

Abstract
The first enantioselective Pd-catalyzed construction of all-carbon quaternary stereocenters via 1,4-addition of arylboronic acids to β-substituted cyclic enones is reported. Reaction of a wide range of arylboronic acids and cyclic enones using a catalyst prepared from Pd(OCOCF3)2 and a chiral pyridinooxazoline ligand yields enantioenriched products bearing benzylic stereocenters. Notably, this transformation is tolerant to air and moisture, providing a practical and operationally simple method of synthesizing enantioenriched all-carbon quaternary stereocenters.
The catalytic enantioselective construction of all-carbon quaternary stereocenters remains a difficult problem in synthetic chemistry.1 A reliable approach toward this challenge has been the asymmetric conjugate addition of carbon-based nucleophiles to suitable α,β-unsaturated carbonyl acceptors.2 Pioneered by the groups of Feringa, Alexakis, and Hoveyda, the majority of asymmetric conjugate additions for the synthesis of quaternary centers involve the use of highly reactive organometallic reagents (e.g., diorganozinc,3 triorganoaluminum,4 and organomagnesium reagents5) to a variety of unsaturated electrophiles with copper catalysts.6 These reactions uniformly involve air and moisture sensitive organometallic reagents that require rigorously anhydrous reaction conditions. Alternatively, Hayashi has championed the use of chiral rhodium catalysts in combination with air stable, easily handled nucleophilic organoboron reagents to produce a wide array of conjugate addition adducts in exceptional yield and ee.7,8 In contrast to the copper systems, relatively few examples in the rhodium series lead to the formation of products containing quaternary centers.9 Notably, Hayashi and Shintani have recently developed a rhodium•diene-catalyzed conjugate addition of sodium tetraaryl borates (Ar4BNa) and arylboroxines ((ArBO)3) to β,β-disubstituted enones to provide ketone products bearing β-chiral all-carbon quarternary stereocenters.10,11 Unfortunately, in these examples commercially available arylboronic acids (ArB(OH)2) are not competent nucleophiles.10,11,12
The conjugate addition of arylboronic acids and their derivatives to enones with palladium catalysis has been investigated for some time and has resulted in the development of addition reactions that produce enantioenriched tertiary β-substituted ketones.13 In 2010, Lu reported the use of a dicationic bipyridine-derived palladium catalyst for additions to β-substituted enones that deliver racemic products containing the quaternary center in high yield.14 Notably absent from the Lu report and from the work of others in the area are examples of Pd-catalyzed enantioselective conjugate addition reactions that forge a quaternary center.13 Herein, we report the first palladium-catalyzed asymmetric conjugate addition of arylboronic acids to β-substituted cyclic enones employing an easily accessible chiral pyridinooxazoline (PyOX) ligand.15 These reactions generate a wide array of benzylic all-carbon quaternary stereocenters while exhibiting extraordinary tolerance to both air and water.
To achieve the desired enantioselective conjugate addition, the reaction of 3-methylcyclohexen-2-one (1) with phenylboronic acid (2) was investigated in the presence of various palladium catalysts and chiral ligands (Table 1). After a preliminary ligand search that included an array of standard chiral ligand frameworks,16 we discovered that t-BuPyOX (4)15 provided high levels of enantioselection. In the presence of ligand 4, a range of Pd(II) sources were capable of catalyzing the desired reaction (entries 1–5), with carboxylate counterions leading to the highest levels of asymmetric induction and chemical yield. A catalyst derived from Pd(OCOCF3)2 and pyridinooxazoline 4 produced the desired ketone product 317 in 87% yield and 91% ee (entry 5).18 By using 1,2-dichloroethane in place of dichloromethane as solvent and increasing the reaction temperature from 40 to 60 °C, ketone 3 was isolated in 99% yield and 93% ee (entry 6).19,20 Remarkably, the high yield and enantioselectivity were maintained even upon addition of 10 equiv of water (entry 7). Furthermore, the amount of phenylboronic acid was reduced to 1.1 equiv with no detrimental effects (entry 8). Finally, we have performed the reaction on multi-gram scale without complication (entry 9). It should be noted that (S)-t-BuPyOX (4), can be easily prepared in only two synthetic steps from commercially available materials.15,18,21
Table 1.
Optimization of Reaction Conditions.a
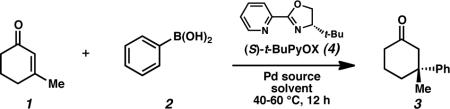
| entry | Pd source | solvent | temp (°C) | yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | PdCl2 | CH2Cl2 | 40 | – | – |
| 2 | Pd(MeCN)2Cl2 | CH2Cl2 | 40 | – | – |
| 3d | Pd(MeCN)2Cl2, AgOTf | CH2Cl2 | 40 | 69 | 17 |
| 4 | Pd(OAc)2 | CH2Cl2 | 40 | 65 | 92 |
| 5 | Pd(OCOCF3)2 | CH2Cl2 | 40 | 87 | 91 |
| 6 | Pd(OCOCF3)2 | ClCH2CH2Cl | 60 | 99 | 93 |
| 7e | Pd(OCOCF3)2 | ClCH2CH2Cl | 60 | 99 | 91 |
| 8f | Pd(OCOCF3)2 | ClCH2CH2Cl | 60 | 99 | 93 |
| 9g | Pd(OCOCF3)2 | ClCH2CH2Cl | 60 | 97 | 91 |
Conditions: Reactions were performed with phenylboronic acid (0.50 mmol), 3-methylcyclohexen-2-one (0.25 mmol), Pd(OCOCF3)2 (5 mol%), and ligand 4 (6 mol%) in solvent (1 mL) for 12 h, unless otherwise noted.
Isolated yield.
ee was determined by chiral HPLC, see Supporting Information.
12 mol% AgOTf.
Reaction performed in the presence of added H2O (2.5 mmol, 10 equiv).
Phenylboronic acid loading reduced to 1.1 equiv.
Multi gram scale-up reaction performed with 3-methylcyclohexen-2-one (2.42 g, 22.0 mmol), phenylboronic acid (44.0 mmol), H2O (5 equiv), Pd(OCOCF3)2 (5 mol%), and ligand 4 (6 mol%) in solvent (88 mL) for 12 h.
To investigate the reaction scope, we explored various arylboronic acids as nucleophiles for this process (Table 2). Generally, para-substituted arylboronic acids are well tolerated (entries 1–9). Reactions with 4-methyl and 4-ethylphenylboronic acid proceeded well to give high yields of the desired products with good asymmetric induction (entries 1 and 2). While electron-rich nucleophiles tend to be reliable reaction partners, they often furnish products in moderate enantioselectivity (entries 3–5). Electron-deficient nucleophiles fared particularly well, producing ketone products in excellent ee (entries 6–9). Specifically, these electron-poor nucleophiles can possess a wide range of functional groups, such as ketone (entry 6), halide (entries 7 and 8) and a trifluoromethyl group (entry 9). Reactions involving meta-substituted arylboronic acids were also broadly successful with alkyl (entry 10), halide (entries 11 and 12), ester (entry 13) and even nitro (entry 14) groups on the nucleophile.22,23,24
Table 2.
Addition of Arylboronic Acids to 3-Methylcyclohexen-2-one.a
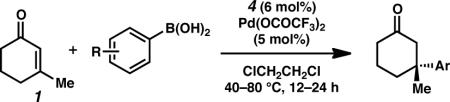
| entry | R = | temp (°C) | time (h) | yield (%)b | ee (%)c |
|---|---|---|---|---|---|
| 1 | 4-Me– | 60 | 12 | 99 | 87 |
| 2 | 4-Et– | 60 | 12 | 90 | 85 |
| 3 | 4-MeO– | 40 | 24 | 58 | 69 |
| 4 | 4-BnO– | 60 | 18 | 96 | 74 |
| 5 | 4-TBSO– | 40 | 24 | 52 | 82 |
| 6 | 4-Ac– | 60 | 18 | 99 | 96 |
| 7 | 4-Cl– | 60 | 12 | 94 | 95 |
| 8 | 4-F– | 80 | 12 | 84 | 92 |
| 9 | 4-F3C– | 60 | 12 | 99 | 96 |
| 10 | 3-Me– | 60 | 24 | 99 | 91 |
| 11 | 3-Cl– | 60 | 18 | 55 | 96 |
| 12 | 3-Br– | 60 | 24 | 44 | 85 |
| 13 | 3-MeO2C– | 60 | 24 | 91 | 95 |
| 14 | 3-O2N– | 60 | 18 | 40 | 92 |
Conditions: Reactions were performed with phenylboronic acid (0.50 mmol), 3-methylcyclohexen-2-one (0.25 mmol), Pd(OCOCF3)2 (5 mol%), and ligand 4 (6 mol%) in (ClCH2)2 (1 mL) at 40–80 °C for 12–24 h.
Isolated yield.
ee was determined by chiral HPLC, see Supporting Information.
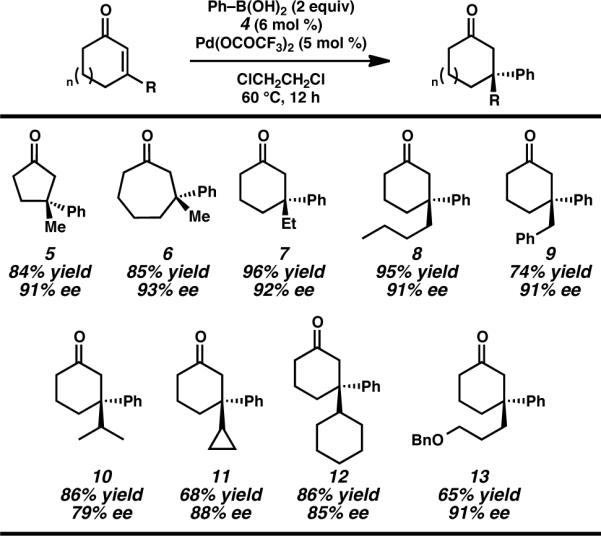
We sought to further examine the scope of the reaction by exploring cyclic enones of different ring sizes and with a range of β-substitution (Table 3). Importantly, altering the ring size to the 5- or 7-membered ring series had no deleterious effect on the transformation and fashioned ketones 4 and 5 in high yield and ee. To the best of our knowledge this represents the first time that quaternary centers have been constructed by asymmetric conjugate addition of boronic acids to these differing ring sized enones using a single catalyst.2 Cyclohexenones bearing other β-alkyl substituents, such as ethyl, n-butyl, and benzyl furnished ketone products (i.e., 6–8) in good yield and excellent ee as well. In addition to linear alkyl substitution, ketones with branched β-alkyl substituents such as iso-propyl (7) and cyclopropyl (8) and cyclohexyl (12) are produced in good yield and enantioselectivity. Finally, products containing functionalized side-chains, such as benzyl ether 12, are readily obtained, providing a useful chemical handle for further transformations.25
Table 3.
Asymmetric Synthesis of β,β-Disubstituted Cyclic Ketones.a
|
Conditions: Reactions were performed with phenylboronic acid (0.50 mmol), cycloalkenone (0.25 mmol), Pd(OCOCF3)2 (5 mol%), and ligand 4 (6 mol%) in (ClCH2)2 (1 mL) at 60 °C for 12 h.
In summary, we report the first palladium-catalyzed enantioselective conjugate addition of arylboronic acids to β-substituted cyclic enones to deliver products containing an all-carbon quaternary stereocenter. Critically, 5-, 6-, and 7-membered ring enones function well in the process, delivering products of uniformly high ee using a single catalyst. A wide variety of commercially available arylboronic acids and substituted enones can be employed in the asymmetric transformation, while exhibiting broad functional group tolerance. Furthermore, the reaction displays a remarkable tolerance to water and oxygen, and reactions are typically performed under an atmosphere of air in screw-top vials and without the need for purification or distillation of any commercially obtained materials. Finally, the optimal chiral ligand, (S)-t-BuPyOX (4), is expediently prepared, rendering this process an experimentally simple, practical method for enantioselective construction of all-carbon quaternary stereocenters. Continuing investigations of this method and application of this chemistry in the context of natural product synthesis is currently underway and will be reported in due course.
Supplementary Material
Acknowledgment
This publication is based on work supported by Award No. KUS-11-006-02, made by King Abdullah University of Science and Technology (KAUST). The authors wish to thank NIH-NIGMS (R01GM080269-01), Amgen, Abbott, Boehringer Ingelheim, and Caltech for financial support. K. K. acknowledges the Japan Society for the Promotion of Science for a postdoctoral fellowship. M. G. is grateful to the Swiss National Science Foundation for financial support through a postdoctoral fellowship.
Footnotes
Supporting Information Available. Experimental details are available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For reviews on the synthesis of quaternary stereocenters, see: Denissova I, Barriault L. Tetrahedron. 2003;59:10105. Douglas CJ, Overman LE. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5363. doi: 10.1073/pnas.0307113101. Christoffers J, Baro A. Adv. Synth. Catal. 2005;347:1473. Trost BM, Jiang C. Synthesis. 2006:369. Mohr JT, Stoltz BM. Chem. Asian J. 2007;2:1476. doi: 10.1002/asia.200700183. Cozzi PG, Hilgraf R, Zimmermann N. Eur. J. Org. Chem. 2007;36:5969.
- (2).For an excellent comprehensive review, see: Hawner C, Alexakis A. Chem. Commun. 2010;46:7295. doi: 10.1039/c0cc02309d.
- (3).(a) Feringa BL. Acc. Chem. Res. 2000;33:346. doi: 10.1021/ar990084k. [DOI] [PubMed] [Google Scholar]; (b) Wu J, Mampreian DM, Hoveyda AH. J. Am. Chem. Soc. 2005;127:4584. doi: 10.1021/ja050800f. [DOI] [PubMed] [Google Scholar]; (c) Hird AW, Hoveyda AH. J. Am. Chem. Soc. 2005;127:14988. doi: 10.1021/ja0553811. [DOI] [PubMed] [Google Scholar]; (d) Wilsily A, Fillion E. J. Am. Chem. Soc. 2006;128:2774. doi: 10.1021/ja056692e. [DOI] [PubMed] [Google Scholar]; (e) Lee K-S, Brown MK, Hird AW, Hoveyda AH. J. Am. Chem. Soc. 2006;128:7182. doi: 10.1021/ja062061o. [DOI] [PubMed] [Google Scholar]; (f) Brown MK, May TL, Baxter CA, Hoveyda AH. Angew. Chem., Int. Ed. 2007;46:1097. doi: 10.1002/anie.200604511. [DOI] [PubMed] [Google Scholar]; (g) Wilsily A, Fillion E. Org. Lett. 2008;10:2801. doi: 10.1021/ol800923q. [DOI] [PubMed] [Google Scholar]; (h) Wilsily A, Fillion E. J. Org. Chem. 2009;74:8583. doi: 10.1021/jo901559d. [DOI] [PubMed] [Google Scholar]; (i) Dumas AM, Fillion E. Acc. Chem. Res. 2010;43:440. doi: 10.1021/ar900229z. [DOI] [PubMed] [Google Scholar]
- (4).(a) d'Augustin M, Palais L, Alexakis A. Angew. Chem., Int. Ed. 2005;44:1376. doi: 10.1002/anie.200462137. [DOI] [PubMed] [Google Scholar]; (b) Fuchs N, d'Augustin M, Humam M, Alexakis A, Taras R, Gladiali S. Tetrahedron: Asymm. 2005;16:3143. [Google Scholar]; (c) Vuagnoux-d'Augustin M, Alexakis A. Chem.Eur. J. 2007;13:9647. doi: 10.1002/chem.200701001. [DOI] [PubMed] [Google Scholar]; (d) Vuagnoux-d'Augustin M, Kehrli S, Alexakis A. Synlett. 2007:2057. [Google Scholar]; (e) Palais L, Mikhel IS, Bournaud C, Micouin L, Falciola CA, Vuagnoux-d'Augustin M, Rosset S, Bernardinelli G, Alexakis A. Angew. Chem., Int. Ed. 2007;46:7462. doi: 10.1002/anie.200702186. [DOI] [PubMed] [Google Scholar]; (f) May TL, Brown MK, Hoveyda AH. Angew. Chem., Int. Ed. 2008;47:7358. doi: 10.1002/anie.200802910. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hawner C, Li K, Cirriez V, Alexakis A. Angew. Chem., Int. Ed. 2008;47:8211. doi: 10.1002/anie.200803436. [DOI] [PubMed] [Google Scholar]; (h) Ladjel C, Fuchs N, Zhao J, Bernardinelli G, Alexakis A. Eur. J. Org. Chem. 2009:4949. [Google Scholar]; (i) Palais L, Alexakis A. Chem. Eur. J. 2009;15:10473. doi: 10.1002/chem.200901577. [DOI] [PubMed] [Google Scholar]; (j) Müller D, Hawner C, Tissot M, Palais L, Alexakis A. Synlett. 2010:1694. [Google Scholar]; (k) Hawner C, Müller D, Gremaud L, Felouat A, Woodward S, Alexakis A. Angew. Chem., Int. Ed. 2010;49:7769. doi: 10.1002/anie.201003300. [DOI] [PubMed] [Google Scholar]
- (5).(a) Martin D, Kehrli S, d'Augustin M, Clavier H, Mauduit M, Alexakis A. J. Am. Chem. Soc. 2006;128:8416. doi: 10.1021/ja0629920. [DOI] [PubMed] [Google Scholar]; (b) Kehrli S, Martin D, Rix D, Mauduit M, Alexakis A. Chem.Eur. J. 2010;16:9890. doi: 10.1002/chem.201000471. [DOI] [PubMed] [Google Scholar]; (c) Hénon H, Mauduit M, Alexakis A. Angew. Chem., Int. Ed. 2008;47:9122. doi: 10.1002/anie.200803735. [DOI] [PubMed] [Google Scholar]; (d) Matsumoto Y, Yamada K-I, Tomioka K. J. Org. Chem. 2008;73:4578. doi: 10.1021/jo800613h. [DOI] [PubMed] [Google Scholar]
- (6).Recently, an asymmetric conjugate addition of cyanide in the presence of a catalyst derived from Sr(Oi-Pr)3 has been reported, see: Tanaka Y, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2010;132:8862. doi: 10.1021/ja1035286.
- (7).Takaya Y, Ogasawara M, Hayashi T, Sakai M, Miyaura N. J. Am. Chem. Soc. 1998;120:5579. Hayashi T, Yamasaki K. Chem Rev. 2003;103:2829. doi: 10.1021/cr020022z.
- (8).For selected recent examples, see: Hayashi T, Ueyama K, Tokunaga N, Yoshida K. J. Am. Chem. Soc. 2003;125:11508. doi: 10.1021/ja037367z. Fischer C, Defieber C, Suzuki T, Carreira EM. J. Am. Chem. Soc. 2004;126:1628. doi: 10.1021/ja0390707. Shintani R, Ueyama K, Yamada I, Hayashi T. Org. Lett. 2004;6:3425. doi: 10.1021/ol048421z. Otomaru Y, Okamoto K, Shintani R, Hayashi T. J. Org. Chem. 2005;70:2503. doi: 10.1021/jo047831y. Paquin J-F, Defieber C, Stephenson CRJ, Carreira EM. J. Am. Chem. Soc. 2005;127:10850. doi: 10.1021/ja053270w.
- (9).(a) Mauleón P, Carretero JC. Chem. Commun. 2005:4961. doi: 10.1039/b508142d. [DOI] [PubMed] [Google Scholar]; (b) Shintani R, Duan W-L, Hayashi T. J. Am. Chem. Soc. 2006;128:5628. doi: 10.1021/ja061430d. [DOI] [PubMed] [Google Scholar]
- (10).(a) Shintani R, Tsutsumi Y, Nagaosa M, Nishimura T, Hayashi T. J. Am. Chem. Soc. 2009;131:13588. doi: 10.1021/ja905432x. [DOI] [PubMed] [Google Scholar]; (b) Shintani R, Takeda M, Nishimura T, Hayashi T. Angew. Chem., Int. Ed. 2010;49:3969. doi: 10.1002/anie.201000467. [DOI] [PubMed] [Google Scholar]
- (11).The same group also reported additions to β,β-disubstituted α,β-unsaturated esters, see: Shintani R, Hayashi T. Org. Lett. 2011;13:350. doi: 10.1021/ol102674z.
- (12).A recent paper describing the use of a Rh•OlefOX (olefin-oxazoline) complex provided a single example of a phenylboronic acid addition to 3-methylcyclohexen-2-one (i.e., 1 + 2 → 3). Unfortunately, ketone 3 was isolated in only 36% yield and 85% ee, see: Hahn BT, Tewes F, Fröhlich R, Glorius F. Angew Chem., Int. Ed. 2010;49:1143. doi: 10.1002/anie.200905712.
- (13).For excellent review articles, see: Gutnov A. Eur. J. Org. Chem. 2008:4547. Christoffers J, Koripelly G, Rosiak A, Rössle M. Synthesis. 2007:1279. Xu Q, Zhang R, Zhang T, Shi M. J. Org. Chem. 2010;75:3935. doi: 10.1021/jo1006224.
- (14).Lin S, Lu X. Org. Lett. 2010;12:2536. doi: 10.1021/ol100767u. [DOI] [PubMed] [Google Scholar]
- (15).Brunner H, Obermann U. Chem. Ber. 1989;122:499. [Google Scholar]
- (16).Our preliminary ligand search was conducted under a range of conditions that varied solvent, temperature, additives, and palladium source. Ligand frameworks tested included a variety of chiral bis(oxazolines) (BOX), pyridino(bis)oxazolines (PyBOX), phosphinooxazolines (PHOX), and quinolinooxazolines (QuinOX).
- (17).The absolute stereochemistry for all products shown was assigned by analogy to the product from Table 2, entry 2 as described in ref 3c.
- (18).See Supporting Information.
- (19).Other solvents proved to be inferior to dichloroethane.18
- (20).Under the optimized conditions it was found that other PyOX ligands were inferior with respect to enantiocontrol. For instance, use of (S)-Ph-PyOX and (S)-i-Pr-PyOX as ligand under the reaction conditions outlined in Table 1, entry 6 provided 3 in 99% yield in each case, but 52% and 40% ee, respectively.
- (21).It should be noted that (R)-t-Bu-PyOX can be prepared from (R)-t-leucine, a relatively expensive commercial compound that is now readily available by the Strecker method of Jacobsen, see: Zuend SJ, Coughlin MP, Lalonde MP, Jacobsen EN. Nature. 2009;461:968–970. doi: 10.1038/nature08484.
- (22).Substituents at the 2-position of the arylboronic acid were detrimental to the yields and stereoselectivity of the reaction with enone 1, although 2-fluorophenylboronic acid underwent the desired reaction to provide a product in 32% yield and 77% ee.18
- (23).Although the reactions are generally high yielding, the impurity profile is dominated by homocoupling of the arylboronic acid to the corresponding biaryl.
- (24).To date, reactions involving either heteroarylboronic acids or β-aryl substituted enones have been unsuccessful.
-
(25).We have conducted the Pd-catalyzed reaction of PhB(OH)2 with racemic bicyclic enone (±)-i, under our standard conditions (Ph−B(OH)2 (2 equiv), 4 (6 mol %), Pd(OCOCF3)2 (5 mol %), ClCH2CH2Cl, 60 °C, 12 h). This process results in a modest kinetic resolution of the starting enone (krel(fast/slow) = 3.4, (+)-i isolated in 95% ee at 89% conversion).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.