

Abstract
Wacker-type oxidative cyclization reactions have been the subject of extensive research for several decades, but few systematic mechanistic studies of these reactions have been reported. The present study features experimental and DFT computational studies of Pd(OAc)2/pyridine-catalyzed intramolecular aerobic oxidative amination of alkenes. The data support a stepwise catalytic mechanism that consists of (1) steady-state formation of a PdII-amidate-alkene chelate with release of one equivalent of pyridine and AcOH from the catalyst center, (2) alkene insertion into a Pd–N bond, (3) reversible β-hydride elimination, (4) irreversible reductive elimination of AcOH, and (5) aerobic oxidation of palladium(0) to regenerate the active trans-Pd(OAc)2(py)2 catalyst. Evidence is obtained for two energetically viable pathways for the key C–N bond-forming step, featuring a pyridine-ligated and a pyridine-dissociated PdII species. Analysis of natural charges and bond lengths of the alkene-insertion transition state suggest that this reaction is best described as an intramolecular nucleophilic attack of the amidate ligand on the coordinated alkene.
Introduction
Palladium-catalyzed intramolecular aza-Wacker reactions provide access to a number of nitrogen-containing hetercycles such as pyrrolidine, piperazine, oxazolidin-2-one, pyridine, pyrimidyldione and urea deriviatives.1 These heterocyclization reactions have been a subject of considerable attention, with recent efforts focused especially on the develoment new synthetic transformations (e.g. 1,2-difunctionalization of alkenes),2–6 enantioselective reactions,7,8 and methods compatible with the use of molecular oxygen as the terminal oxidant.9
Seminal early work by Hegedus10 and others11 demonstrated that weakly coordinating nitrogen nucleophiles, especially sulfonamides, are particularly compatible with intramolecular oxidative amination reactions (e.g. eq 1). Early examples of these reactions used benzoquinone or CuCl2 as the stoichiometric oxidant.10b Later, the groups of Larock11d and Hiemstra11c demonstrated that molecular oxygen can be used as an effective oxidant if the reaction is carried out in DMSO as the solvent (eq 2).
 |
(1) |
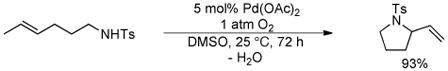 |
(2) |
In 2002, we reported the use of a very simple and efficient Pd(OAc)2/pyridine catalyst system for the intramolecular oxidative amination of alkenes to produce pyrrolidine and pyrroline heterocycles in high yield (Scheme 1).12,13 This catalyst system, originally reported by Uemura and coworkers for alcohol oxidation, 14 is capable of using molecular oxygen as the stoichiometric oxidant, and achieves unprecedented catalytic activity for such reactions. The p-toluenesulfonyl (Ts) group proved to be the most effective nitrogen substituent. Substrates bearing the p-nitrophenylsulfonyl (Ns) or Cbz (PhCH2OC(O)–) groups were also effective, albeit requiring somewhat longer reaction time. The reaction proceeds well with aromatic and aliphatic tosylamides and tolerates wide variations in solvent polarity, ranging from DMSO to heptane. Non-polar solvents permit the reaction to be performed at significantly reduced catalyst loading. With 0.2 mol % Pd(OAc)2 and 0.4 mol% pyridine in p-xylene, the hexenyltosylamide substrate reacted with a turnover rate of 70 h−1 during the first two hours of the reaction and turnover numbers up to 250–300 were attained. These values are significantly higher than those observed with previous catalyst systems.10,11 Moreover, this catalyst system has served as the starting point for the development of enantioselective oxidative heterocyclization reactions.2b,13b,c
Scheme 1.

Intramolecular Aerobic Oxidative Amination of Alkenes Catalyzed by Pd(OAc)2/Pyridine.12
Despite the extensive history of Pd-catalyzed oxidative amination reactions, systematic mechanistic investigations have been quite limited. The stereochemical course of the C–N bond forming step, aminopalladation of alkene, has been a focus of some attention, and evidence for both cis-2a,4b,15 and trans-aminopalladation10a,16 pathways have been obtained (Scheme 2). A recent study from our group demonstrated that the Pd(OAc)2/pyridine-catalyzed intramolecular aerobic oxidative amination reactions (cf. Scheme 1) proceed exclusively via cis-aminopalladation of the alkene, consistent with alkene insertion into a palladium-nitrogen bond (Scheme 2). 17 Here, we present experimental and computational studies that provide insights into other important aspects of the catalytic mechanism of Pd(OAc)2/pyridine-catalyzed aerobic oxidative intramolecular amination of alkene. Results include determination of the turnover-limiting step and catalyst resting state, and insights into the reversibility of catalytic steps, including those that take place after the turnover-limiting step, and the impact of O2 pressure on the catalyst stability. These results and their implications for Pd-catalyzed oxidative heterocyclization reactions are presented below.18
Scheme 2.

Possible Pathways for Intramolecular Aminopalladation of Alkenes.
Results and Discussion
Kinetics Studies: Substrate and Catalyst Effects
Intramolecular aerobic oxidative hetero-cyclization of (Z)-4-hexenyltosylamide 1 is catalyzed by Pd(OAc)2:pyridine (2:8 mol%) (eq 3), and the reaction proceeds to completion within 5 hours at 80 °C.19,20 A computer-interfaced gas-uptake apparatus was used to probe the kinetics of the catalytic reaction by monitoring the change in oxygen pressure within a sealed, temperature-controlled reaction vessel. The reaction time-course exhibits a monotonic decrease in pressure (Figure 1), and the lack of an induction period allowed us to obtain much of our kinetic data via initial-rates methods.
Figure 1.

A representative kinetic time-course for Pd(OAc)2/pyridine catalyzed intramolecular oxidative amination of (Z)-4-hexenyltosylamide obtained by gas-uptake methods. Data sampling occurred at a rate of 1 s−1 (not all data are shown). Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [amide] = 100 mM, 4.0 ml of toluene, 80 °C.
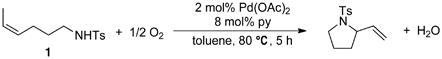 |
(3) |
The initial kinetic studies focused on the influence of the concentration of the primary reaction components (tosylamide, O2 and catalyst) on the initial turnover rate. The data revealed a saturation rate dependence on the tosylamide (Figure 2A) catalyst (Figure 2B) concentrations. The rate dependence on the O2 pressure was found to be dependent on the Pd(OAc)2:pyridine ratio. At a Pd:py ratio of 1:4, no dependence on O2 pressure was observed above 300 torr (Figure 3A). At a Pd:py ratio of 1:2, an apparent O2-dependence was observed, and the rate observed at the maximum pressure of the reaction vessel was approximately two-fold higher than observed in the reaction with a 1:4 Pd:py ratio (Figure 3B). In the reaction with Pd:py = 1:2, significant quantities of palladium black were observed at lower O2 pressures.
Figure 2.

(A) Dependence of the initial rate on amide concentration. The curve fit results from a nonlinear least-squares fit to a hyperbolic function of [amide]. Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [amide] = 0–240 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C. (B) Dependence of the initial rate on catalyst concentration, where the “catalyst” is a 4:1 mixture of pyridine and Pd(OAc)2. Conditions: [Pd(OAc)2] = 0–12.0 mM, [pyridine] = 0–48.0 mM [amide] = 100 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
Figure 3.

(A) Dependence of the initial rate on the initial oxygen pressure (with Pd/py ratio of 1/4). Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [tosylamide] = 100 mM, 4.0 ml of toluene, initial pO2 = 300–700 torr, 80 °C. (B) Dependence of the initial rate on the initial oxygen pressure (with Pd/py ratio of 1/2). Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 4.0 mM, [tosylamide] = 100 mM, 4.0 ml of toluene, initial pO2 = 300–700 torr, 80 °C.
Pyridine and Acetic Acid Effect
The initial turnover rates exhibit a sharp dependence on [pyridine] that maximizes at a Pd:py ratio of approximately 1:1–1:1.5 (Figure 4A), and beyond which significant inhibition is observed. A rate dependence on [acetic acid] concentration is also observed. The catalytic turnover rate maximizes at a 1:1 ratio of added acetic acid to Pd(OAc)2 (Figure 4B).
Figure 4.

(A) Dependence of the initial rate on pyridine concentration with the [Pd(OAc)2] held constant. Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 0–10 mM, [amide] = 100 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C. (B) Dependence of the initial rate on acetic acid concentration. Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [amide] = 100 mM, [AcOH] = 0–100 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
If excess [pyridine] is added to the reaction mixture (120 mM; 10–240 equiv relative to Pd(OAc)2), the intial rate exhibits a square-root dependence on [Pd(OAc)2] (Figure 5A). This result contrasts the hyperbolic dependence observed when the catalyst concentration is varied at a constant py:Pd ratio of 4:1 (Figure 2B). The rate dependence on [Pd(OAc)2] changes again if both pyridine and acetic acid are added to the reaction mixture (120 mM and 5 mM, respectively). Under these conditions, a linear dependence of the rate on [Pd(OAc)2] is observed (Figure 5B).
Figure 5.

(A) Dependence of the initial rate on the square root of Pd(OAc)2 concentration in the presence of large excess of pyridine. Conditions: [pyridine] = 120 mM, [amide] = 100 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C. (B) Dependence of the initial rate on the concentration of Pd(OAc)2 in the presence of excess pyridine and acetic acid. Conditions: [pyridine] = 120 mM, [AcOH] = 5 mM, [amide] = 100 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
Electronic Effects
A series of para-substituted benzenesulfonamides (p-X = OMe, H, CH3, Cl, NO2) were used to examine electronic effects on the catalytic turnover rate. The Hammett plot (Figure 6) reveals that electron-rich substrates react more rapidly than those bearing electron-withdrawing groups, and a linear fit of the data exhibits a slope (ρ) of −0.22. These results expand upon the qualitative data acquired with the p-CH3 and -NO2 derivatives (cf. Scheme 1), which showed that the Ns-substituted derivative required a longer reaction time.
Figure 6.

Hammett plot derived from the relative initial rates of catalytic oxidative amination conducted with a series of para-substituted benzenesulfonamides. Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [amide] = 100 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
Kinetic Isotope Effects and Isotopic Labeling Studies
Kinetic isotope effects were determined by comparing the independent rates of reaction for tosylamide substrates bearing terminal CH3- and CD3-groups (eq 4, Figure 7), as well as NH- vs. ND-labelled tosyl amides (eq 5, Figure 8). Small, secondary kinetic isotope effects were evident in both cases (1.20 ± 0.05 and 1.15 ± 0.05, respectively).
Figure 7.

Dependence of the initial O2 uptake rate on amide concentration. CH3- vs. CD3-labelled tosylamides. Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [amide] = 0–240 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
Figure 8.

Dependence of the initial O2 uptake rate on amide concentration. NH- vs. ND-labelled tosylamides. Conditions: [Pd(OAc)2] = 2.0 mM, [pyridine] = 8.0 mM, [amide] = 0–240 mM, 4.0 ml of toluene, initial pO2 = 700 torr, 80 °C.
 |
(4) |
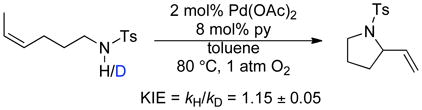 |
(5) |
Deuterium incorporation (~20%) into the β-vinyl position was observed in the reaction of CD3-labelled tosylamide (eq 4). The scrambling of deuterium label into the internal vinylic position suggests the β-hydride elimination from the palladium-alkyl intermediate is reversible. Crossover experiments demonstrate that this isotopic scrambling takes place exclusively in an intramolecular fashion. No intermolecular deuterium scrambling was observed when the reaction was carried out with a 1:1 mixture of CH3- and CD3-labelled tosylamide (eq 7).21
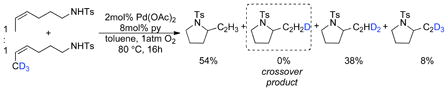 |
(6) |
The partially deuterated tosylamide substrates 2 and 3 were utilized to probe the intramolecular kinetic isotope effect associated with β-hydride versus β-deuteride elimination from the palladium-alkyl intermediate 4 (Scheme 3).22 1H NMR spectroscopic analysis of final product mixtures reveals an approximately 3:1 distribution between β-hydride elimination and β-deuteride elimination products.
Scheme 3.

Intramolecular Selectivity Between β-Hydride and β-Deuteride Elimination.
The prospect of reversible aminopalladation was examined with substrate 5, which features two symmetrical alkenes with isotopically substituted methyl groups (eq 7). Two limiting scenarios are possible with this substrate: 1) if aminopalladation is reversible, the ratio of cyclic products 6 and 7 could approach 3:1, as dictated by the relative rates of β-H versus β-D elimination; 2) if aminopalladation is irreversible, the yield of 6 and 7 should be identical (Scheme 4). When this reaction was performed, a 1.04:1 ratio of 6: 7 was obtained, implicating irreversible aminopalladation.
Scheme 4.

Probing the Reversibility of Aminopalladation of Alkene with Diene Substrate 5.
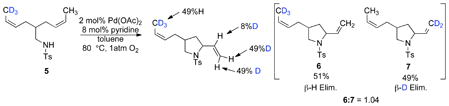 |
(7) |
1H NMR Spectroscopic Studies: Characterization of the Catalyst Resting
1H NMR spectroscopic studies were conducted to gain insights into the identity of the catalyst resting state. Trans-(py)2Pd(OAc)2 forms upon the addition of 2 equiv of pyridine to Pd(OAc)2, and was previously characterized in our mechanistic study of Pd(OAc)2/pyridine catalyzed aerobic alcohol oxidation.23 Two sets of pyridine 1H resonances are observed if more then 2 equiv. of pyridine are present, corresponding to coordinated and free pyridine. Titration of (Z)-4-hexenyltosylamide (from 0 to 77 equiv. relative to Pd(OAc)2) into a solution of the catalyst (1:2.2 Pd(OAc)2:py) results in essentially no change of the chemical shifts associated with the coordinated and free pyridine at temperatures ranging from −40 to 40 °C.22
Catalytic Mechanism Based on Experimental Observations
Intramolecular aerobic oxidative amination of alkenes catalyzed by Pd(OAc)2/pyridine proceeds via a PdII/Pd0 catalytic cycle in which palladium(II)-mediated substrate oxidation and aerobic oxidation of the catalyst occur in two independent, sequential stages. The kinetic studies described above, which reveal no dependence of the rate on [O2] and a dependence on [catalyst] and [amide], indicate that a step associated with PdII-mediated substrate oxidation is turnover-limiting. Consistent with this proposal, trans-(py)2Pd(OAc)2 has been identified as the catalyst resting state, and this species is capable of promoting kinetically competent stoichiometric substrate oxidation in the absence of molecular oxygen.
The catalytic cycle shown in Scheme 5 illustrates a mechanism that is consistent with all of the experimental data presented above. Key features of this mechanism include: (1) a two-step sequence for formation of the palladium(II) amidate-alkene chelate 9, via release of one equivalent of pyridine and acetic acid (steps i and ii); (2) turnover-limiting alkene insertion into Pd–N bond (step iii); (3) β-hydride elimination from the alkyl-palladium(II) intermediate 10 (step iv); and (4) dissociation of product and reductive elimination of AcOH (both included in step v). Reoxidation of a pyridine-ligated Pd0 species completes the catalytic cycle (step vi).
Scheme 5.

Proposed Catalytic Mechanism for Pd(OAc)2/Pyridine-Catalyzed Aerobic Oxidative Intramolecular Amination of Alkenes.
The precise nature and sequence of the first two steps (Scheme 5, steps i and ii) cannot be established from the available data; however, reversible formation of intermediate 9 is consistent with the small N–H/N–D isotope effect (Figure 8), and it rationalizes the inhibitory effect of pyridine and AcOH. The amidate-alkene chelate 9 is the starting point for cis-aminopalladation of the alkene.17
This mechanism also accounts for the variable [catalyst] dependence observed under different reaction conditions: hyperbolic dependence with a 1:4 Pd:py catalyst system; half-order dependence in the presence of excess pyridine (py:Pd > 10:1); and first-order dependence in the presence of excess pyridine and AcOH (Figure 2B, Figure 5). Nearly identical kinetic behaviour was characterized previously for Pd(OAc)2/pyridine-catalyzed alcohol oxidation.20 These observations are attributed to the kinetic influence of the small pre-equilibrium and/or steady-state quantities of pyridine and AcOH, whose concentrations will be directly proportional to the concentration of the Pd intermediates, 8 and 9, respectively. If sufficient quantities of pyridine and AcOH are added to ensure that a constant excess concentration of these species is present, a first-order dependence on [Pd(OAc)2] is observed. A more-thorough discussion of this effect has been presented previously,20 and rate laws, based on steady-state and pre-equilibrium formation of intermediates 8 and 9 are presented in the Supporting Information.
The neutral ligand, pyridine, plays a key role in the stabilization and aerobic oxidation of palladium(0). At pyridine:Pd ratio < 4:1 (e.g. 2:1), the catalytic turnover rate exhibits an O2 dependence and significant palladium-black formation is observed (Figure 3B), indicating a competition between catalyst oxidation and decomposition upon formation of palladium(0). The beneficial rate effect of pyridine, which maximizes at a ~1:1–1:1.5 Pd:pyridine ratio (Figure 4A), probably reflects several factors. Palladium acetate exists as a trimer in non-polar solvents,24 and the presence of coordinating ligands lead to the formation of lower nuclearity Pd species that are probably more electrophilic and/or reactive with organic substrates.25 In addition, pyridine can stabilize the palladium(0) intermediate 12, either by preventing aggregation or enhancing the reaction rate between 12 and molecular oxygen.23 At high concentrations, however, pyridine inhibits the catalytic turnover rate because it competes with the substrate for coordination sites at the Pd center.26
The addition of excess AcOH strongly inhibits the catalytic turnover rates at high concentrations (Figure 4B). This observation is readily explained by a competition between alkene insertion into palladium-amidate bond (k3, Scheme 5) and protonation of palladium-amidate intermediate 9 (k-2, Scheme 5). Elevated [AcOH] will favor protonation of 9 and lead to inhibition of catalytic turnover. The small, but noticable, beneficial effect of AcOH at low concentrations (Pd:AcOH = 1:1) is less certain but may be related to the ability of AcOH to promote the pyridine-dissociation equilibrium in step i via hydrogen bonding to the pyridine (Figure 4B).27
The proposed turnover-limiting alkene insertion into a Pd–Namidate bond (k3, Scheme 5) is consistent with the observed Hammett correlation (Figure 6). Alkene insertion into the Pd–N bond is expected to be more facile with more-electron-rich (i.e., nucleophilic) amidates.28 β-Hydride elimination (step iv, Scheme 5) takes place after the turnover limiting step and, therefore, the proposed mechanism is consistent with the small secondary kinetic isotope effect determined by comparing independent rates of tosylamide substrates bearing terminal CH3- versus CD3-groups (Figure 7).
The isotope-labelling studies reveal three key features of the reaction: (1) β-hydride elimination is reversible (cf. eq 4, Scheme 3); (2) deuterium scrambling among the three vinyl positions occurs exclusively in an intramolecular fashion (cf. eq 6); and (3) aminopalladation of alkene is irreversible (cf. Scheme 4). All three observations are accommodated by the proposed mechanism in Scheme 5. Turnover-limiting alkene insertion into palladium-amidate bond renders the overall aminopalladation of alkene irreversible, and the exclusively intramolecular deuterium scrambling suggests that the product dissociation is irreversible.
Computational studies
With the mechanistic framework in Scheme 5 as a starting point, we performed computational studies of the oxidative amination mechanism using density functional theory (DFT) methods in order to gain further insights into the energetics of the reaction pathway. These studies employed trans-(py)2Pd(OAc)2 as the catalyst with a model substrate, (Z)-4-hexenylmesylamide 1M (eq 8), in which the experimental toluenesulfonyl group was replaced with a methanesulfonyl group in the computational studies (note: for experimental compounds modeled by computed analogs differing only in the identity of the sulfonyl group, we use the experimental compound number with an “M” suffix for the computed structure). The calculations were performed using the B3LYP functional and the experimental solvent, toluene, was described with a continuum solvent model. Free energies (the only energies discussed here) were calculated for all intermediates and transition states at 80 °C.29 All calculated free energies presented below are reported relative to the energy of trans-(py)2Pd(OAc)2 + 1M.30
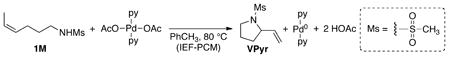 |
(8) |
The experimental studies indicate that aminopalladation of the alkene is the turnover-limiting step of the catalytic cycle. The PdII-amidate-alkene chelate 9M is the starting point for the aminopalladation step, and the calculated pathway for formation of this species from trans-(py)2Pd(OAc)2 + 1M is presented in the Supporting Information. Two energetically similar pathways were identified for alkene insertion into the palladium-amidate bond, a pyridine-ligated and pyridine-dissociated pathway (Figure 9). The first pathway reflects the proposed mechanism in Scheme 5 and proceeds via transition state 13AP-TS for C–N bond formation, in which pyridine is coordinated to the PdII center (Scheme 5, solid line). Alkene insertion into the Pd–N bond exhibits a barrier of 22.0 kcal/mol. In the other pathway, pyridine dissociates from the Pd center to form a PdII-amidate-alkene species with a κ2-acetate ligand, κ2-16M. Subsequent alkene insertion via the pyridine-dissociated transition state 17AP-TS exhibits a much lower barrier, ΔG‡ = 14.7 kcal/mol (ΔΔG‡ = −5.8 kcal/mol). In this pathway, pyridine dissociation from PdII via [15/py]TS is calculated to be the rate-limiting step, with a calculated barrier slightly higher (1.2 kcal/mol) than transition state 13AP-TS. The pyridine-ligated alkyl-PdII metallacycle 10M is 4.7 kcal/mol higher in energy than the corresponding pyridine-dissociated species 18M, which has a κ2-acetate ligand.
Figure 9.

Lowest free energy pathways for formation of aminopalladative intermediates with and without pyridine ligation. All reactions are relative to trans-(py)2Pd(OAc)2 + 1M; free py and ½[AcOH]2.
Several other aminopalladation transition states were considered in the course of these studies, but all proved to be higher in energy than those shown in Figure 9. For example, a transition state isomer of 13AP-TS, in which the amidate nitrogen atom and alkene are trans to acetate and pyridine ligands, respectively, is 2.3 kcal/mol higher in energy (Scheme 6, iso-13AP-TS). And, pathways that proceed via a six-membered C–N bond-forming transition state, in which a sulfonyl oxygen atom is coordinated to the PdII center proved to be much higher in energy (> 35 kcal/mol; see Scheme 6).
Scheme 6.

Calculated Energies for Alternative Aminopalladation Transition States.
The metrical parameters for the Pd-amidate-alkene intermediates 9M and κ2-16M and the alkene insertion transition states 13AP-TS and 17AP-TS are shown in Figure 10. The bond lengths together with the calculated natural charges for these species are consistent with a reaction pathway that can be described as an intramolecular nucleophilic attack of the amidate ligand onto a PdII-activated alkene. For example, transition state 13AP-TS features a lengthened carbon-carbon bond relative to 9M (1.42 vs. 1.39 Å, respectively), and the alkene is unsymmetrically coordinated to the palladium center in 13AP-TS, reflecting a progression toward the 5-exo-trig cyclization transition-state geometry. Natural charges (NC) on the alkene reflect a highly polarized carbon-carbon bond, NC = −0.38 and +0.07 for the terminal and internal carbon atoms of the alkene, respectively.29 In addition, the palladium-coordinated nitrogen atom of the sulfonamide has substantial negative charge in 13AP-TS: NC = −0.86.
Figure 10.
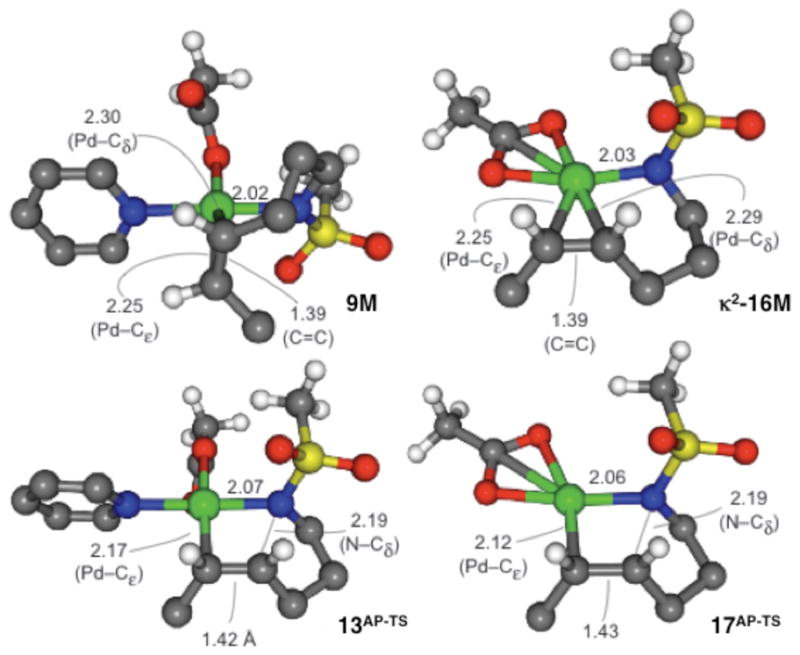
Ball-and-stick models with bond-length metrics for the Pd-amidate-alkene intermediates, 9M and κ2-16M, and alkene insertion transition states, 13AP-TS and 17AP-TS.
The experimental data reveal that β-hydride elimination and subsequent AcOH-reductive-elimination take place after the turnover-limiting step in the catalytic mechanism. Nevertheless, these are important steps in the overall reaction.31,32 The “pyridine-ligated” intermediate 10M and the “pyridine-dissociated” intermediate 18M can both undergo β-hydride elimination, and the energy profiles originating from these species are shown in Figure 11. In both pathways, the energy barriers of vinyl pyrrolidine product dissociation (via structures 22 and 31; red pathway) are large relative to the barriers associated with isomerisation between 1° and 2° palladium-alkyl intermediates (blue pathway). Therefore, both pathways are consistent with the experimentally observed intramolecular deuterium scrambling into β-vinyl position (eq 6). The overall barriers evident on these profiles, however, suggests the “pyridine-ligated” pathway (Figure 11A) is unlikely. The barrier for product dissociation via 22 is calculated to be higher in energy than the barrier calculated for turnover-limiting aminopalladation (Figure 9). Turnover-limiting product dissociation is not consistent with the Hammett correlation observed with the different sulfonamide nucleophiles (Figure 6) and the pyridine-dissociated pathway (Figure 11B) provides an energetically viable alternative for the β-hydride elimination and product dissociation steps.
Figure 11.

Low-energy pathways for the β-hydride elimination and AcOH-reductive-elimination. All reactions are relative to (py)2Pd(OAc)2 + 1M; free py and ½[AcOH]2. (A) Pyridine-ligated pathway; (B) Pyridine-dissociated pathway.
Modified Proposed Catalytic Mechanism
The collection of experimental and computational data presented above provide the basis for a slightly revised catalytic cycle for the Pd(OAc)2/pyridine-catalyzed aerobic oxidative intramolecular amination of alkenes (Scheme 7). This mechanism reflects the two possible aminopalladation pathways, in which the PdII-amidate-alkene intermediate 9 can react via a pyridine-ligated pathway or a pyridine-dissociated pathway. The former pathway features direct insertion of the alkene into the palladium-amidate bond (Scheme 7, 9 → 10), followed by dissociation of the pyridine from PdII (10 → 18). The latter pathway features dissociation of pyridine prior to the insertion of alkene into palladium-amidate bond (Scheme 7, 9 → 16 → 18). The two proposed pathways feature different turnover limiting steps, alkene insertion (9 → 10) vs. pyridine disscociation (9 → 16), both of which are consistent with the experimental data, including (a) the rate law, (b) the Hammett plot (pyridine dissociation should proceed more rapidly with more-electron-rich trans amidate ligands), and (c) reversible β-hydride elimination after the turnover-limiting step of the mechanism.
Scheme 7.

Refined Catalytic Mechanism for Pd(OAc)2/Pyridine-Catalyzed Aerobic Oxidative Intramolecular Amination of Alkenes.
The pyridine-dissociated pathway for β-hydride elimination (Scheme 7, 18 → 29) is favored over the originally proposed pyridine-ligated pathway (Scheme 5, 10 to 11) based on computational analysis. The regeneration of palladium(II) catalyst from palladium-hydride intermediate 29 proceeds via irreversible reductive elimination of AcOH. Facile reoxidation of palladium(0) complex 37 takes place in competition with catalyst decomposition.
Conclusion
The mechanistic study presented in this paper provides a thorough analysis of palladium-catalyzed Wacker-type aerobic oxidative amination reactions. Experimental and computational data suggest a catalytic mechanism that consists of (1) steady-state formation of a PdII-amidate-alkene intermediate, (2) alkene insertion into a Pd–N bond, (3) reversible β-hydride elimination, (4) irreversible AcOH reductive elimination, and (5) aerobic oxidation of palladium(0) to regenerate the active catalyst trans-Pd(OAc)2(py)2. Two energetically-viable pathways, including a pyridine-ligated and a pyridine-dissociated pathways, have been identified for the key C–N bond-forming step. The difference between these two nearly-isoenergetic pathways lies in the coordination environment at Pd when the alkene inserts into the palladium-amidate bond. Analysis of natural charges and bond lengths of the alkene-insertion transition state reveal that this formal migratory insertion process can be described as an intramolecular attack of the nitrogen nucleophile onto the coordinated alkene. The possible involvement of ligand-dissociated pathways for nucleopalladation of alkenes has important practical implications for the development of enantioselective reactions. Dissociation of a chiral ligand from PdII will result in formation of an achiral catalyst. Depending on the relative energies of the other steps in the mechanism, this process could account for some of the historical difficulty in developing enantioselective Wacker-type oxidative cyclization reactions.
Experimental Section
Representative procedure for gas-uptake kinetics
A typical reaction was conducted as follows. A 25 mL round-bottom flask with a stirbar was attached to an apparatus with a calibrated volume and a pressure transducer designed to measure the gas pressure within the sealed reaction vessel. The apparatus was evacuated to 10 Torr and filled with oxygen to 800 Torr and this cycle was repeated 10 times. The pressure was established at 675 Torr. When the pressure stablilzed in the apparatus, stock solutions of Pd(OAc)2 (2.5 mM, in 3.2 mL toluene) and pyridine (1.2M, in 0.4 mL toluene) were added via syringe through a septum. The flask was heated to 80 °C. When the temperature stabilized, stock solution of substrate (1.0 M, in 0.4 mL toluene) was added via syringe through a septum. Data was acquired using custom software written within LabVIEW™. Correlations between oxygen uptake and conversion were made by analysis by 1H NMR spectroscopy with 1,3,5-trimethoxybenzene as an internal standard.
Representative procedure for reactions with isotopically-labeled substrates
Pd(OAc)2 (0.4 mg, 2 μmol) was added to 13×100 mm disposable culture tubes. The reaction tubes were placed into a custom 48-well parallel reactor mounted on a Large Capacity Mixer and the headspace was purged with molecular oxygen for ca. 15 min. Solutions of pyridine (8 μmol in 0.5 mL toluene) and substrate (0.1 mmol in 0.5 mL toluene) were added to the tubes. The reactions were carried out for 24 h under an oxygen atmosphere (1 atm) at 80 °C. Following removal of the solvent under vacuum, the crude oxidative amination product was purified via column chromatography with ethyl acetate/hexanes and analyzed by 1H NMR spectroscopy.
Substrate syntheses
(Z)-4-hexenylbenzenesulfonamide 1
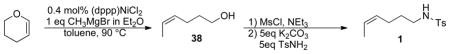
Cis-4-Hexen-1-ol 38 was prepared by a modification of the literature procedure.33 A 3.0 M ethereal solution of MeMgBr (100 mL, 0.3 mmol) was added to a stirring suspension of (dppp)NiCl2 (610 mg, 1.1 mmol) in 200 mL dry toluene under an N2 atmosphere. The stirring was continued at room temperature for 20 min. 4,5-Dihydropyran (25 g, 0.3 mol) was added, and the solution was heated to 90 °C and stirred overnight. The cooled reaction mixture was poured into a saturated ammonium chloride solution and extracted with ether. The extract was dried with MgSO4 and the solvent was removed under vaccum. The residue was purified by distillation to afford cis-4-hexen-1-ol (27 g, 90% yield). The identity and purity of cis-4-hexen-1-ol was confirmed by comparison of the 1H NMR spectrum to that reported in the literature.33
(Z)-4-Hexenyltoluenesulfonamide 1 was prepared via sequential formation of the mesylate from cis-4-hexen-1-ol and SN2 substitition of the mesylate by tosylamide according to literature procedures.17
(Z)-4-hexenyltoluenesulfonamide-d3 (6-d3-1)

The procedure for preparation of CD3-labeled (Z)-4-hexenyltoluenesulfonamide 6-d3-1 was the same as that of (Z)-4-hexenyltoluenesulfonamide, with the exception that CD3MgI (1.0 M in diethyl ether) was used instead of CH3MgBr.33 (Z)-4-Hexenylbenzenesulfonamide-d3: 1H NMR (CDCl3) δ 7.74 (dt, J = 8.4, 1.8 Hz, 2H), 7.31 (dt, J = 8.4, 1.8 Hz, 2H), 5.44 (dt, J = 10.8, 0.9 Hz, 1H), 5.27 (dt, J = 7.2, 10.8 Hz, 1H), 4.50 (t, J = 6.0 Hz, 1H), 2.95 (dt, J = 6.9, 6.3 Hz, 2H), 2.43 (s, 3H), 2.03 (dq, J = 7.2, 1.2 Hz, 2H), 1.54 (m, 2H); 2H NMR (CHCl3) δ 1.52 (s, 3D);13C NMR (CDCl3) δ 143.5, 137.2, 129.9, 129.2, 127.3, 125.2, 43.1, 29.5, 24.1, 21.7, 12.1 (heptad, J = 19.2 Hz); HRMS (ESI) calculated for C13H16D3NO2SNa, 279.1223; measured, 279.1211.
CH2D-labeled (Z)-4-hexenyltosylamide 2
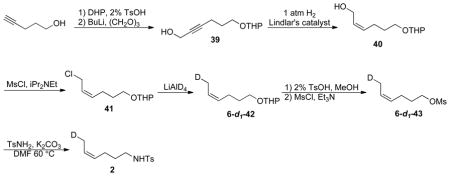
40: Alkyne 39 was synthesized according to literature procedures.34 To a solution of 39 (4 g, 20 mmol) in MeOH (50 mL), Lindlar’s catalyst (100 mg) and quinoline (1 mL, 8.5 mmol) were added. The mixture was agitated under an H2 atmosphere (8 psig) at room temperature for 2 hours. The catalyst was filtered, and the solvent was removed under vacuum. The residue was purified by silica gel chromatography (hexane/ethyl acetate) to afford 40 (3.23 g, 79% yield over three steps). 1H NMR (CDCl3) δ 5.66 (m, 1H), 5.50 (m, 1H), 4.53 (dd, J = 4.2, 2.7 Hz, 1H), 4.24 (ddd, J = 12.6, 7.2, 1.2 Hz, 1H), 4.10 (ddd, J = 12.6, 7.2, 1.2 Hz, 1H), 3.86 (m, 1H), 3.74 (dt, J = 9.6, 6.6 Hz, 1H), 3.48 (M, 1H), 3.39 (dt, J = 9.6, 6.6 Hz, 1H), 3.12 (br, 1H), 2.20 (m, 2H), 1.81-1.50 (m, 8H).
41: To a solution of allylic alcohol 40 (3 g, 15 mmol) in methylene dichloride (50 mL), iPr2NEt (2.6 g, 20 mmol) and MsCl (1.8 g, 16 mmol) were added at 0 °C via syringe. The reaction was monitored by TLC. After the reaction was complete, water (40 mL) was added. The mixture was extracted with diethyl ether. The extract was dried with MgSO4 and the solvent was removed under vaccum. The residue was purified by silica gel chromatography (hexane/ethyl acetate) to afford 41 (1.9 g, 59% yield). 1H NMR (CDCl3) δ 5.66 (m, 2H), 4.57 (dd, J = 4.2, 2.7 Hz, 1H), 4.16 (d, J = 5.1 Hz, 2H), 3.88 (m, 1H), 3.75 (dt, J = 9.6, 6.6 Hz, 1H), 3.50 (m, 1H), 3.39 (dt, J = 9.6, 6.6 Hz, 1H), 2.23 (m, 2H), 1.71-1.50 (m, 8H).
6-d1-42: Under an N2 atmosphere, LiAlD4 (150 mg, 4 mmol) was suspended in dry diethyl ether (20 mL). 41 (600mg, 2.8mmol) was added slowly via a syringe to this solution. The resulting suspension was refluxed for 2 hours. Excess LiAlD4 was quenched by water, and then by an aqueous solution of NaOH (40%, 10 ml). The mixture was stirred for 1 h. The white solid was filtered, and the solvent was removed under vacuum to afford crude 6-d1-42 (471 mg, 91%). 1H NMR (CDCl3) δ 5.44 (m, 2H), 4.58 (dd, J = 4.2, 2.7 Hz, 1H), 3.87 (m, 1H), 3.75 (dt, J = 9.6, 6.6 Hz, 1H), 3.50 (M, 1H), 3.39 (dt, J = 9.6, 6.6 Hz, 1H), 2.12 (m, 2H), 1.71-1.50 (m, 10H).
6-d1-43: To a solution of crude 6-d1-42 (471 mg, ~2.6 mmol) in MeOH (5 mL), TsOH·H2O (10 mg, 0.05 mmol) was added. The mixture was stirred for 1 h. Then Na2CO3 (50 mg, 0.5 mmol) was added, and the mixture was stirred for 30 min. The solid was filtered, and the solvent was removed under low vacuum. The crude oil was dissolved in CH2Cl2 (20 mL), and Et3N (1 mL) and MsCl (400 mg, 3.5 mmol) were then added slowly via syringe. The mixture was stirred overnight at room temperature. After the reaction was complete (by TLC), water (20 mL) was added. The mixture was extracted with diethyl ether. The extract was dried with MgSO4 and the solvent was removed under vaccum. The residue was purified by silica gel chromatography (hexane/ethyl acetate) to afford 6-d1-43 (214 mg, 78% yield). 1H NMR (CDCl3) δ 5.52 (m, 1H), 5.37 (m, 1H), 4.23 (t, J = 6.6 Hz, 2H), 3.01 (s, 3H), 2.18 (q, J = 7.2 Hz, 2H), 1.83 (m, 2H), 1.60 (m, 2H). 13C NMR (CDCl3) δ 128.4, 125.9, 69.7, 37.5, 29.1, 22.8, 12.7 (t, J = 19.6 Hz).
2: This compound was prepared according to the literature procedure used for 1.17 (60% yield, colorless oil) 1H NMR (CDCl3) δ 7.76 (dt, J = 8.4, 1.8 Hz, 2H), 7.31 (dt, J = 8.4, 1.8 Hz, 2H), 5.45 (m, 1H), 5.28 (m, 1H), 4.42 (t, J = 1.8 Hz, 1H), 2.95 (dt, J = 6.9, 6.3 Hz, 2H), 2.43 (s, 3H), 2.03 (q, J = 7.2 Hz, 2H), 1.54 (m, 4H); 2H NMR (CHCl3) δ 1.53 (s, 1D); 13C NMR (CDCl3) δ 143.6, 137.3, 129.9, 129.1, 127.4, 125.4, 43.1, 29.6, 24.2, 21.7, 12.9 (t, J = 19.8 Hz); HRMS (ESI) calculated for C13H18DNO2S, 254.1199; measured, 254.1192.
CHD2-labeled (Z)-4-hexenyltosylamide 3
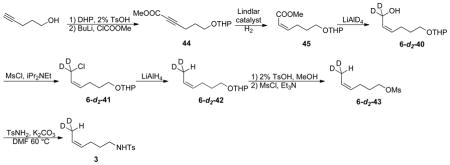
45: The compound 44 was synthesized from 4-pentyn-1-ol according to literature procedures.35 44 was hydrogenated according to the procedure used for compound 40 to afford 45 (95%, colorless oil). 1H NMR (CDCl3) δ 6.28 (dt, J = 11.7, 7.5 Hz, 1H), 5.79 (dt, J = 11.7, 1.8 Hz, 1H), 4.58 (dd, J = 4.2, 2.7 Hz, 1H), 3.88 (m, 1H), 3.75 (dt, J = 9.6, 6.6 Hz, 1H), 3.70 (s, 3H), 3.50 (m, 1H), 3.41 (dt, J = 9.6, 6.6 Hz, 1H), 2.75 (m, 2H), 1.81-1.45 (m, 8H).
6-d2-40: Under an N2 atmosphere, LiAlD4 (1.1 g, 27 mmol) was suspended in dry diethyl ether (40 mL). 45 (3.4 g, 15 mmol) was added to this suspension at −20 °C via syringe. The mixture was warmed to 0 °C and stirred for 2 hours. The excess LiAlD4 was quenched by water, and then an aqueous solution of NaOH (40%, 10 ml) was added. The mixture was stirred for 1 h. The white solid was filtered and the solvent was removed under vacuum to afford crude 6-d2-40 (2.8 g, 93%). 1H NMR (CDCl3) 5.70 (d, J = 10.8 Hz, 1H), 5.50 (ddd, J = 10.8, 8.4, 6.6 Hz, 1H), 4.52 (dd, J = 4.2, 2.7 Hz, 1H), 3.86 (m, 1H), 3.75 (dt, J = 9.6, 6.6 Hz, 1H), 3.49 (m, 1H), 3.41 (dt, J = 9.6, 6.6 Hz, 1H), 2.30 (m, 2H), 2.19 (s, 1H), 1.79-1.51 (m, 8H).
6-d2-41: This compound was prepared according to the procedure used for 41 (see above). (62% yield). 1H NMR (CDCl3) δ 5.41 (m, 2H), 4.58 (dd, J = 4.2, 2.7 Hz, 1H), 3.88 (m, 1H), 3.75 (dt, J = 9.6, 6.6 Hz, 1H), 3.50 (m, 1H), 3.39 (dt, J = 9.6, 6.6 Hz, 1H), 2.13 (m, 2H), 1.71-1.50 (m, 8H).
6-d2-42: Under an N2 atmosphere, LiAlH4 (500 mg, 13 mmol) was suspended in dry diethyl ether (40 mL). Then 6-d2-41 (1.5 g, 6.8mmol) was added via a syringe. The mixture solution was refluxed for 2 hours. The excess LiAlH4 was quenched by water, and then an aqueous solution of NaOH (40%, 10 ml) was added. The mixture was stirred for 1 h. The white solid was filtred and the solvent was removed under vacuum to afford crude 6-d2-42 (1.18 g, 94%). 1H NMR (CDCl3) δ 5.65 (m, 2H), 4.57 (dd, J = 4.2, 2.7 Hz, 1H), 3.86 (m, 1H), 3.76 (dt, J = 9.6, 6.6 Hz, 1H), 3.52 (m, 1H), 3.39 (dt, J = 9.6, 6.6 Hz, 1H), 2.23 (m, 2H), 1.71-1.50 (m, 9H).
6-d2-43: This compound was synthesized according to the procedure used for 6-d1-43 (see above). (75% yield over two steps). 1H NMR (CDCl3) δ 5.57 (m, 1H), 5.39 (m, 1H), 4.27 (t, J = 6.6 Hz, 2H), 3.06 (s, 3H), 2.21 (q, J = 7.2 Hz, 2H), 1.86 (m, 2H), 1.63 (m, 1H). 13C NMR (CDCl3) δ 128.5, 125.9, 69.7, 37.6, 29.1, 22.9, 12.4 (p, J = 19.6 Hz).
3: The compound 3 was prepared according to the literature procedure used for 1.17 (60% yield, colorless oil). 1H NMR (CDCl3) δ 7.76 (dt, J = 8.4, 1.8 Hz, 2H), 7.30 (dt, J = 8.4, 1.8 Hz, 2H), 5.45 (dd, J = 10.8, 6.6 Hz, 1H), 5.27 (m, 1H), 4.52 (t, J = 1.8 Hz, 1H), 2.96 (dt, J = 6.9, 6.6 Hz, 2H), 2.43 (s, 3H), 2.03 (q, J = 7.2 Hz, 2H), 1.54 (m, 3H 2H NMR (CDCl3)); δ 1.51 (s, 2D); 13C NMR (CDCl3) δ 143.6, 137.3, 129.9, 129.1, 127.3, 125.3, 43.1, 29.6, 24.2, 21.7, 12.4 (t, J = 19.6 Hz); HRMS (ESI) calculated for C13H17D2NO2S, 255.1262; measured, 255.1259.
Diene 5
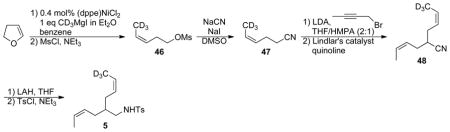
46: The compound was prepared by a modification of the literature procedure.36 A 1.0 M ethereal solution of CD3MgI (35 mL, 35 mmol) was added to a stirring suspension of (dppe)NiCl2 (526 mg, 1 mmol) in 40 mL dry benzene under N2, and the stirring was continued at room temperature for 20 min. 4,5-Dihydropyran (2.8 g, 40 mmol) was added and the solution was refluxed for 2 h. The cooled reaction mixture was poured into a solution of saturated ammonium chloride and was extracted with ether. The extract was dried with MgSO4 and the solvent was removed under reduced pressure. The crude product was dissolved in toluene, and Et3N (6 mL) and MsCl (0.44 mL, 4.5 mmol) were added slowly via syringe to the toluene solution. The mixture was stirred overnight at room temperature. After the reaction was completed, water (20 mL) was added. The mixture was extracted with diethyl ether. The extract was dried with MgSO4 followed by the removal of solvent under reduced pressure. The residue was purified by silica gel chromatography (hexane/ethyl acetate) to afford 46 (5.4g, 92% yield over two steps). 1H NMR (CDCl3) δ 5.64 (d, J = 10.8 Hz, 1H), 5.38 (dt, J = 10.8, 7.2 Hz, 1H), 4.22 (t, J = 6.9 Hz, 2H), 3.00 (s, 3H), 2.51 (dq, J = 6.9, 1.5 Hz, 2H).
47: To a solution of 46 (5.4g, 3.2 mmol) in DMSO (20 mL), NaCN (1.6 g, 3.2 mmol) and NaI (480 mg, 3.2 mmol) were added under an N2 atmosphere. The mixture was stirred at 55 °C for 12 hours. After the reaction was complete, water (50 mL) was added. The mixture was extracted with diethyl ether. The extract was dried with MgSO4 and the solvent was removed under vaccum. The residue was purified by distallation to give 47 (2.8 g, 89% yield, colorless liquid). 1H NMR (CDCl3) δ 5.64 (d, J = 10.8 Hz, 1H), 5.41 (m, 1H), 2.40 (m, 4H).
48: The compound was prepared by a modification of the literature procedure.37 To a solution of 47 (1.1 g, 11 mmol) in a 2:1 (volume ratio) mixture of THF and HMPA, a solution of LDA (6.1 mL 1.8 M in THF, 11 mmol), was added slowly via syringe at −78 °C under an N2 atmosphere. After stirring for 30 min at −78 °C, 1-Bromo-2-butyne (1.6 g, 15 mmol) was added to the mixture solution.38 The mixture was stirred for 3 hours at −78 °C, and then was warmed to room temperature. After the reaction was complete, water (40 mL) was added. The mixture was extracted with diethyl ether. The extract was dried with MgSO4 and the solvent was removed under vaccum. The residue was purified by silica gel chromatography (hexane/ethyl acetate) to give 48 (750 mg, 45% yield over two steps). 1H NMR (CDCl3) δ 5.69 (m, 2H), 5.44 (m, 2H), 2.58 (p, J = 7.2 Hz, 1H), 2.39 (m, 4H), 1.66 (dp, J = 6.9, 0.9 Hz, 3H). 13C NMR (CDCl3) δ 128.4, 128.2, 125.1, 125.0, 122.1, 32.0, 29.13, 29.11, 13.2. HRMS (ESI) calculated for C10H12D3NNa, 175.1290; measured, 175.1295.
5: Under an N2 atmosphere, 48 (350 mg, 2.3 mmol) was added to a suspension of LiAlH4 (150 mg, 4 mmol) in dry THF (10 mL) at 0 °C, and the mixture was stirred overnight at room temperature. After the reaction was complete, water (0.5 mL) was added. To the crude reaction mixture, Et3N (1 mL) and TsCl (570 mg, 3 mmol) were added via a syringe. The mixture was stirred for 12 hours. After the reaction was complete, water (20 mL) was added. The mixture was extracted with diethyl ether. The extract was dried with MgSO4 and the solvent was removed under vaccum. The residue was purified by silica gel chromatography (ethyl acetate/hexanes) to afford 5 (550 mg, 77% yield, colorless oil). 1H NMR (CDCl3) δ 7.74 (dt, J = 8.4, 1.8 Hz, 2H), 7.31 (dt, J = 8.4, 1.8 Hz, 2H), 5.51 (m, 2H), 5.29 (m, 2H), 4.54 (t, J = 1.8 Hz, 1H), 2.87 (t, J = 6.6 Hz, 2H), 2.43 (s, 3H), 2.01 (t, J = 6.9 Hz, 4H), 1.60 (m, 1H), 1.55 (dt, J = 6.9, 0.9 Hz, 3H). 2H NMR (CHCl3) δ 1.49 (s, 3D). 13C NMR (CDCl3) δ 143.5, 137.2, 129.9, 127.9, 127.8, 127.3, 126.3, 126.2, 46.9, 39.1, 29.43, 29.41, 21.7, 13.1. HRMS (ESI) calculated for C17H22D3NO2SNa, 333.1692; measured, 333.1705.
Computational Studies
All computations were performed with the Gaussian 03 (G03) program39 using resources provided by NSF TeraGrid partners. Spin-restricted density functional theory (RDFT)40 calculations were performed with the hybrid density-functional, B3LYP.41,42 A combination of the Stuttgart RSC 1997 ECP43 for Pd and the all-electron 6-31+G(d) basis sets (Basis I)44 for all other atoms were used for gas-phase geometry optimization and normal mode analyses. Full geometry optimizations were carried out in internal coordinates using the Berny algorithm.45 Frequency calculations were performed at optimized geometries and transition states, confirming that each optimized minimum has zero imaginary frequencies and each optimized transition state has exactly one imaginary frequency. When visual inspection of the single negative eigenvalue defining a saddle point did not clearly confirm the reaction trajectory, IRC calculations were performed to verify that the identified transition state corresponded to the appropriate reactant/product potential energy surface.46 Zero-point energy, thermal corrections, and entropic corrections were estimated from normal-mode analysis. Charge analyses were carried out on converged spin-restricted density matrices using the Natural Population Analysis (NPA) method47 as implemented within NBO 3.1 in G03.
At the calculated stationary points, solvation-corrected single-point total energy calculations were carried out with the Pd basis detailed above and the 6-311+G(d, p) basis (Basis II) on all other atoms with electrostatic and non-electrostatic solvation effects evaluated using the integral-equation-formalism polarizable-continuum model (IEF-PCM).48 These calculations were used to predict the solvation free energy under typical catalytic conditions (i.e., toluene solvent at 80° C (353 K)). The solvation cavity was generated using UFF radii, explicitly treating hydrogen atoms, and the radii were scaled by a factor of 1.2. The PCM input was modified with the following parameters to define the physical characteristics of the solvent (e = 2.24, ρ = 0.810 g•cm−1, r = 2.82 Å). The dielectric constant (e) used here was determined with (eq 1).49 The temperature range over which (eq 9) is reported is 207–316K; however, we find that (eq 9) reproduces, with necessary accuracy, dielectric constants at the higher temperature examined here.50 The density (ρ) of toluene at 353K was determined using a 24-parameter empirical model that is valid for T = 223–423 K and P = 1–30 atm.51
| (9) |
We report Gibbs’ free energies at 353 K (ΔG353K) (eqs 10–13). Since the reported free energies are corrected for a solvated environment, an additional energy correction (Scorr) to the translation entropy component of the gas-phase entropy was included. This correction (eq 13) is necessary to account for the standard state change from gas (1 atm) to solution (1 M).52
| (10) |
| (11) |
where n = # of rotational and translational modes
| (12) |
| (13) |
where R = 0.001987 kcal•mol−1•K−1 and T = 353.15 K and Q°/Q = 28. 977.
Supplementary Material
Acknowledgments
We are grateful to the NIH for financial support of this work (R01 GM67163) and the the NSF for partial support of the computation studies through Teragrid resources provided by NCSA and The Pittsburgh Supercomputing Center (TG-CHE070040N).
Footnotes
Supporting Information Available: Experimental details, characterization for all new compounds, and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Minatti A, Muñiz K. Chem Soc Rev. 2007;36:1142–1152. doi: 10.1039/b607474j. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ney JE, Wolfe JP. J Am Chem Soc. 2005;127:8644–8651. doi: 10.1021/ja0430346. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yip KT, Yang M, Law KL, Zhu NY, Yang D. J Am Chem Soc. 2006;128:3130–3131. doi: 10.1021/ja060291x. [DOI] [PubMed] [Google Scholar]; (c) Zeng W, Chemler SR. J Am Chem Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Rosewall CF, Sibbald PA, Liskin DV, Michael FE. J Am Chem Soc. 2009;131:9488–9489. doi: 10.1021/ja9031659. [DOI] [PubMed] [Google Scholar]; (e) Yip KT, Zhu NY, Yang D. Org Lett. 2009;11:1911–1914. doi: 10.1021/ol900355h. [DOI] [PubMed] [Google Scholar]; (f) Sherman ES, Fuller PH, Kasi D, Chemler SR. J Org Chem. 2007;72:3896–3905. doi: 10.1021/jo070321u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Alexanian EJ, Lee C, Sorensen EJ. J Am Chem Soc. 2005;127:7690–7691. doi: 10.1021/ja051406k. [DOI] [PubMed] [Google Scholar]; (b) Fuller PH, Kim JW, Chemler SR. J Am Chem Soc. 2008;130:17638–17639. doi: 10.1021/ja806585m. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Muñiz K, Iglesias A, Fang YW. Chem Commun. 2009:5591–5593. doi: 10.1039/b912139k. [DOI] [PubMed] [Google Scholar]
- 4.(a) Streuff J, Hövelmann CH, Nieger M, Muñiz K. J Am Chem Soc. 2005;127:14586–14587. doi: 10.1021/ja055190y. [DOI] [PubMed] [Google Scholar]; (b) Muñiz K, Hövelmann CH, Streuff J. J Am Chem Soc. 2007;130:763–773. doi: 10.1021/ja075041a. [DOI] [PubMed] [Google Scholar]; (c) Hövelmann CH, Streuff J, Brelot L, Muñiz K. Chem Commun. 2008:2334–2336. doi: 10.1039/b719479j. [DOI] [PubMed] [Google Scholar]; (d) Sibbald PA, Michael FE. Org Lett. 2009;11:1147–1149. doi: 10.1021/ol9000087. [DOI] [PubMed] [Google Scholar]
- 5.(a) Helaja J, Göttlich R. Chem Commun. 2002:720–721. doi: 10.1039/b201209j. [DOI] [PubMed] [Google Scholar]; (b) Manzoni MR, Zabawa TP, Kasi D, Chemler SR. Organometallics. 2004;23:5618–5621. [Google Scholar]; (c) Lei AW, Lu XY, Liu GS. Tetrahedron Lett. 2004;45:1785–1788. [Google Scholar]; (d) Michael FE, Sibbald PA, Cochran BM. Org Lett. 2008;10:793–796. doi: 10.1021/ol702922c. [DOI] [PubMed] [Google Scholar]; (e) Wu T, Yin GY, Liu GS. J Am Chem Soc. 2009;131:16354–16355. doi: 10.1021/ja9076588. [DOI] [PubMed] [Google Scholar]
- 6.(a) Tamaru Y, Hojo M, Yoshida Z. J Org Chem. 1988;53:5731–5741. [Google Scholar]; (b) Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J Org Chem. 1997;62:2113–2122. doi: 10.1021/jo961988b. [DOI] [PubMed] [Google Scholar]; (c) Shinohara T, Arai MA, Wakita K, Arai T, Sasai H. Tetrahedron Lett. 2003;44:711–714. [Google Scholar]; (d) Tsujihara T, Shinohara T, Takenaka K, Takizawa S, Onitsuka K, Hatanaka M, Sasai H. J Org Chem. 2009;74:9274–9279. doi: 10.1021/jo901778a. [DOI] [PubMed] [Google Scholar]
- 7.For a general review on this subject, See: Chemler SR. Org Biomol Chem. 2009;7:3009–3019. doi: 10.1039/B907743J.
- 8.See ref. 2b, 2e, 6d and the following: Overman LE, Remarchuk TP. J Am Chem Soc. 2001;124:12–13. doi: 10.1021/ja017198n.
- 9.For recent reviews describing direct dioxygen-coupled palladium-catalyzed oxidative cyclization reactions, see: Stahl SS. Angew Chem, Int Ed. 2004;43:3400–3420. doi: 10.1002/anie.200300630.Stoltz BM. Chem Lett. 2004;33:362–367.Sigman MS, Schultz MJ. Org Biomol Chem. 2004;2:2551–2554. doi: 10.1039/B409127M.Stahl SS. Science. 2005;309:1824–1826. doi: 10.1126/science.1114666.Kotov V, Scarborough CC, Stahl SS. Inorg Chem. 2007;46:1910–1923. doi: 10.1021/ic061997v.
- 10.(a) Hegedus LS, Allen GF, Waterman EL. J Am Chem Soc. 1976;98:2674–2676. [Google Scholar]; (b) Hegedus LS, Allen GF, Bozell JJ, Waterman EL. J Am Chem Soc. 1978;100:5800–5807. [Google Scholar]; (c) Hegedus LS, Allen GF, Olsen DJ. J Am Chem Soc. 1980;102:3583–3587. [Google Scholar]; (d) Hegedus LS, McKearin JM. J Am Chem Soc. 1982;104:2444–2451. [Google Scholar]
- 11.(a) Tamaru Y, Hojo M, Higashimura H, Yoshida Z. J Am Chem Soc. 1988;110:3994–4002. [Google Scholar]; (b) Tamaru Y, Hojo M, Yoshida Z. J Org Chem. 1988;53:5731–5741. [Google Scholar]; (c) van Benthem R, Hiemstra H, Longarela GR, Speckamp WN. Tetrahedron Lett. 1994;35:9281–9284. [Google Scholar]; (d) Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584–3585. doi: 10.1021/jo952088i. [DOI] [PubMed] [Google Scholar]; (e) Harayama H, Okuno H, Takahashi Y, Kimura M, Fugami K, Tanaka S, Tamaru Y. Tetrahedron Lett. 1996;37:7287–7290. [Google Scholar]; (f) Harayama H, Abe A, Sakado T, Kimura M, Fugami K, Tanaka S, Tamaru Y. J Org Chem. 1997;62:2113–2122. doi: 10.1021/jo961988b. [DOI] [PubMed] [Google Scholar]; (g) Tamaru Y, Kimura M. Synlett. 1997:749–757. [Google Scholar]
- 12.Fix SR, Brice JL, Stahl SS. Angew Chem, Int Ed. 2002;41:164–166. doi: 10.1002/1521-3773(20020104)41:1<164::aid-anie164>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 13.Concurrent with our work, related carbo- and heterocyclization reactions were developed by Stoltz and coworkers: Ferreira EM, Stoltz BM. J Am Chem Soc. 2003;125:9578–9579. doi: 10.1021/ja035054y.Trend RM, Ramtohul YK, Ferreira EM, Stoltz BM. Angew Chem Int Ed. 2003;42:2892–2895. doi: 10.1002/anie.200351196.Trend RM, Ramtohul YK, Stoltz BM. J Am Chem Soc. 2005;127:17778–17788. doi: 10.1021/ja055534k.
- 14.Nishimura T, Onoue T, Ohe K, Uemura S. J Org Chem. 1999;64:6750–6755. doi: 10.1021/jo9906734. [DOI] [PubMed] [Google Scholar]
- 15.(a) Brice JL, Harang JE, Timokhin VI, Anastasi NR, Stahl SS. J Am Chem Soc. 2005;127:2868–2869. doi: 10.1021/ja0433020. [DOI] [PubMed] [Google Scholar]; (b) Liu G, Stahl SS. J Am Chem Soc. 2006;128:7179–7181. doi: 10.1021/ja061706h. [DOI] [PubMed] [Google Scholar]; (c) Isomura K, Okada N, Saruwatari M, Yamasaki H, Taniguchi H. Chem Lett. 1985:385–388. [Google Scholar]; (d) Ney JE, Wolfe JP. Angew Chem, Int Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060. [DOI] [PubMed] [Google Scholar]; (e) Nakhla JS, Kampf JW, Wolfe JP. J Am Chem Soc. 2006;128:2893–2901. doi: 10.1021/ja057489m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Åkermark B, Bäckvall JE, Siirala-Hansén K, Sjöberg K, Zetterberg K. Tetrahedron Lett. 1974;15:1363–1366. [Google Scholar]; (b) Åkermark B, Bäckvall JE, Hegedus LS, Zetterberg K, Siirala-Hansén K, Sjöberg K. J Organomet Chem. 1974;72:127–138. [Google Scholar]; (c) Bäckvall JE. Acc Chem Res. 1983;16:335–342. [Google Scholar]; (d) Bäckvall JE, Björkman EE. Acta Chem Scand B. 1984;38:91–93. [Google Scholar]; (e) Sibbald PA, Rosewall CF, Swartz RD, Michael FE. J Am Chem Soc. 2009;131:15945–15951. doi: 10.1021/ja906915w. [DOI] [PubMed] [Google Scholar]
- 17.Liu GS, Stahl SS. J Am Chem Soc. 2007;129:6328–6335. doi: 10.1021/ja070424u. [DOI] [PubMed] [Google Scholar]
- 18.For an analogous study of aerobic oxidative amination reactions, catalyzed by an N-heterocyclic carbene-PdII catalyst system, see the following: Rogers MM, Wendlandt JE, Guzei IA, Stahl SS. Org Lett. 2006;8:2257–2260. doi: 10.1021/ol060327q.(b) Ye, X., Stahl, S. S. unpublished results.
- 19.(Z)-4-hexenyltosylamide was selected instead of (E)-4-hexenyltosylamide as the model substrate for this mechanistic study because cis-alkenes usually afford better yields in palladium-catalyzed aerobic oxidative amination reactions, possibly due to better coordinating ability of cis-alkene to the palladium center.
- 20.In our original report of Pd(OAc)2/pyridine-catalyzed intramolecular aerobic oxidative amination of alkenes, a 1:2 ratio of Pd(OAc)2:pyridine was employed. However, in the present study, a 1:4 ratio of Pd(OAc)2:pyridine was used as the standard catalyst mixture because the palladium catalyst is more stable and less susceptible to decomposition in the presence of 4 equiv. of pyridine ligand.
- 21.The intramolecular nature of deuterium scrambling into the β-vinyl position of the pyrrolidine product has been independently confirmed by crossover experiment with 1:1 mixture of CD3-labeled tosylamide and all-protio pyrrolidine products. For detailed experimental description, see Supporting Information.
- 22.For detailed experimental description, see Supporting Information.
- 23.(a) Steinhoff BA, Stahl SS. Org Lett. 2002;4:4179–4181. doi: 10.1021/ol026988e. [DOI] [PubMed] [Google Scholar]; (b) Steinhoff BA, Guzei IA, Stahl SS. J Am Chem Soc. 2004;126:11268–11278. doi: 10.1021/ja049962m. [DOI] [PubMed] [Google Scholar]
- 24.Stephenson TA, Morehouse SM, Powell AR, Heffer JP, Wilkinson G. J Chem Soc. 1965:3632–3640. [Google Scholar]
- 25.In a recent demonstration of this principle, pyridine ligands have been shown to activate Pd-carboxylate catalysts toward C–H activation in the aerobic oxidative coupling of o-xylene: Izawa Y, Stahl SS. Adv Synth Catal. 2010;352:3223–3229. doi: 10.1002/adsc.201000771.
- 26.The apparent kinetic dependence on [O2] when the reaction is carried out in the presence of 1:2 Pd:py (Figure 3B) reflects the fact that at low [O2] the catalyst decomposes more rapidly, thereby removing active catalyst from the reaction mixture. At higher [O2], the catalyst is more stable, and the higher concentration of active catalyst contributes to a faster rate. The O2-pressure effects on catalyst stablility have been analyzed in considerable detail in the following: Steinhoff BA, Stahl SS. J Am Chem Soc. 2006;128:4348–4355. doi: 10.1021/ja057914b.
- 27.A similar “up/down” dependence on [AcOH] has also been observed by Sigman and coworkders in Pd(IiPr)(OAc)2(H2O)-catalyzed aerobic alcohol oxidation. In this case, the observation was attributed to a balance between the relative rate of HOAc-inhibited alcohol oxidation and HOAc-promoted palladium catalyst regeneration by O2. See: Mueller JA, Goller CP, Sigman MS. J Am Chem Soc. 2004;126:9724–9734. doi: 10.1021/ja047794s.
- 28.For insights into electronic effects on alkene insertion into Pd–N bonds, see: Hanley PS, Markovic D, Hartwig JF. J Am Chem Soc. 2010;132:6302–6303. doi: 10.1021/ja102172m.Neukom JD, Perch NS, Wolfe JP. J Am Chem Soc. 2010;132:6276–6277. doi: 10.1021/ja9102259.
- 29.For further computational details, see the Experimental Section and Supporting Information.
- 30.For stuctures lacking pyridine or AcOH, the energies account for the presence of free py and a half-equivalent of [AcOH]2, respectively.
- 31.Although pathways involving direct reaction of a Pd–H species with molecular oxygen have been proposed to initiate catalyst regeneration, recent computational work provides compelling evidence that this catalytic system must proceed through Pd0, see: Popp BV, Stahl SS. Chem Eur J. 2009;15:2915–2922. doi: 10.1002/chem.200802311.Keith JM, Goddard WA. J Am Chem Soc. 2009;131:1416–1425. doi: 10.1021/ja8040459.
- 32.For a thorough description of β-hydride elimination and HOAc-reductive elimination pathways, see Supporting Information.
- 33.Wenkert E, Michelotti EL, Swindell CS, Tingoli M. J Org Chem. 1984;49:4894–4899. [Google Scholar]
- 34.Heitz MP, Wagner A, Mioskowski C, Noel JP, Beaucourt JP. J Org Chem. 1989;54:500–503. [Google Scholar]
- 35.Kita Y, Okunaka R, Honda T, Shindo M, Taniguchi M, Kondo M, Sasho M. J Org Chem. 1991;56:119–125. [Google Scholar]
- 36.Wadman S, Whitby R, Yeates C, Kocienski P, Cooper K. J Chem Soc, Chem Comm. 1987:241–243. [Google Scholar]
- 37.Petschen I, Parrilla A, Bosch MP, Amela C, Botar AA, camps F, Guerrero A. Chem Eur J. 1999;5:3299–3309. [Google Scholar]
- 38.Kurth MJ, Decker OHW. J Org Chem. 1985;50:5769–5775. [Google Scholar]
- 39.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03, Revision D.01. Gaussian, Inc; Wallingford CT: 2004. [Google Scholar]
- 40.Koch W, Holthausen MC. A Chemist’s Guide to Density Functional Theory. Wiley-VCH; Weinheim: 2000. [DOI] [PubMed] [Google Scholar]
- 41.Becke AD. J Chem Phys. 1993;98:1372–1377. [Google Scholar]
- 42.Lee C, Yang W, Parr RG. Phys Rev B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 43.(a) The Stuttgart RSC 1997 ECP basis set for Pd was obtained from the Extensible Computational Chemistry Environment Basis Set Database, Version 02/25/04, as developed and distributed by the Molecular Science Computing Facility, Environmental and Molecular Sciences Laboratory which is part of the Pacific Northwest Laboratory, P. O. Box 999, Richland, Washington 99352, USA, and funded by the U. S. Department of Energy. The Pacific Northwest Laboratory is a multiprogram laboratory operated by Battelle Memorial Institute for the U. S. Department of Energy under contract DE-AC06-76RLO 1830. Contact Karen Schuchardt for further information; Andrae D, Häußermann U, Dolg M, Stoll H, Preuß H. Theor Chim Acta. 1990;77:123–141.
- 44.Hehre WJ, Radom L, Schleyer LPvR, Pople JA. Ab Initio Molecular Orbital Theory. Wiley; New York: 1986. [Google Scholar]
- 45.(a) Peng CY, Ayala PY, Schlegel HB, Frisch MJ. J Comp Chem. 1996;17:49–56. [Google Scholar]; (b) Peng CY, Schlegel HB. Israel J Chem. 1993;33:449–454. [Google Scholar]
- 46.(a) Gonzalez C, Schlegel HB. J Chem Phys. 1989;90:2154–2161. [Google Scholar]; (b) Gonzalez C, Schlegel HB. J Chem Phys. 1990;94:5523–5527. [Google Scholar]
- 47.Reed AE, Weinstock RB, Weinhold F. J Chem Phys. 1985;83:735–746. [Google Scholar]
- 48.For an overview of solvation models and reviews on PCM methods, see: Tomasi J, Mennucci B, Cammi R. Chem Rev. 2005;105:2999–3093. doi: 10.1021/cr9904009.Cramer CJ, Truhlar DG. Chem Rev. 1999;99:2161–2200. doi: 10.1021/cr960149m.For references on our current approach, see: Cancès E, Mennucci B, Tomasi J. J Chem Phys. 1997;107:3032–3041.Mennucci B, Tomasi J. J Chem Phys. 1997;106:5151–5158.Mennucci B, Cancès E, Tomasi J. J Phys Chem B. 1997;101:10506–10517.Tomasi J, Mennucci B, Cancès E. J Mol Struct (Theochem) 1999;464:211–226.
- 49.Lide DR. Handbook of Chemistry and Physics. 88. CRC Press; Boca Raton: 2007. [Google Scholar]
- 50.Kandil ME, Marsh KN, Goodwin ARH. J Chem Eng Data. 2008;53:1056–1065. [Google Scholar]
- 51.McLinden MO, Splett JD. J Res Natl Inst Stan. 2008;113:29–67. doi: 10.6028/jres.113.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cramer CJ. Essentials of Computational Chemistry: Theories and Models. J. Wiley; New York: 2002. pp. 341–342. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.