

Abstract
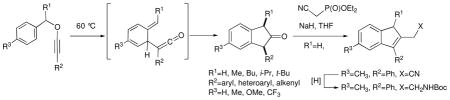
Substituted benzyl alkynyl ethers, prepared from the corresponding α-alkoxy ketones in a two-step sequence involving enol triflate formation and KOtBu-induced E2 elimination, undergo [3,3]-sigmatropic rearrangement/intramolecular 5-exo-dig cyclization at 60°C to form substituted 2-indanones in good overall yields. 1,3-cis-disubstituted-2-indanones are formed preferentially when the benzylic substituent R1 is bulky. Substituted indenes may be prepared from 2-indanones in high yields by Horner-Wadsworth-Emmons reaction.
Ynol ethers are functional groups that possess significant potential in organic chemistry for the formation of carbon-carbon bonds.1–3 Due to their linear geometry, alkynyl ethers are relatively unhindered to approach by functional groups present in the same or different molecules; furthermore, alkynyl ethers can prospectively form up to three new bonds in a single reaction.4
We have recently uncovered a straightforward methodology for the preparation of alkynyl ethers (Scheme 1)2 involving the coupling of alcohols 2 with diazoketones 1 in the presence of indium triflate as a catalyst; the α-alkoxy ketone product 3 is readily transformed into the enol triflate or phosphate at low temperatures, which then undergoes base-induced elimination.3 Allyl-1-alkynyl ethers 7 are thermally unstable and undergo a rapid [3,3] sigmatropic rearrangment5 upon generation at −78 °C to form allyl ketene intermediate 8, which is subsequently trapped by an alcohol or amine to form γ,δ-unsaturated carboxylic acid derivatives 9 in high yields.4
SCHEME 1.

1-alkynyl ether synthesis and [3,3]-sigmatropy
Guided by the pioneering studies of Arens6 and Schmidt,7 we have also shown that benzyl-1-alkynyl ether 12, which is stable at ambient temperatures, undergoes sigmatropic rearrangement and intramolecular cyclization at 60 °C to form 1-phenyl-2-indanone 13 in 98% yield. Due to the efficiency of this process, we decided to further explore its scope and utility, and in this Note we report our findings on the diastereoselectivity of the benzyl alkynyl ether sigmatropic rearrangement/intramolecular cyclization reaction and its applicability to the preparation of substituted 2-indanones and indenes.
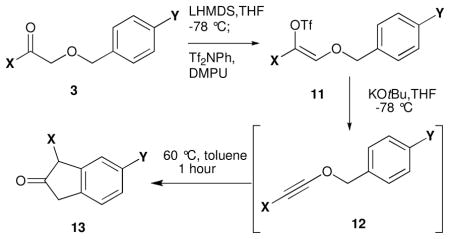
The α-alkoxy ketones 3 (Scheme 2 and Table 1) necessary for the synthesis of benzyl alkynyl ethers 12 were prepared either by coupling of the appropriate diazoketones 18 with benzyl alcohol 2 in the presence of 10 mol% In(OTf)3 (path A, Scheme 2; Table 1, entries 1, 2, and 5),9 or by reaction of aryloxyacetic acid10-derived Weinreb amide11 10 with the appropriate organolithium or Grignard reagent (path B, Scheme 2; Table 1, entries 3–4 and 6–10). Pathway B proved to be more general for the synthesis of α-alkoxy ketones; indeed, attempted synthesis of electron-rich substrates 3g and 3i by diazoketone-alcohol coupling led to extensive decomposition and minimal yields of the desired products.
SCHEME 2.

Two methods for the synthesis of ketones 3
TABLE 1.
Scope of benzyl alkynyl ether [3,3]sigmatropy/5-exo-dig cyclization
 | |||||
|---|---|---|---|---|---|
| entry | X | Y | 3 | % yield 11d | % yield 13e |
| 1 |
|
H | aa | 75 | 98 |
| 2 |
|
CH3 | ba | 73 | 93 |
| 3 |
|
OCH3 | cb | 68 | 65 |
| 4 |
|
CF3 | db | 81 | 72 |
| 5 |
|
H | ea | 85 | 95 |
| 6 |
|
H | fb | 68 | 90 |
| 7 |
|
H | gb | 81 | 88 |
| 8 |
|
H | hb | 78 | 91 |
| 9 |
|
H | ib | 67 | 62 |
| 10 |
|
H | jb | 82 | ––c |
α-Alkoxy ketone 3 prepared by method A (Scheme 2).
α-Alkoxy ketone 3 prepared by method B (Scheme 2).
A complex mixture of products was formed.
Yield (from 3) based on weight obtained after rapid filtration of the crude triflation reaction mixture through silica gel (see text).
Isolated yield (from 11) after silica gel chromatography.
Treatment of α-alkoxy ketones 3 with LHMDS at −78 °C, followed by addition of NPhTf2 in THF/DMPU, led to enol triflates 11a–j, which were sensitive to trace amounts of acid and heat. After rapid filtration of the crude reaction mixture through silica gel, the enol triflates were immediately treated with potassium tert-butoxide (1M in THF) at −78 °C to effect elimination of triflate ion, affording benzyl alkynyl ethers 12a–i. The crude benzyl alkynyl ethers were also sensitive to heat and silica gel chromatography, so they were directly warmed to 60 °C in toluene to effect [3,3]-sigmatropic rearrangement/5-exo-dig cyclization to provide the substituted indanones 13a–i in good overall yields. This process was successfully employed for alkynyl ether substrates derived from electron-rich (Table 1, entries 2 and 3) and electron-deficient (entry 4) benzylic alcohols and electron-rich (entries 6 and 7) and electron deficient (entry 8) aryl ketones.
Alkynyl ethers derived from α,β-unsaturated ketones (entry 5) and heteroaryl ketones (entry 9) also smoothly underwent the rearrangement/cyclization process, furnishing indanones 13e and 13i in 95 and 62% yields, respectively. However, attempted elimination and thermolysis of alkyne-substituted enol triflate 11j (entry 10) led to a complex mixture of products, likely arising from the intermediacy of various allenic species3 under the basic conditions employed for elimination.
Monitoring the rates of rearrangement/cyclization12 for alkynyl ether substrates 12a–d by 1H NMR spectroscopy revealed interesting trends (Table 2). Whereas unsubstituted parent compound 12a underwent first-order decay to 13a at 57 °C in CDCl3 with a half-life of 4.8 minutes, alkynyl ether 12b under the same conditions displayed a half-life of only 0.8 minutes, and methoxy-substituted substrate 12c could not be isolated at room temperature, having completely rearranged to indanone 13c upon warming the KOtBu elimination reaction of 11c from -78 °C to ambient. In contrast, CF3-substituted alkynyl ether 12d displayed a half-life of 13.3 minutes at 57 °C. Assuming that the rate-determining step of the transformation 12→13 is the [3,3] sigmatropic rearrangement, the rate acceleration observed for substrates bearing electron-donating substituents (Y) on the benzyl moiety suggests the development of charge deficiency (either a radical or cationic intermediate)13 at the benzylic carbon atom during the transition state for sigmatropic rearrangement.
TABLE 2.
Kinetic data for the first-order decay of 12 to 13 at 57 °C in CDCl3
| Substrate | t1/2(min) | k (min−1) |
|---|---|---|
| 12a | 4.8 | 0.145 |
| 12b | 0.8 | 0.905 |
| 12d | 13.3 | 0.052 |
| 12f | 3.6 | 0.191 |
| 12h | 4.6 | 0.149 |
A similar analysis of the rates of rearrangement of 12f–h compared to 12a revealed again that electron-donating substituents accelerated the rearrangement/cyclization process: whereas 12f displayed a half life of 3.6 minutes at 57 °C, substrate 12g had completely rearranged to 13g upon reaching room temperature after generation at −78 °C. Intriguingly, however, it was found that CF3-substituted alkynyl ether 12h underwent first-order decay slightly faster than 12a, displaying a half-life of 4.6 minutes at 57 °C. These observations are consistent with a change in mechanism14 upon proceeding from electron-donating to electron-withdrawing substituents on the aromatic moiety X, with a radical mechanism (pathway A, figure 1) likely operative for the former and a polar mechanism (pathway B, figure 1) for the latter.
FIGURE 1.

Possible TS charge development for benzyl alkynyl ether sigmatropy
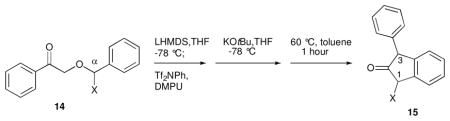
Next, we sought to examine the stereoselectivity of the rearrangement/cyclization reaction when α-substituted benzyl ethers are employed. Ketones 14a–e were prepared by protocol A (Scheme 2) and subjected to enol triflate formation, tert-butoxide induced triflate elimination, and thermal rearrangement/cyclization to furnish 1,3-disubstituted-2-indanones 15 (Table 3). Only in the case of the phenyl-substituted ketone 14e did the reaction sequence fail to afford the desired indanone product; enol triflate formation from 14e led to complicated mixtures due to the heightened acidity of the benzylic proton and the basic conditions involved.
Table 3.
Rearrangement/cyclization of α–substituted benzyl alkynyl ethersa
 | ||||
|---|---|---|---|---|
| entry | X | 14 | % yield 15 (from 14)b | d.r. |
| 1 | CH3 | a | 61 | 1:1 |
| 2 | CH3(CH2)3 | b | 53 | 4:1 |
| 3 | (CH3)2CH | c | 48 | 2:1 |
| 4 | (CH3)3C | d | 69 | >95:5 |
| 5 |
|
e | –– | –– |
Ketones 14 prepared by method A (Scheme 2).
Isolated yields.
Low to moderate diastereoselectivity was observed for methyl-, butyl-, and isopropyl-substituted indanones 15a–c; however, excellent diastereoselectivity (>95:5) was obtained for 1-tert-butyl-3-phenyl-indanone 15d. The presence of crosspeaks between the C.1 and C.3 protons (Hb and Ha, respectively) observed in the NOESY spectrum of 15d (as well as correlations between Ha and Hc, Hc and Hd, and Hd and Hb) confirmed the cis stereochemistry (Scheme 3). Mechanistically, we propose that the large tert-butyl substituent prefers to be oriented trans to the ketene in the nonaromatic intermediate 16a;15 5-exo dig cyclization, followed by a syn-1,2-proton migration to the enolate carbon atom, would give rise to the product with the observed stereochemistry.
SCHEME 3.

Mechanistic proposal for the formation of indanone 15d.
Finally, we sought to transform our 2-indanone products 13 into synthetically useful substituted indenes. Indenes are building blocks in the synthesis of biologically active pharmaceutical agents16 and are often used as ligands in olefin polymerization catalysts.17 Addition of indanone 13b to a solution of diethyl(cyanomethyl)phosphonoacetate and NaH in THF and stirring for 4 hours at room temperature resulted in clean transformation to indene 18b (91% isolated yield), arising from carbonyl olefination and base-mediated isomerization (Scheme 4). Reduction of the nitrile functional group and in situ primary amine protection as a tert-butyl carbamate was accomplished in a single step by employing Caddick’s protocol18 (cat. NiCl2•H2O, NaBH4, Boc2O, MeOH) to afford indene 19b in 60% yield.
SCHEME 4.

Transformation of 2-indanones into substituted indenes
In conclusion, we have shown that the [3,3]-sigmatropic rearrangement of benzyl alkynyl ethers may be successfully employed for a variety of substrates bearing electron- deficient and electron-rich aromatic and heteroaromatic ring systems, and the rates of rearrangement vary depending upon the electronic nature of the aromatic substituents. Furthermore, 1,3-cis-disubstituted-2-indanones are formed in excess when α-substituted benzyl alkynyl ethers are employed as substrates. The 2-indanones obtained by this method may be easily transformed into useful substituted indene products.
Experimental Section
Representative procedure for the synthesis of ketones 3a, 3b, 3e, 14a–e. 2-(Benzyloxy)-1-phenylethanone 3a
A mixture of the diazoacetophenone (136 mg, 1 mmol) and benzyl alcohol (162 mg, 1.5 mmol) were dissolved in dry toluene (4 mL) under argon and stirred at room temperature for 2 minutes. Indium (III) trifluoromethanesulfonate (56.2 mg, 0.1 mmol, 10 mol%) was added and a rapid evolution of nitrogen gas was observed. The reaction was monitored by TLC, and when complete consumption of the starting material was observed, saturated NaHCO3 (10 mL) was added and the reaction mixture was diluted with ether (20 mL). The phases were separated and the aqueous phase was back-extracted with ether (1 × 20 mL). The combined organic extracts were then dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography (SiO2) afforded α-alkoxyketone 3a.
3a3 (87%): 1H NMR (400 MHz, CDCl3) δ 7.93 (d, J=7.6 Hz, 2H), 7.57 (t, J=7.6 Hz, 1H), 7.46 (t, J=8.0 Hz, 2H), 7.40–7.27 (m, 5H), 4.82 (s, 2H), 4.70 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 197.2, 136.9, 134.6, 133.9, 129.4, 128.8, 128.6, 128.2, 127.9, 127.1, 123.6, 73.4, 72.4; HRMS (ESI) calculated for C15H14NaO2 249.0892, found 249.0854 (M+Na)+. 3b (82%): 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J=7.6 Hz, 2H), 7.51 (t, J=7.6 Hz, 1H), 7.46–7.27 (m, 6H), 4.72 (s, 2H), 4.65 (s, 2H), 2.34 (s, 3H); 13C NMR: (100 MHz, CDCl3) δ 196.3, 137.6, 135.0, 134.4, 133.5, 129.2, 128.7, 128.2, 128.0, 73.2, 72.5, 21.2; HRMS (ESI) calculated for C16H16NaO2 263.1048, found 263.1107 (M+Na)+.
3e (61%): 1H NMR (400 MHz, CDCl3) δ 7.83 (dd, J=8.4, 1.2 Hz, 2H), 7.45–7.25 (m, 5H), 6.83 (d, J=8.8 Hz, 2H), 4.62 (s, 2H), 4.54 (s, 2H), 3.67 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 159.4, 134.9, 133.4, 129.7, 129.4, 128.6, 128.3, 127.8, 113.8, 72.9, 72.2, 55.1; HRMS (ESI) calculated for C16H16NaO3 279.0997, found 279.1098 (M+Na)+.
14a (75%):1H NMR (400 MHz, CDCl3) δ 7.82 (d, J= 7.6 Hz, 2H), 7.46 (t, J= 7.6 Hz, 1H), 7.36–7.25 (m, 7H), 4.65 (d, J=16.8 Hz, 1H), 4.54 (q, J=6.4 Hz, 1H), 4.52 (d, J=16.8 Hz, 1H), 1.54 (d, J=6.4Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 195.9, 142.9, 135.0, 131.1, 128.6, 128.5, 128.2, 127.8, 127.7, 126.3, 78.3, 77.9, 71.2, 24.0, 23.9; HRMS (ESI) calculated for C16H16NaO2 263.1048 found 263.1632 (M+Na)+.
14b (78%): 1H NMR: (400 MHz, CDCl3) δ 7.84 (d, J= 8.4 Hz, 2H), 7.50 (t, J= 7.6 Hz, 1H), 7.40–7.25 (m, 7H), 4.63 (d, J=16.8 Hz, 1H), 4.49 (d, J=16.8 Hz, 1H), 4.40 (t, J=6.8 Hz, 1H), 1.97 (m, 1H), 1.72 (m, 1H), 1.43 (m, 1H), 1.30 (m, 1H), 0.86 (t, J=7.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.4, 141.7, 135.1, 133.3, 128.6, 128.5, 128.3, 127.9, 127.0, 83.1, 71.3, 37.9, 28.0, 22.6, 14.0; HRMS (ESI) calculated for C19H22NaO2 305.1517, found 305.1343 (M+Na)+.
14c (73%): 1H NMR (400 MHz, CDCl3) δ 7.76 (d, J= 8.0 Hz, 2H), 7.43 (t, J= 7.8 Hz, 1H), 7.33–7.21 (m, 7H), 4.52 (d, J=16.0 Hz, 1H); 4.35 (d, J=16.0 Hz, 1H); 3.97 (d, J=8.0 Hz, 1H); 1.96 (m, J=8.0 Hz, 1H); 0.98 (d, J=8.0 Hz, 3H); 0.65 (d, J=8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 196.6, 140.3, 135.2, 133.3, 128.6, 128.5, 128.2, 128.0, 127.8, 127.7, 88.6, 71.6, 34.8, 19.2, 19.0; HRMS (ESI) calculated for C18H20NaO2 291.1361, found 291.1440 (M+Na)+.
14d (66%): 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J= 8.0 Hz, 2H), 7.49 (t, J= 7.8 Hz, 1H); 7.37 (t, J= 7.8 Hz, 2H). 7.32–7.25 (m, 5H), 4.62 (d, J=16.0 Hz, 1H), 4.41 (d, J=16.0 Hz, 1H), 4.15 (s, 1H), 0.96 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 196.5, 138.8, 135.3, 133.3, 128.6, 128.5, 128.0, 127.7, 127.6, 90.6, 71.9, 35.8; 26.4; HRMS (ESI) calculated for C19H22NaO2 305.1517, found 305.1962 (M+Na)+.
14e (71%): 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J= 7.6 Hz, 2H), 7.55 (t, J= 7.4 Hz, 1H), 7.46–7.41 (m, 6H), 7.35 (t, J=7.6 Hz, 4H), 7.28 (t, J=7.8 Hz, 2H), 5.64 (s, 1H), 4.77 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 196.3, 141.3, 135.1, 133.5, 128.7, 128.5, 128.0, 127.8, 127.4, 83.8, 77.4, 77.1, 76.8, 71.4; HRMS (ESI) calculated for C21H18NaO2 325.1204, found 325.1541 (M+Na)+.
Representative procedure for the synthesis of ketones 3c, 3d, 3f–j
To a solution of the aryloxy acetic acid10 (1 g, 6 mmol) in 11 mL dichloromethane at 0 °C was added 1,1’-carbonyl diimidazole (1.26 g, 7.82 mmol) at 0 °C. The solution bubbled and upon completion it was warmed to room temperature for 30 minutes. The solution was cooled to 0 °C and triethylamine (1.1 mL, 8.4 mmol) was added, followed by the Weinreb amine hydrochloride salt (0.82 g, 8.4 mmol). The solution was allowed to warm to room temperature and was stirred overnight. The mixture was diluted with 1M HCl (10 mL) and EtOAc (20 mL). The phases were separated and the aqueous phase was back-extracted with EtOAc (1 × 20 mL). The combined organic extracts were then dried over Na2SO4, filtered, and concentrated in vacuo. Crude amide 10 was taken directly to the next step without further purification.
To a solution of Weinreb amide 10 (5 mmol) in THF (5 mL) at −78 °C under argon was added a solution of aryllithium reagent (generated from the corresponding aryl bromide 1M in THF by the addition of 0.95 equiv n-BuLi) dropwise. Upon completion of the addition, TLC indicated complete conversion to the aryl ketone. The mixture was allowed to warm to −20 °C and was quenched with saturated NH4Cl solution (10 mL). The mixture was diluted with ether (3 mL) and was allowed to stir at room temperature for one hour. The phases were separated and the aqueous phase was back-extracted with EtOAc (1 × 20 mL). The combined organic extracts were then dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography (SiO2) afforded α-alkoxyketones 3.
3c (71%): 1H NMR (400 MHz, CDCl3) δ 7.83 (dd, J=8.4, 1.2 Hz, 2H), 7.45–7.25 (m, 5H), 6.83 (d, J=8.8 Hz, 2H), 4.62 (s, 2H), 4.54 (s, 2H), 3.67 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 196.3, 159.4, 134.9, 133.4, 129.7, 129.4, 128.6, 128.3, 127.8, 113.8, 72.9, 72.2, 55.1; HRMS (ESI) calculated for C16H16NaO3 279.0997, found 279.1098 (M+Na)+.
3d (68%): 1H NMR (400 MHz, CDCl3) δ 7.91 (dd, J=9.6 Hz, 2H), 7.61–7.42 (m, 7H), 4.81 (s, 2H), 4.72 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 196.0, 141.6, 134.7, 133.7, 129.5, 128.8, 127.9, 127.8, 125.4, 125.3, 115.4, 72.8, 72.5; HRMS (ESI) calculated for C16H13 F3NaO2 317.0765, found 317.0852 (M+Na)+.
3f (69%):1H NMR (400 MHz, CDCl3) δ 7.83–7.80 (d, J=8.4 Hz, 2H), 7.41–7.23 (m, 7H), 4.73 (s, 2H), 4.68 (s, 2H), 2.39 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 195.9, 144.4, 137.4, 132.4, 129.3, 128.5, 128.1, 128.0, 73.3, 72.5, 21.7; HRMS (ESI) calculated for C16H16NaO2 263.1048, found 263.1055 (M+Na)+.
3g (82%): 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J=8.8 Hz, 2H), 7.40–7.32 (m, 5H), 6.88 (d, J=8.8 Hz, 2H), 4.69 (s, 2H), 4.66 (s, 2H), 3.81 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 194.9, 163.8, 150.6, 137.5, 130.3, 128.5, 128.0, 127.9, 116.2, 114.7, 113.9, 73.3, 72.5, 55.4; HRMS (ESI) calculated for C16H16NaO3 279.0997, found 279.0780 (M+Na)+.
3h (73%): 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J=8.0 Hz, 2H), 7.74 (d, J=8.4 Hz, 2H), 7.39–7.33 (m, 5H), 4.76 (s, 2H), 4.70 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 195.6, 137.5, 136.9, 134.8, 128.6, 128.4, 128.2, 128.1, 125.7, 73.5, 72.8; HRMS (ESI) calculated for C16H13F3NaO2 317.0765, found 317.0923 (M+Na)+.
3i (85%): 1H NMR (400 MHz, CDCl3) δ 7.53 (s, 1H), 7.52–7.24 (m, 6H), 6.47 (m, 1H), 4.63 (s, 2H), 4.54 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 185.5, 150.9, 146.7, 137.3, 128.5, 128.3, 128.0, 127.9, 118.2, 112.3, 73.4, 72.2; HRMS (ESI) calculated for C13H12NaO3 239.0684, found 239.0717 (M+Na)+.
3j (92%): 1H NMR (400 MHz, CDCl3) δ 7.36–7.25 (m, 5H), 4.62 (s, 2H), 4.19 (s, 2H), 2.37 (t, J=6.8 Hz, 2H), 1.56 (m, J=7.2 Hz, 2H), 1.38–1.26 (m, 5H), 0.89 (t, J=6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 185.0, 137.1, 128.4, 127.9, 97.5, 82.7, 75.8, 73.3, 31.2, 28.5, 27.5, 22.5, 22.4, 19.0, 14.0; HRMS (ESI) calculated for C17H22NaO2 281.1517, found 281.1582 (M+Na)+.
Representative procedure for enol triflate (11a–i) formation
Hexamethyldisilazane (0.29 mL, 2.5 mmol) dissolved in dry THF (1 mL) under argon was cooled to 0 °C in an ice bath and n-BuLi (0.75 mL), 2M in cyclohexane, 1.5 mmol) was added dropwise. After 10 minutes, the mixture was cooled to −78 °C and a solution of α-alkoxyketone (1 mmol) in THF (1 mL) was added dropwise. The reaction mixture was stirred for one hour at −78 °C and then a solution of N-phenyl triflimide (1.5 mmol) in 1:1 THF : DMPU (1 mL) was added rapidly (1 s). The mixture was allowed to warm to room temperature and then was stirred for one hour, at which time TLC indicated >90% consumption of starting material. The phases were separated and the aqueous phase was back-extracted with ether (1 × 20 mL). The combined organic extracts were then dried over Na2SO4, filtered, and concentrated in vacuo. The crude enol triflate was filtered over silica gel employing 1% Et3N in the eluant (95:5 hexanes:ether), and the material obtained was used directly in the next reaction.
Representative procedure for alkynyl ether (12a–i) formation and sigmatropic rearrangement/cyclization to indanones 13a–i and 15a–d
The enol triflate obtained from the above procedure (~1 mmol) was dissolved in THF (1 mL) under argon and cooled to −78 °C. A solution of potassium tert-butoxide (1M in THF, 3.0 mL, 3.0 mmol) was added dropwise, and the reaction mixture darkened in color. After 20 minutes, a solution of saturated NaHCO3 (10 mL) was added and the mixture was allowed to warm to room temperature with stirring. Ether (10 mL) was added, and the phases were separated. The aqueous later was back-extracted with ether (1 × 20 mL), and the combined organic extracts were then dried over Na2SO4, filtered, and concentrated in vacuo. The crude alkynyl ether was then dissolved in toluene (2 mL) and heated on an oil bath under argon for one hour, at which time TLC indicated complete conversion to the indanone product. Concentration in vacuo and purification of the residue by flash chromatography (SiO2) afforded indanones 13a–i and 15a–d.
13a3 (98%): 1H NMR (400 MHz, CDCl3) δ 7.43–7.22 (m, 7H), 7.17 (d, J=7.6 Hz, 2H), 4.71 (s, 1H), 3.68 (s, 2H); 13C NMR (100 MHz, C6D6) δ 213.8, 141.4, 138.2, 137.3, 128.8, 128.5, 128.0, 127.9, 127.3, 126.0, 124.9, 59.8, 43.0; HRMS (ESI) calculated for C15H12NaO 231.0786, found 231.0777 (M+Na)+.
13b (93%): 1H NMR (400 MHz, CDCl3) δ 7.36–7.24 (m, 3H), 7.17–7.10 (m, 3H), 7.01 (s, 1H), 4.63 (s, 1H), 3.62 (s, 2H), 2.33 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 216.8, 143.3, 140.0, 137.7, 128.8, 128.5, 128.0, 127.9, 126.7, 124.3, 123.1, 60.2, 47.1, 23.5; HRMS (ESI) calculated for C16H14NaO 245.0942, found 245.1399 (M+Na)+.
13c (65%): 1H NMR (400 MHz, CDCl3) δ 7.36–7.27 (m, 4H), 7.15 (d, J=7.6 Hz, 2H), 6.93 (m, 1H), 6.74 (m, 1H), 4.66 (s, 1H), 3.78 (s, 3H), 3.62 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 214.2, 159.5, 142.4, 138.0, 129.1, 128.8, 128.4, 127.3, 125.7, 114.7, 110.7, 60.2, 55.4, 42.3; HRMS (ESI) calculated for C16H14NaO2 261.0891, found 261.0922 (M+Na)+.
13d (72%): 1H NMR (400 MHz, CDCl3) δ 7.63–7.26 (m, 7H), 7.11 (dd, J=9.2 Hz, 1H), 4.75 (m, 1H), 3.72 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 212.3, 142.2, 137.2, 130.5, 130.2, 129.0, 128.4, 128.1, 127.2, 126.4, 125.0, 123.0, 122.9, 122.7, 59.6, 42.8; HRMS (ESI) calculated for C16H11F3NaO 299.0660, found 299.0598 (M+Na)+.
13e (95%): 1H NMR (400 MHz, CDCl3) δ 7.30–7.24 (m, 4H), 5.54 (s, 1H), 3.98 (s, 1H), 3.53 (s, 2H), 1.67–1.49 (m, 8H); 13C NMR (100 MHz, CDCl3) δ 215.8; 141.3; 137.3; 134.7; 127.6; 127.5; 127.0; 125.3; 124.7; 62.3; 43.4; 26.1; 25.4; 22.7; 22.1; HRMS (ESI) calculated for C15H16NaO 235.1099, found 235.1174 (M+Na)+.
13f (90%): 1H NMR (400 MHz, CDCl3) δ 7.38–7.30 (m, 3H), 7.21 (d, J=7.2 Hz, 1H), 7.14 (d, J=7.6 Hz, 2H), 7.02 (d, J=8.0 Hz, 2H), 4.63 (s, 1H), 3.65 (s, 2H), 2.34 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 214.2, 141.5, 137.2, 137.0, 135.2, 129.5, 128.5, 128.3, 127.9, 127.8, 126.0, 124.8, 59.4, 42.9, 21.1; HRMS (ESI) calculated for C16H14NaO 245.0942, found 245.0921 (M+Na)+.
13g (88%): 1H NMR (400 MHz, CDCl3) δ 7.38–7.20 (m, 5H), 7.05 (d, J=6.4 Hz, 2H), 6.87 (d, J=6.4 Hz, 2H), 4.63 (s, 1H), 3.78 (s, 3H), 3.66 (s, 2H); 13C NMR (100 MHz, CDCl3) δ 214.4, 158.9, 141.6, 137.2, 130.3, 129.5, 128.3, 127.9, 127.8, 126.0, 124.8, 114.2, 58.9, 55.2, 42.8; HRMS (ESI) calculated for C16H14NaO2 261.0891, found 261.0664 (M+Na)+.
13h (91%): 1H NMR: (400 MHz, CDCl3) δ 8.06 (d, J=8.0 Hz, 1H), 7.75 (d, J=8.4 Hz, 2H), 7.61 (d, J=8.0 Hz, 2H), 7.45–7.18 (m, 5H), 4.71 (s, 1H), 3.73 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 212.9, 140.2, 136.7, 129.8, 129.4, 128.9, 128.5, 128.2, 126.9, 125.9, 125.7, 125.6, 125.4, 59.4, 42.8; HRMS (ESI) calculated for C16H11F3NaO 299.0659, found 299.0721 (M+Na)+.
13i (62%): 1H NMR (400 MHz, CDCl3) δ 7.39–7.27 (m, 5H), 6.35 (m, 1H), 6.16 (m, 1H), 4.82 (s, 1H), 3.72 (d, J=8.0 Hz, 2H); 13C NMR (100 MHz, CDCl3) δ 211.4, 150.5, 142.7, 136.9, 128.3, 127.8, 125.6, 124.9, 110.4, 107.8, 53.3, 42.9; HRMS (ESI) calculated for C13H10NaO2 221.0578, found 221.0842 (M+Na)+.
15a (61%; dr=1:1): 1H NMR (400 MHz, CDCl3) δ 7.41–7.26 (m, 6H); 7.18–7.10 (m, 3H); 4.70 (s, 1H); 3.61 (q, J=4.2 Hz, 1H); 1.48 (d, J=5.6 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 216.4, 143.2, 140.1, 138.3, 128.9, 128.6, 128.0, 127.6, 126.0, 124.2, 59.0, 46.7, 15.8; HRMS (ESI) calculated for C16H14NaO 245.0942, found 245.4017 (M+Na)+.
15b (53%; dr=4:1): 1H NMR (400 MHz, CDCl3) δ 7.33–7.21 (m, 9H), 4.42 (s, 1H), 3.34 (t, J=5.4Hz, 1H), 1.86 (m, 2H), 1.41–1.19 (m, 4H), 0.80 (t, J=6.0, 3H); 13C NMR (100 MHz, CDCl3) δ 214.2, 142.3, 141.0, 128.9, 128.5, 128.0, 127.7, 127.5, 58.9, 52.0, 31.2, 28.8, 22.7, 19.6; HRMS (ESI) calculated for C19H20NaO 287.1412, found 287.1361 (M+Na)+.
15c (48%; dr=2:1): 1H NMR (400 MHz, CDCl3) δ 7.24–7.01 (m, 9H), 4.38 (s, 1H), 3.25 (d, J=3.6Hz, 1H), 2.29 (m, J=4.0Hz, 1H), 1.01 (d, J=6.4Hz, 3H), 0.89 (d, J=6.4Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 213.8, 141.3, 140.9, 138.0, 128.9, 128.4, 128.0, 127.7, 127.5, 126.9, 126.1, 124.7, 58.9, 58.0, 31.2, 19.9, 19.6; HRMS (ESI) calculated for C18H18NaO 273.1255, found 273.1271 (M+Na)+.
15d (69%): 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J=10.0 Hz, 2H); 7.31 (m, 1H); 7.20–7.09 (m, 6H); 4.40 (s, 1H), 3.09 (s, 1H), 0.99 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 213.3, 141.3, 140.7, 138.6, 128.5, 128.0, 127.9, 127.6, 127.3, 126.6, 62.3, 58.6, 35.0; 30.5, 28.6; HRMS (ESI) calculated for C19H20NaO 287.1412, found 287.1438 (M+Na)+.
Synthesis of 2-(5-methyl-3-phenyl-1H-inden-2-yl)ethanenitrile 18b
To a solution of diethyl(cyanomethyl)phosphonate (531 mg, 3.0 mmol) in THF (1 mL) was added NaH (100 mg, 2.5 mmol) at 0 °C and the solution was allowed to stir for 30 minutes. Then a solution of 2-indanone 13b (100 mg, 0.45 mmol) in THF (1 mL) was added and the mixture was allowed to stir for 4 hours at room temperature. The mixture was diluted with saturated NaHCO3 (10 mL). Ether (20 mL) was added, and the phases were separated. The aqueous later was back-extracted with ether (1 × 20 mL), and the combined organic extracts were then dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography (SiO2) afforded indene 18b. 18b (91%): 1H NMR (400 MHz, CDCl3) δ 7.53–7.35 (m, 6H), 7.09–7.05 (m, 2H), 3.63 (s, 2H), 3.55 (s, 2H), 2.35 (s, 3H); 13C NMR: (100 MHz, CDCl3) δ 144.8, 139.1, 136.4, 133.6, 130.5, 128.9, 128.8, 128.1, 126.5, 123.6, 121.2, 40.2, 21.5, 17.9; HRMS (ESI) calculated for C18H15NNa 268.1102, found 268.1099 (M+Na)+.
Synthesis of tert-butyl 2-(5-methyl-3-phenyl-1H-inden-2-yl)ethylcarbamate 19b
To a solution of nitrile 18b (35.0 mg, 0.142 mmol) in methanol (1.2 mL) at 0 °C were added Boc2O (69.0 mg, 0.32 mmol) and NiCl2•6H2O (3.8 mg, 0.016 mmol). Then NaBH4 (42.4 mg, 1.12 mmol) was added in small portions over 30 minutes. After warming to room temperature and stirring for 2 hours, the mixture was diluted with saturated NaHCO3 (10 mL) and concentrated in vacuo to remove methanol. Ethyl acetate (10 mL) was added and the phases were separated. The aqueous later was back-extracted with ethyl acetate (1 × 10 mL), and the combined organic extracts were then dried over Na2SO4, filtered, and concentrated in vacuo. Purification of the residue by flash chromatography (SiO2) afforded indene 19b.
19b (60%): 1H NMR (400 MHz, CDCl3) δ 7.49–7.35 (m, 6H), 7.00 (m, 2H), 3.47 (s, 2H), 3.35 (m, 2H), 2.71 (t, J=6.8 Hz, 2H) 2.34 (s, 3H), 1.41 (s, 9H); 13C NMR (100 MHz, CDCl3) δ 155.7, 146.3, 141.1, 138.0, 135.2, 129.1, 128.5, 128.3, 127.3, 125.3, 123.3, 120.4, 100.0, 79.2, 40.2, 40.0, 29.4, 28.3, 21.5; HRMS (ESI) calculated for C23H27NNaO2 372.1939, found 372.1763 (M+Na)+.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (SC2 GM081064-01) and the Henry Dreyfus Teacher-Scholar Award for their generous support of our research program.
Footnotes
Supporting Information Available: Experimental details, characterization data, 1H, 13C spectra of all products, and 1H NMR kinetic data at 330K. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For reviews of the chemistry of ynol ethers and the methods for the synthesis of ynol ethers, see: Brandsma L, Bos HJ, Arens JF. In: The Chemistry of Acetylenes. Viehe HG, editor. Marcel Dekker; New York: 1969. pp. 751–60.Stang PJ, Zhdankin VV. In: The Chemistry of Triple-Bonded Functional Groups. Patai S, editor. Vol. 19 John Wiley & Sons; New York: 1994.
- 2.(a) Bruckner D. Synlett. 2000;10:1402–404. [Google Scholar]; (b) Moyano A, Charbonnier F, Greene AE. J Org Chem. 1987;52:2919–922. [Google Scholar]; (c) Pericas MA, Serratosa F, Valenti E. Tetrahedron. 1987:2311–316. [Google Scholar]; (d) Smithers RH. Synthesis. 1985:556–58. [Google Scholar]; (e) Himbert G, Loffler A. Synthesis. 1992:495–98. [Google Scholar]
- 3.Sosa JR, Tudjarian AA, Minehan TG. Org Lett. 2008;10:5091–094. doi: 10.1021/ol802147h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Christopher A, Brandes B, Kelly S, Minehan TG. Org Lett. 2006;8:451–54. doi: 10.1021/ol052685j. [DOI] [PubMed] [Google Scholar]
- 5.For reviews of the Claisen Rearrangement, see: Ziegler FE. Chem Rev. 1988;88:1423.Ito H, Taguchi T. Chem Soc Rev. 1999;28:43.Hiersmann M, Abraham L. Eur J Org Chem. 2002:1461.Nubbemeyer U. Synthesis. 2003:961.Hiersemann M, Nubbemeyer U, editors. Claisen Rearrangement. Wiley VCH; Weinheim: 2007.
- 6.Olsman H, Graveland A, Arens JF. Rec Trav Chim Pays-Bas. 1964;83:301. [Google Scholar]
- 7.Wunderli A, Zsindely J, Hansen HJ, Schmid H. Chimia. 1972;26:643. [Google Scholar]
- 8.The diazoketones were prepared by the procedure of Danheiser: Danheiser RL, Miller RF, Brisbois RG, Park SZ. J Org Chem. 1990;55:1959.
- 9.The protocol of Muthusamy et al. was followed for diazoketone-alcohol coupling: Muthusamy S, Babu SA, Gunanathan C. Tetrahedron Lett. 2002;43:3133.
- 10.Synthesis of aryloxyacetic acids: Yu H, Ballard CE, Boyle PD, Wang B. Tetrahedron. 2002;58:7663.Solladie G, Adamy M, Colobert F. J Org Chem. 1996;61:4369. doi: 10.1021/jo960134o.
- 11.Nahm S, Weinreb SM. Tetrahedron Lett. 1981;22:3815. [Google Scholar]
- 12.For seminal papers on substitutent effects on the aliphatic Claisen Rearrangment, see: Burrows CJ, Carpenter BK. J Am Chem Soc. 1981;103:6983.Burrows CJ, Carpenter BK. J Am Chem Soc. 1981;103:6984.
- 13.A Hammett plot of ln(k/ko) vs. σ for the data for substrates 12a, b, and d shows a break in slope at the data point for 12a, with a strongly negative slope (ρ= −10.5) to 12b and a moderately negative slope (ρ= −1.9) to 12d. The break in slope is suggestive of a change in mechanism upon proceeding from substrates bearing electron-donating substituents (12b) to substrates bearing electron-withdrawing subsituents (12d). See: Ritchie CD, Sager WF. Prog Phys Org Chem. 1964;2:323.See supporting information for data graphs.
- 14.A Hammett plot of ln(k/ko) vs. σ for the data for substrates 12a, f, and h shows a break in slope at the data point for 12a, with a negative slope (ρ= −1.62) to 12f and a slightly positive slope (ρ=+0.05) to 12h. Again, the break in slope is suggestive of a change in mechanism upon proceeding from substrates bearing electron-donating substituents (12f) to substrates bearing electron-withdrawing subsituents (12h). See supporting information for data graphs.
- 15.(a) We thank a reviewer for mentioning the allylic strain apparent in the anti disposition of the tert-butyl group in structure 16a. We believe that steric interactions between the tert-butyl group and the phenyl-substituted ketene in the corresponding syn form (16s, see Supporting Information) hinder alignment of the ketene carbonyl carbon with the benzylic carbon atom (which is necessary for 5-exo-dig cyclization to occur) by favoring rotamers in which the ketene is directed away from the benzylic carbon atom. Thus, despite the allylic strain present in 16a, it appears that the reaction proceeds preferentially through 16a to 15d. (b) To assess if the product ratios obtained for 15a-c represent an equilibrium mixture arising from a reversible process, we have attempted to obtain pure major or minor isomer from these reactions and resubject them individually to the reaction conditions. Chromatographic separation of the cis and trans indanone products has proven quite difficult since the diasteremers co-elute in most solvents. Thus, a mixture enriched in the major isomer of indanone 15c (dr=~5:1) was heated to 57°C in an NMR tube (CDCl3 as solvent) for 30 minutes, and essentially no change in the isomer ratio was observed. This suggests that the 5-exo- dig cyclization is irreversible under these reaction conditions and that the isomer distributions are under kinetic control.
- 16.(a) Guillon J, Dallemagne P, Leger JM, Sopkova J, Bovy PR, Jarry C, Rault S. Bioorg Med Chem. 2002;10:1043. doi: 10.1016/s0968-0896(01)00360-1. [DOI] [PubMed] [Google Scholar]; (b) Kolanos R, Siripurapu U, Pullagurla M, Riaz M, Setola V, Roth BL, Dukat M, Glennon RA. Bioorg Med Chem Lett. 2005;15:1987. doi: 10.1016/j.bmcl.2005.02.070. [DOI] [PubMed] [Google Scholar]; (c) Watanabe N, Nakagava H, Ikeno A, Minato H, Kohayakawa C, Tsuji J. Bioorg Med Chem Lett. 2003;13:4317. doi: 10.1016/j.bmcl.2003.09.049. [DOI] [PubMed] [Google Scholar]; (d) Karaguni IM, Glusenkamp KH, Langerak A, Geisen C, Ullrich V, Winde G, Moroy T, Muller O. Bioorg Med Chem Lett. 2002;12:709. doi: 10.1016/s0960-894x(01)00839-3. [DOI] [PubMed] [Google Scholar]
- 17.Alt HG, Koppl A. Chem Rev. 2000;100:1205. doi: 10.1021/cr9804700. [DOI] [PubMed] [Google Scholar]
- 18.Caddick S, Judd DB, Lewis AK, Reich MT, Williams MRV. Tetrahedron. 2003;59:5417. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.