

In order to begin an exact determination of hydrogen positions in proteins, a neutron diffraction study of bovine gamma-chymotrypsin has been conducted. This paper details the data collection of the protein at pD (pH*) 7.1.
Keywords: neutron diffraction, γ-chymotrypsin
Abstract
The overarching goal of this research project is to determine, for a subset of proteins, exact hydrogen positions using neutron diffraction, thereby improving H-atom placement in proteins so that they may be better used in various computational methods that are critically dependent upon said placement. In order to be considered applicable for neutron diffraction studies, the protein of choice must be amenable to ultrahigh-resolution X-ray crystallography, be able to form large crystals (1 mm3 or greater) and have a modestly sized unit cell (no dimension longer than 100 Å). As such, γ-chymotrypsin is a perfect candidate for neutron diffraction. To understand and probe the role of specific active-site residues and hydrogen-bonding patterns in γ-chymotrypsin, neutron diffraction studies were initiated at the Protein Crystallography Station (PCS) at Los Alamos Neutron Science Center (LANSCE). A large single crystal was subjected to H/D exchange prior to data collection. Time-of-flight neutron diffraction data were collected to 2.0 Å resolution at the PCS with ∼85% completeness. Here, the first time-of-flight neutron data collection from γ-chymotrypsin is reported.
1. Introduction
Hydrogen is perhaps the most important atom in a protein, yet is the most difficult to visualize via an X-ray diffraction experiment. Hydrogen plays a key role in such fields as mechanistic enzymology, protein folding, protein engineering and rational drug design. However, it can be difficult to assign protonation states (hydrogen sites) in enzyme active sites, as it is well established that the pK a values of the active-site residues can differ greatly from those in solution (Johnson et al., 1981 ▶; Parsons & Raftery, 1972 ▶; Schmidt & Westheimer, 1971 ▶). In the field of rational drug design, it has been shown that significant improvements in the scoring functions of programs for computational ligand docking are possible when hydrogen sites can be explicitly assigned (Ferrara et al., 2004 ▶; Zou et al., 1999 ▶). As can be seen, H atoms are crucially important in the above areas, yet published structures are routinely presented without them, even at ultrahigh resolutions (<1.2 Å). Furthermore, difficulties can arise owing to the majority of structures published today being solved at cryogenic temperatures, leading to artifacts such as the alteration of pK a values (Halle, 2004 ▶; Hazemann et al., 2005 ▶). These artifacts need to be examined further. The overarching goal of this research project is to develop a means of improving protein models with respect to H-atom placement, such that they might be better used in various computational methods that depend critically upon the accurate and precise placement of H atoms.
As stated, over time, our aim is to identify the positions of H atoms for a small set of proteins at a variety of pH values to observe changes in protonation states. This can only be accomplished directly via neutron diffraction, as H atoms (or D atoms) are readily visible at moderate resolution (2–2.5 Å). In these experiments, we propose to identify the positions of H (D) atoms in γ-chymotrypsin as a model protein at three different pH values. Data have already been collected at pH ∼5.6 and a report has been published (Novak et al., 2009 ▶). As such, we wish to collect data at pH ∼7.0, the subject of this preliminary work, as well as at pH ∼9.0. As the various pK a values of important catalytic residues are known from previous enzymological studies, we hope to validate these measurements by direct visualization of protonation (deuteration) sites on the enzyme at the active site. We will pay particularly close attention to the residues of the catalytic triad: His57, Asp102 and Ser195.
X-ray crystallography is the routine method of choice for high-resolution structure determination of proteins. X-rays are scattered by the clouds of electrons around atomic nuclei, with a resulting increase in the magnitude of scattering as a function of atomic number Z. This means that while the atom types found in proteins (C, N, O and S) are readily visible in electron-density maps, the functionally important H atoms are usually invisible, except in extreme cases where a subset of H atoms can be seen at ultrahigh resolution (Blakeley, Ruiz et al., 2008 ▶). In sharp contrast to this, neutrons are diffracted strongly by H (scattering length −3.7 fm), similar to other atom types found in proteins (scattering lengths for C, 6.6 fm; O, 5.8 fm; N, 9.4 fm; S, 2.8 fm). This feature makes the assignment of H-atom locations at medium resolution viable (Niimura & Bau, 2008 ▶; Blakeley, Langan et al., 2008 ▶).
Substitution of H by its isotope D (scattering length 6.7 fm) leads to a dramatic improvement in the coherent scattering as D has a large positive scattering length and a smaller incoherent component compared with H (incoherent scattering length of 4.04 fm for D and 25.3 fm for H). As such, it has now become routine to subject crystals to H/D exchange prior to data collection. This strategy dramatically decreases the background arising from the presence of H, effectively increasing signal to noise. It also provides additional information such as solvent accessibility, levels of H/D exchange and thermal fluctuations and can identify minimal folding domains in proteins (Bennett et al., 2008 ▶).
In conventional electron-density maps water molecules usually appear as spherical peaks indicating the position of the O atom. In nuclear density peaks waters appear as extended shapes with clearly defined density for both D atoms and this feature makes it possible to differentiate between OH−, H3O+ and H2O (or OD−, D3O+ and D2O) species. In the same way, it is possible to visualize and place H/D atoms on amino-acid residues to determine their protonation states and hydrogen-bonding interactions. In this way X-rays and neutrons are highly compatible and can be combined during model refinement in a joint approach. This leads to more accurate structures owing to the inclusion of all of the atoms present in the crystal (Adams et al., 2009 ▶; Afonine et al., 2010 ▶). Accurate information on the positions and locations of H/D atoms in enzymes can result in a detailed analysis of amino-acid side-chain orientations and their interactions with each other and with solvent. This is a very useful approach for the elucidation of enzyme mechanisms and also contributes information about the hydration of proteins, hydrogen bonds, solvent accessibility and minimal folding domains (Kossiakoff & Spencer, 1981 ▶; Bennett et al., 2006 ▶; Blakeley, Langan et al., 2008 ▶; Coates et al., 2008 ▶; Blum et al., 2009 ▶; Adachi et al., 2009 ▶; Yagi et al., 2009 ▶; Tomanicek et al., 2010 ▶; Fisher et al., 2010 ▶; Kovalevsky, Hanson et al., 2010 ▶).
The Protein Crystallography Station at Los Alamos Neutron Science Center is rather unique as it uses spallation neutrons as well as time-of-flight (TOF) techniques. In particular, neutrons are produced from protons striking the 1L tungsten target in microsecond pulses at a frequency of 20 Hz. The resulting high-energy neutrons are reduced to thermal energies in a moderator and sent down a flight path of 28 m in vacuum pipes with collimator inserts, thus resulting in a final beam divergence of 0.12° (Schoenborn & Langan, 2004 ▶; Langan et al., 2008 ▶). The characteristics of the resulting beam allow data to be collected using time-of-flight (wavelength-resolved) Laue techniques. This results in the efficient collection of data with good signal-to-noise ratios using all available neutrons (∼0.7–7.0 Å). The data are seen on the position-sensitive detector as a three-dimensional TOF Laue pattern. The TOF technique has allowed smaller crystals with larger unit-cell volumes to be used, thus increasing the complexity of the biological problems that can be tackled (Langan et al., 2008 ▶; Kovalevsky, Fisher et al., 2010 ▶).
2. Crystallization of γ-chymotrypsin for neutron data collection
γ-Chymotrypsin was purchased from Sigma–Aldrich (St Louis, Missouri, USA) and was used without further purification. Crystals were grown with minor modifications using a previously outlined method (Novak et al., 2009 ▶). Briefly, γ-chymotrypsin was dissolved to 30 mg ml−1 in water. 250 µl protein solution was added to 200 µl buffer solution consisting of 45% saturated ammonium sulfate, 10 mM sodium cacodylate pH 6.0 and 0.75% saturated cetyltrimethylammonium bromide. To this solution, 50 µl of a 1 M sodium iodide solution was added, a reagent that is necessary for the production of large crystals of γ-chymotrypsin. In order to achieve crystals that were large enough to be suitable for use with neutron diffraction, the above 500 µl solution (hereafter referred to as the inner solution) was added to a 0.5 dram vial that was left uncapped. This vial was placed inside a scintillation vial in which the solution was equilibrated against 3 ml 65% saturated ammonium sulfate (hereafter referred to as the outer solution) and which was tightly sealed with Parafilm. Crystals of a size large enough for neutron diffraction grew over the course of two to three months. Once crystals appeared, stepwise transfer to a deuterated buffer solution (45% saturated ammonium sulfate, 10 mM sodium cacodylate pH 6.0 and 0.75% saturated cetyltrimethylammonium bromide, first prepared in water, placed on a RotoVap and then prepared in deuterated water) took place by adding the deuterated buffer in 100 µl increments to the outer well solution until it was fully replaced. The entire outer solution was then exchanged (a total of 3 ml) in one sitting. A small volume (∼25 µl) of the inner solution was then exchanged for deuterated buffer solution. This entire exchange of both inner and outer solutions lasted for approximately two months. After this time, the large γ-chymotrypsin crystal that was chosen for neutron diffraction was mounted in a 2.0 mm quartz capillary with a plug of deuterated buffer solution.
The crystals used for X-ray structure determination were grown using similar solutions, but with much smaller volumes and using traditional setups. Specifically, the sitting-drop method was employed using 10 µl protein solution, 8 µl buffer solution (as described above) and 2 µl 1 M sodium iodide solution to give a 20 µl total drop size. These crystals appeared after only a few days, were transferred stepwise into the deuterated buffer solution in 2 µl steps and were then mounted in either 0.5 or 0.7 mm quartz capillaries containing a plug of the deuterated buffer solution. A room-temperature X-ray data set was collected to 1.05 Å resolution on the X-ray Operations and Research beamline 23-ID-B at the Advanced Photon Source, Argonne National Laboratory.
3. Room-temperature neutron data collection and reduction
A suitable H/D-exchanged crystal of γ-chymotrypsin was selected for neutron data collection (Fig. 1 ▶). After a 24 h test exposure, it was clear that the diffraction quality and signal-to-noise ratio were appropriate for full data collection and structure determination (Fig. 2 ▶).
Figure 1.

Optical photograph of a large single γ-chymotrypsin crystal (∼1 × 1 × 1.5 mm).
Figure 2.

Neutron Laue time-of-flight diffraction images of γ-chymotrypsin collected at the PCS at two different ϕ settings at κ = 30° and 90°. For this representation, the time-of-flight data were overlaid to produce a conventional Laue pattern.
Time-of-flight wavelength-resolved Laue diffraction data were collected at room temperature on a Huber κ-circle goniometer at 18 usable settings. Each setting was collected for 16 h and the data extended to better than 2.0 Å resolution. The crystal-to-detector distance was fixed at 730 mm, which corresponds to the cylindrical radius of the curved detector. The detector has a limited vertical area (120° horizontal span, 16° vertical span) and to cover reciprocal space the crystal was re-oriented five times using the κ and ω goniometer circles. We performed 30° steps in ϕ at each orientation for a total of 18 images (Fig. 2 ▶; Table 1 ▶).
Table 1. Room-temperature neutron data-collection statistics.
Values in parentheses are for the highest resolution shell.
| Source/detector | LANSCE 1L target/PCS 3He detector |
| Settings | 18 |
| Wavelength range used (Å) | 0.75–6.4 |
| Space group | P42212 |
| Unit-cell parameters (Å) | a = b = 69.5, c = 97.4 |
| Unit-cell volume (Å3) | 470466 |
| Resolution (Å) | 29.0–2.00 (2.11–2.00) |
| No. of reflections (measured/unique) | 51999 (3632)/13865 (1633) |
| Multiplicity | 3.8 (2.2) |
| Mean I/σ(I) | 4.3 (2.0) |
| Rmerge† (%) | 25.0 (28.8) |
| Rp.i.m.‡ (%) | 4.7 (20.9) |
| Completeness (%) | 84.5 (70.5) |
R
merge = 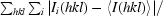
 × 100.
× 100.
R
p.i.m. = 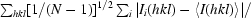 × 100.
× 100.
Each image was processed with a version of d*TREK that had been modified in-house for use with wavelength-resolved Laue neutron data (Pflugrath, 1999 ▶; Langan & Greene, 2004 ▶). Integrated reflections were wavelength-normalized using LAUENORM and merged using SCALA from the CCP4 suite of crystallographic programs (Helliwell et al., 1989 ▶; Evans, 2006 ▶; Diederichs & Karplus, 1997 ▶; Winn et al., 1994 ▶). We removed the least accurate data by reducing the wavelength spectrum from 0.6–7.0 Å to 0.75–6.4 Å. Restricting the wavelength range in this way leads to more accurate data and improves the data statistics by using only the most accurately measured thermal neutrons. The overall completeness for the data set is ∼85% to 2.0 Å resolution (Table 1 ▶). The crystal diffracted to better than 2.0 Å resolution; however, at present we do not have sufficient data in the higher resolution bins.
A room-temperature X-ray data set from a similar crystal is available and will be used for joint neutron and X-ray structure refinement. Structure refinement is in progress using a modified version of CNS (called nCNS) that has been designed to handle neutron and X-ray data simultaneously (Adams et al., 2009 ▶). Initial neutron and X-ray density maps after only one round of rigid-body refinement already reveal some of the details of protonation and hydrogen bonding in the active site of γ-chymotrypsin. Analysis of the neutron structure will involved detailed mapping of H atoms, overall H/D exchange and hydrogen-bonding patterns in the enzyme active site.
4. Results and discussion
The Laue time-of-flight neutron diffraction study of γ-chymotrypsin reported here is the first of its kind and represents a significant accomplishment. After initial refinement using only neutron data and not including room-temperature X-ray data there is significant density for catalytically important residues. In particular, we can see that the catalytic histidine is doubly protonated (deuterated). The serine and aspartate that make up the remainder of the catalytic triad do not show density corresponding to the presence of deuterium; in addition, we also see deuteration of backbone NH at terminal positions of β-sheets (data not shown). Sample size and the length of time required to acquire neutron data are the major impediments in the accessibility of neutrons to most structural biologists. Here, we show that it is possible to obtain a complete neutron data set from a medium-sized crystal in under two weeks. These data will reveal many important details about the γ-chymotrypsin active site and the ionization states of specific residues as well as the hydrogen-bonding patterns in the active site that support catalysis.
Acknowledgments
The PCS is funded by the Department of Energy Office of Biological and Environmental Research (DOE-OBER). Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-06CH11357. This research was supported by National Institutes of Health Grants GM32415 and GM26788 (to GAP and DR). LL was supported in part by Fidelity Grant No. 16-28100-40000-401591 as well as NIH Grant GM32415.
References
- Adachi, M. et al. (2009). Proc. Natl Acad. Sci. USA, 106, 4641–4646.
- Adams, P. D., Mustyakimov, M., Afonine, P. V. & Langan, P. (2009). Acta Cryst. D65, 567–573. [DOI] [PMC free article] [PubMed]
- Afonine, P. V., Mustyakimov, M., Grosse-Kunstleve, R. W., Moriarty, N. W., Langan, P. & Adams, P. D. (2010). Acta Cryst. D66, 1153–1163. [DOI] [PMC free article] [PubMed]
- Bennett, B. C., Gardberg, A. S., Blair, M. D. & Dealwis, C. G. (2008). Acta Cryst. D64, 764–783. [DOI] [PubMed]
- Bennett, B., Langan, P., Coates, L., Mustyakimov, M., Schoenborn, B., Howell, E. E. & Dealwis, C. (2006). Proc. Natl Acad. Sci. USA, 103, 18493–18498. [DOI] [PMC free article] [PubMed]
- Blakeley, M., Langan, P., Niimura, N. & Podjarny, A. (2008). Curr. Opin. Struct. Biol. 18, 593–600. [DOI] [PMC free article] [PubMed]
- Blakeley, M. P., Ruiz, F., Cachau, R., Hazemann, I., Meilleur, F., Mitschler, A., Ginell, S., Afonine, P., Ventura, O. N., Cousido-Siah, A., Haertlein, M., Joachimiak, A., Myles, D. & Podjarny, A. (2008). Proc. Natl Acad. Sci. USA, 105, 1844–1848. [DOI] [PMC free article] [PubMed]
- Blum, M. M., Mustyakimov, M., Rüterjans, H., Kehe, K., Schoenborn, B. P., Langan, P. & Chen, J. C. (2009). Proc. Natl Acad. Sci. USA, 106, 713–718. [DOI] [PMC free article] [PubMed]
- Coates, L., Tuan, H.-F., Tomanicek, S., Kovalevsky, A., Mustyakimov, M., Erskine, P. & Cooper, J. (2008). J. Am. Chem. Soc. 130, 7235–7237. [DOI] [PMC free article] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Ferrara, P., Gohlke, H., Price, D. J., Klebe, G. & Brooks, C. L. (2004). J. Med. Chem. 47, 3032–3047. [DOI] [PubMed]
- Fisher, S. Z., Kovalevsky, A. Y., Domsic, J. F., Mustyakimov, M., McKenna, R., Silverman, D. N. & Langan, P. A. (2010). Biochemistry, 49, 415–421.
- Halle, B. (2004). Proc. Natl Acad. Sci. USA, 101, 4793–4798. [DOI] [PMC free article] [PubMed]
- Hazemann, I., Dauvergne, M. T., Blakeley, M. P., Meilleur, F., Haertlein, M., Van Dorsselaer, A., Mitschler, A., Myles, D. A. A. & Podjarny, A. (2005). Acta Cryst. D61, 1413–1417. [DOI] [PubMed]
- Helliwell, J. R., Habash, J., Cruickshank, D. W. J., Harding, M. M., Greenhough, T. J., Campbell, J. W., Clifton, I. J., Elder, M., Machin, P. A., Papiz, M. Z. & Zurek, S. (1989). J. Appl. Cryst. 22, 483–497.
- Johnson, F. A., Lewis, S. D. & Shafer, J. A. (1981). Biochemistry, 20, 44–48. [DOI] [PubMed]
- Kossiakoff, A. A. & Spencer, S. A. (1981). Biochemistry, 20, 6462–6474. [DOI] [PubMed]
- Kovalevsky, A., Fisher, Z., Johnson, H., Mustyakimov, M., Waltman, M. J. & Langan, P. (2010). Acta Cryst. D66, 1206–1212. [DOI] [PMC free article] [PubMed]
- Kovalevsky, A. Y., Hanson, L., Fisher, S. Z., Mustyakimov, M., Mason, S. A., Forsyth, V. T., Blakeley, M. P., Keen, D. A., Wagner, T., Carrell, H. L., Katz, A. K., Glusker, J. P. & Langan, P. (2010). Structure, 18, 688–699. [DOI] [PMC free article] [PubMed]
- Langan, P., Fisher, Z., Kovalevsky, A., Mustyakimov, M., Sutcliffe Valone, A., Unkefer, C., Waltman, M. J., Coates, L., Adams, P. D., Afonine, P. V., Bennett, B., Dealwis, C. & Schoenborn, B. P. (2008). J. Synchrotron Rad. 15, 215–218. [DOI] [PMC free article] [PubMed]
- Langan, P. & Greene, G. (2004). J. Appl. Cryst. 37, 253–257.
- Niimura, N. & Bau, R. (2008). Acta Cryst. A64, 12–22. [DOI] [PubMed]
- Novak, W. R. P., Moulin, A. G., Blakeley, M. P., Schlichting, I., Petsko, G. A. & Ringe, D. (2009). Acta Cryst. F65, 317–320. [DOI] [PMC free article] [PubMed]
- Parsons, S. M. & Raftery, M. A. (1972). Biochemistry, 11, 1623–1629. [DOI] [PubMed]
- Pflugrath, J. W. (1999). Acta Cryst. D55, 1718–1725. [DOI] [PubMed]
- Schmidt, D. E. & Westheimer, F. H. (1971). Biochemistry, 10, 1249–1253. [DOI] [PubMed]
- Schoenborn, B. P. & Langan, P. (2004). J. Synchrotron Rad. 11, 80–82. [DOI] [PubMed]
- Tomanicek, V. U., Blakeley, M. P., Cooper, J., Chen, Y., Afonine, P. V. & Coates, L. (2010). J. Mol. Biol. 396, 1070–1080. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yagi, D., Yamada, T., Kurihara, K., Ohnishi, Y., Yamashita, M., Tamada, T., Tanaka, I., Kuroki, R. & Niimura, N. (2009). Acta Cryst. D65, 892–899. [DOI] [PubMed]
- Zou, X. Q., Sun, Y. X. & Kuntz, I. D. (1999). J. Am. Chem. Soc. 121, 8033–8043.