH 4.6. However, these crystals only diffracted to ∼8 Å resolution (BM16, ESRF, Grenoble). Optimization using the hanging-drop vapour-diffusion method, followed by an extensive additive screen, revealed that 5 mM HoCl3 improved the diffraction quality of the crystals. After this discovery, plate-shaped crystals suitable for X-ray data collection (Fig. 2 ▶) were routinely obtained from drops consisting of 2 µl protein solution (∼6 mg ml−1) and 2 µl reservoir solution, which were suspended over 800 µl reservoir solution consisting of 10%(w/v) PEG 4000, 100 mM sodium acetate buffer pH 4.6 and 5 mM HoCl3. Crystals with reasonable size (200 × 200 × 50 µm) could be obtained within 4–5 d. They diffracted to 2.6 Å resolution using synchrotron radiation (ESRF, Grenoble). Finally, it appeared to be necessary to include 1 mM HoCl3 in the cryoprotectant solution for reproducible diffraction by the crystals.
Figure 2.

Plate-shaped crystals of mushroom tyrosinase.
The crystals obtained were analyzed by X-ray diffraction (Table 1 ▶). They belonged to either space group P21 or space group P21212. Crystals with space group P21 had unit-cell parameters a = 104.2, b = 105.0, c = 119.1 Å, β = 110.6° and diffracted to 3.0 Å resolution. Crystals with space group P21212 had unit-cell parameters a = 104.0, b = 104.5, c = 108.4 Å and diffracted to 2.6 Å resolution. Assuming the presence of four 60 kDa HL heterodimers per asymmetric unit in the P21 crystals and two HL heterodimers per asymmetric unit in the P21212 crystals gave similar values for the Matthews coefficient and solvent content of ∼2.4 Å3 Da−1 and 49%, respectively. Data-collection statistics are summarized in Table 1 ▶.
Table 1. Summary of crystallographic data collection and processing.
Values in parentheses are for the highest resolution shell.
| Native P21 | Native P21212 | |
|---|---|---|
| Beamline | ESRF ID23-2 | ESRF ID14-4 |
| Wavelength (Å) | 0.8726 | 0.9395 |
| Resolution (Å) | 50.0–3.00 | 54.2–2.60 |
| Unit-cell parameters (Å, °) | a = 104.2, b = 105.0, c = 119.1, β = 110.6 | a = 104.0, b = 104.5, c = 108.4, β = 90 |
| Rmerge† | 0.16 (0.34) | 0.12 (0.45) |
| Rp.i.m.† | 0.11 (0.24) | 0.07 (0.28) |
| Mean I/σ(I) | 7.3 (2.3) | 8.2 (2.3) |
| Completeness (%) | 88.9 (90.5) | 94.8 (81.8) |
| Multiplicity | 2.8 (2.9) | 4.0 (2.7) |
R
merge = 
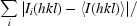
 ; R
p.i.m. =
; R
p.i.m. = 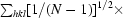
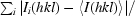 , where Ii(hkl) is the integrated intensity of a reflection, 〈I(hkl)〉 is the mean intensity of multiple corresponding symmetry-related reflections and N is the multiplicity of the given reflections.
, where Ii(hkl) is the integrated intensity of a reflection, 〈I(hkl)〉 is the mean intensity of multiple corresponding symmetry-related reflections and N is the multiplicity of the given reflections.
An SDS–PAGE analysis of a tyrosinase crystal showed the presence of both the 45 and 14 kDa polypeptides, indicating that both the H and L chains are present in the crystal. Furthermore, an X-ray fluorescence scan of a tyrosinase crystal grown in the absence of HoCl3 showed the presence of copper in the protein crystal (Fig. 3 ▶). Finally, significant tyrosinase activity was observed after dissolving a crystal that had been exposed to X-rays. Since the activity of the enzyme relies on the presence of the copper in the active site, this result not only confirmed the presence of copper but also indicates that the copper is still functional. Thus, we have crystallized active mushroom tyrosinase. We are currently trying to solve the structure of the enzyme by molecular replacement with multiple-crystal averaging using as search models the structures of the core domain of Octopus dofleini haemocyanin (PDB entry 1js8; Cuff et al., 1998 ▶) and S. castaneoglobisporus tyrosinase (PDB entry 1wx2; Matoba et al., 2006 ▶), which have sequence identities of 19% and 20% for 274 and 275 aligned amino acids, respectively.
Figure 3.

Energy scan of a tyrosinase crystal. The peak occurs at an energy of ∼8.995 ± 0.001 keV, which is specific for copper in tyrosinase (Della Longa et al., 1996 ▶).
Acknowledgments
We thank the beamline scientists at BM16, ID23-2 and ID14-4 (ESRF, Grenoble, France) for their support during data collection. This work was funded by the Innovation-driven Research Program for Industrial Proteins (project No. IIE00022). Data collection at the ESRF was supported by the ESRF and by the European Molecular Biology Laboratory (EMBL) under the European Community’s Seventh Framework Program (FP7/2007–2013, grant agreement No. 226716).
References
- Cesarini, J. P. (1996). Adv. Space Res. 18, 35–40.
- Collaborative Computational Project, Number 4 (1994). Acta Cryst. D50, 760–763.
- Cuff, M. E., Miller, K. I., van Holde, K. E. & Hendrickson, W. A. (1998). J. Mol. Biol. 278, 855–870. [DOI] [PubMed]
- Della Longa, S., Ascone, I., Bianconi, A., Bonfigli, A., Castellano, A. C., Zarivi, O. & Miranda, M. (1996). J. Biol. Chem. 271, 21025–21030. [DOI] [PubMed]
- Ericsson, U. B., Hallberg, B. M., Detitta, G. T., Dekker, N. & Nordlund, P. (2006). Anal. Biochem. 357, 289–298. [DOI] [PubMed]
- Espin, J. C., Morales, M., Garcia-Ruiz, P. A., Tudela, J. & Garcia-Canovas, F. (1997). J. Agric. Food Chem. 45, 1084–1090.
- Espin, J. C., Soler-Rivas, C. & Wichers, H. J. (2000). Mushroom Sci. 15, 79–86.
- Espin, J. C., Varon, R., Fenoll, L. G., Gilabert, M. A., Garcia-Ruiz, P. A., Tudela, J. & Garcia-Canovas, F. (2000). Eur. J. Biochem. 267, 1270–1279. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Fenoll, L. G., Rodríguez-López, J. N., García-Molina, F., García-Cánovas, F. & Tudela, J. (2002). IUBMB Life, 54, 137–141. [DOI] [PubMed]
- Flurkey, W. H. & Inlow, J. K. (2008). J. Inorg. Biochem. 102, 2160–2170. [DOI] [PubMed]
- Gelder, C. W. van, Flurkey, W. H. & Wichers, H. J. (1997). Phytochemistry, 45, 1309–1323. [DOI] [PubMed]
- Hearing, V. J. (2005). J. Dermatol. Sci. 37, 3–14. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kubo, I., Kinst-Hori, I., Kubo, Y., Yamagiwa, Y., Kamikawa, T. & Haraguchi, H. (2000). J. Agric. Food Chem. 48, 1393–1399. [DOI] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Ley, J. P. & Bertram, H. J. (2001). Bioorg. Med. Chem. 9, 1879–1885. [DOI] [PubMed]
- Matoba, Y., Kumagai, T., Yamamoto, A., Yoshitsu, H. & Sugiyama, M. (2006). J. Biol. Chem. 281, 8981–8990. [DOI] [PubMed]
- Sánchez-Ferrer, A., Rodríguez-López, J. N., García-Cánovas, F. & García-Carmona, F. (1995). Biochim. Biophys. Acta, 1247, 1–11. [DOI] [PubMed]
- Schurink, M., van Berkel, W. J., Wichers, H. J. & Boeriu, C. G. (2007). Peptides, 28, 485–495. [DOI] [PubMed]
- Soler-Rivas, C., Jolivet, S., Arpin, N., Olivier, J. M. & Wichers, H. J. (1999). FEMS Microbiol. Rev. 23, 591–614. [DOI] [PubMed]
- Strothkamp, K. G., Jolley, R. L. & Mason, H. S. (1976). Biochem. Biophys. Res. Commun. 70, 519–524. [DOI] [PubMed]
- Wichers, H. J., Recourt, K., Hendriks, M., Ebbelaar, C. E. M., Biancone, G., Hoeberichts, F. A., Mooibroek, H. & Soler-Rivas, C. (2003). Appl. Microbiol. Biotech. 61, 336–341. [DOI] [PubMed]
- Wu, J., Chen, H., Gao, J., Liu, X., Cheng, W. & Ma, X. (2010). Biotechnol. Lett. 32, 1439–1447. [DOI] [PubMed]
- Zhang, J.-P., Chen, Q.-X., Song, K.-K. & Xie, J.-J. (2006). Food Chem. 95, 579–584.