

Abstract
DNA viruses such as bacteriophages and herpesviruses deliver their genome into and out of the capsid through large proteinaceous assemblies, known as portal proteins. Here we report two snapshots of the dodecameric portal protein of bacteriophage P22. The 3.25 Å resolution structure of the portal protein core bound to twelve copies of gp4 reveals a ~1.1 MDa assembly formed by 24 proteins. Unexpectedly, a lower resolution structure of the full length portal protein unveils the unique topology of the C-terminal domain, which forms a ~200 Å long, α-helical barrel. This domain inserts deeply into the virion and is highly conserved in the Podoviridae family. We propose that the barrel domain facilitates genome spooling onto the interior surface of the capsid during genome packaging and, in analogy to a rifle barrel, increases the accuracy of genome ejection into the host cell.
INTRODUCTION
The icosahedral capsid of tailed bacteriophages and herpesviruses is a tough nano-shell built to contain and protect the viral genome. To allow communication with the outside environment, a unique 5 -fold vertex of the capsid is replaced by a dodecamer of portal protein, which forms a hollow conduit large enough to accommodate the passage of double stranded DNA (dsDNA) 1. The portal protein serves at least two critical functions in the lifecycle of DNA viruses. During infection, it mediates ejection of the viral genome into the host, in a process that is thought to be energized by the highly pressurized genome stored inside the capsid 2. Conversely, during virus assembly, the terminase complex assembles onto the portal protein to actively package DNA into the capsid against a steep concentration and pressure gradient 3. In tailed bacteriophages, the portal protein (or head-to-tail connector) also serves as the attachment point for the tail apparatus, which varies greatly in length and composition. Based on tail morphology, bacteriophages have been classified in three families: Podoviridae, Siphoviridae and Myoviridae, which have short or long non-contractile or long contractile tails, respectively 4.
Bacteriophage P22 infects Salmonella enterica serovar Typhimurium and is a prototypical representative of the Podoviridae family 5. The mature P22 virion presents an icosahedral T=7l capsid about 650 Å in diameter 1,6, interrupted at a unique vertex by ~430 Å long “portal vertex structure” 7. This molecular machine consists of five unique polypeptides, namely the portal protein, gp1; the tail spike, gp9; the tail needle, gp26; and tail factors gp4 and gp10. These five proteins assemble together in various stoichiometries into a macromolecular complex nearly three megadaltons in size, which includes 51 polypeptide chains 7,8. The portal vertex structure rapidly assembles after genome packaging by the sequential addition of gp4, gp10, gp26 and gp9 9. It contains all of the molecular determinants necessary for the virus to adhere and penetrate the host cell surface, as well as providing the conduit to expel the viral genome into the host. The tail needle gp26 functions as the physical ‘plug’ which seals the portal protein channel and stabilizes the highly condensed DNA inside the virion 9–11. Gp26 binds the virion via an N-terminal, highly conserved domain 12, which assembles onto a preformed hexamer of gp10 8,11. The latter binds the portal vertex via gp4, which, although monomeric in solution, oligomerizes upon binding to portal protein, to form a dodecameric assembly 13,14. Gp26 likely plays the dual role of portal protein plug and cell wall penetrating needle 15, thereby controlling the opening of the portal channel and the ejection of the viral genome into the host. Loss or disruption of any of the genes encoding gp4 or gp10/gp26 results in phage particles that package DNA but fail to retain it in the capsid (“DNA-leakage” phenotype) 9,16.
Like the bacteriophage tails, portal proteins also vary greatly in molecular mass and composition from the small connector seen in the phage phi29 (M.W. ~35 kDa) 17 to the large P22 portal protein (M.W. ~83 kDa) 18. In phi29, also a member of the Podoviridae family, the portal protein forms a hollow funnel ~75Å in length with an internal diameter of ~40 Å 17,19,20. A cyclic, likely pentameric 17 RNA molecule of 174-bases, named prohead RNA (pRNA), is bound to the connector and plays a critical role in DNA packaging 21. In SPP1, a phage of the Siphoviridae family, the portal protein structure was determined by cryo-electron microscopy (cryo-EM) 22 and x-ray crystallography 23 as a thirteen-fold symmetric oligomer. Subsequent analysis of the connector in the context of the mature virion 24, or as isolated connector bound to tail factors gp16 and gp15 25 revealed that the biologically relevant oligomeric state of SPP1 portal protein is consistent with a dodecamer. As with the SPP1 connector, many other ectopically purified portal proteins have been reported to be highly polymorphic in vitro 23,24,26,27, although it is believed that the biological active oligomeric state of portal proteins is exclusively dodecameric 1,6,24.
To provide an atomic description of a large portal protein, similar in size and composition to those found in herpesviruses 28, we have studied the structure of the bacteriophage P22 portal protein. This paper describes two independently determined crystallographic snapshots of this dodecameric protein.
RESULTS
Crystallographic studies on bacteriophage P22 portal protein
Crystallization studies on the phage P22 portal protein began nearly a decade ago 26. The limited resolution of full length portal protein crystals, which diffracted weakly to ~7 Å, obstructed high resolution structure determination 26. Complete diffraction data to 3.25 Å resolution was obtained using a large N-terminal chymotryptic fragment of the portal spanning residues 1–602 (referred to as the ‘portal protein core’) 14 bound to twelve copies of tail accessory factor gp4 13,14. Unfortunately, experimental phasing with conventional heavy atoms clusters proved ineffective due to the intrinsic non-isomorphism of portal crystals, which varied greatly in unit cell dimensions and space group. Likewise, all attempts to phase the structure by molecular replacement and non crystallographic symmetry (ncs) averaging using the previously determined cryo-EM reconstruction of the portal protein core bound to gp4 29 were unsuccessful, likely due to the limited quality of this EM-model. When the 9.4 Å resolution structure of P22 tail become available 7, the density for portal protein–gp4 was computationally extracted and used to solve three independent crystal forms of the portal protein core–gp4 complex by molecular replacement, to a resolution of 9.5 Å. Using a combination of multi- and intra-crystal ncs–averaging, we were able to extend this initial set of EM-phases to the resolution of the best diffracting crystal form (3.25 Å), which contains ~18,500 residues in the asymmetric unit (Supplementary Table 1). A complete atomic model of both main and side chains was built and the register confirmed using the 432 seleno-methione sites present in the asymmetric unit. The final portal protein core–gp4 complex model was refined to an Rwork / Rfree of 22.2/ 23.7%, at a resolution of 3.25 Å (Fig. 1a,b, Supplementary Fig. 1, Supplementary Table 1). The atomic model of portal protein core was used to determine the 12-fold averaged structure of full length P22 portal protein (residues 1–725), which was refined to an Rwork / Rfree of 18.6 / 26.3%, at 7.5 Å resolution (Fig. 1c,d, Supplementary Fig. 2, Supplementary Table 1). With the exception of a short loop between residues 464–492, the entire polypeptide chain has been unambiguously traced, allowing for the first time to visualize the entire structure of a prototypical dodecameric portal protein of Podoviridae.
FIG. 1. Crystal structure of the bacteriophage P22 portal protein and the portal protein core–gp4 complex.

Ribbon diagram of the portal protein core bound to twelve copies of gp4 in side (a) and bottom (b) view. The portal protein is colored in blue, red and yellow, while gp4 is in green. In (c) and (d) is the side and top view of the full length portal protein, which includes the barrel domain (res. 603–725) color-coded as in (a,b). See also Supplementary Figs 1, 2 and 3.
Overview of a 1 MDa Genome Translocating Machine
The P22 portal protein folds into a ~0.96 MDa ring of twelve identical subunits, symmetrically arranged around a central channel of variable diameter (Fig. 1a–d), with an overall height of ~300 Å. Gp4 binds to the bottom of portal protein forming a second dodecameric ring ~75 Å in height (Fig. 1a,b). The quaternary structure of the P22 portal protein can be described as a funnel-shaped core ~170 Å in diameter, connected to a ~200 Å long largely α-helical tube formed by the C-terminal residues 603–725, which resembles a rifle barrel. Whereas the portal core is similar in topology to other portal proteins from phage SPP1 23 and phi29 17,20, the helical barrel (Fig. 1c) is the first example of a dodecameric tube in a portal protein, and resembles the α-barrel domain of the bacterial outer membrane protein TolC 30. The connection between the portal protein core and helical tube is readily cleaved by chymotrypsin in solution, suggesting an intrinsic flexibility between these two folded units. The X-ray structure of the full length portal protein determined crystallographically fits accurately inside the asymmetric cryo-EM reconstruction of the mature P22 virion 1 (Fig. 2b). The only region which shows substantial deviations in the two structures is the barrel domain, which in the cryo-EM was determined as four distinct rings of density 1, while it is obviously continuous in the crystal structure (Fig. 2b). The structure of the portal protein determined crystallographically is virtually identical to that seen in the cryo-EM reconstruction, suggesting that binding of gp4 does not induce any dramatic conformational changes in the portal protein. This is also demonstrated by fitting the crystal structure of the portal protein core–gp4 complex in the 9.4 Å symmetrized cryo-EM reconstruction of the isolated portal vertex structure 7 (Fig. 2c). Although in the latter, the barrel domain is not visible and is likely unfolded due to the harsh condition used for extraction 7, the fitting of the portal protein core–gp4 X-ray model in the EM-density is excellent (Fig. 2c,d). Gp4 directly contacts the tail spike head-binding domain 31,32, which flexibly connects the tail spike to the rest of the portal vertex structure (Fig. 2d). The loop in portal protein between residues 464–492, which is not present in our crystallographic model, is also poorly visible in the EM-density, reinforcing the intrinsic flexibility in this moiety (Fig. 2d). This region of P22 portal protein is likely mobile and does not obey dodecameric symmetry. Together the X-ray structures of the gp4-bound and -unbound portal protein presented in this paper, as well as cryo-EM of mature virion and isolated tail apparatus clearly demonstrate that the gp4 assembly to portal protein induces little, if any, difference in the portal core. These structures unambiguously confirm that the conformational changes in the P22 portal channel observed by Zheng et al. as a result of gp4 binding 29 are not physiologically relevant and may reflect an artifact of the cryo-EM reconstruction. Accordingly, fitting the crystal structure of portal protein core (with and without gp4) inside the cryo-EM reconstructions determined by Zheng et al. 29 reveals poor correlation between the X-ray models and cryo-EM density in the DNA pumping channel (Supplementary Fig. 5).
FIG. 2. Fitting of P22 portal protein and gp4 X-ray models into the cryo-EM reconstructions of the mature virion 1 and isolated portal vertex structure 7.

(a) The 17 Å asymmetric reconstruction of bacteriophage P22 mature virion (EMD-1220) 1 with capsid and tail colored in gray and purple, respectively. (b) Blow-up of the five-fold icosahedral axis occupied by the portal protein, with X-ray models of portal protein (in red), gp4 (in green) and tail spike 31,32 (in purple) fit into the EM-density. The density for the barrel domain is partially continuous in the EM model and fades towards the C-terminus of the barrel domain. (c) The 9.4 Å symmetrized cryo-EM reconstruction of the P22 portal vertex structure 7 (EMD-5051), with the X-ray models of portal protein core (in red), gp4 (in green), tail spike 31,32 (in purple) and tail needle gp26 10,11 (in blue) fit into the EM-density. (d) Blow-up of the portal protein core–gp4 complex showing the excellent fit of the X-ray models inside the cryo-EM density.
The Portal Protein Fold in Bacteriophage P22
The structure of the P22 portal protein protomer can be divided into three distinct domains, named here as the leg (I), hip (II) and barrel (III) (Fig. 3a,b, Supplementary Fig. 3). Domains I and III are largely helical and form the majority of the DNA translocating channel. Domain II adopts an α/β-fold characterized by two sheets of eight β-strands, which cross each other to form a β-barrel-like structure. Comparison of P22 with SPP1 and phi29 connectors reveals a fundamentally conserved protein fold despite the relatively low sequence similarity (typically less than 20%) (Fig. 3b–d). The leg domain (res. 317–512) presents the greatest conservation across bacteriophages and likely herpesviruses, while both the hip and barrel domains vary greatly in length and complexity. In the dodecamer, the body of each portal protomer is nearly parallel to the 12-fold axis running along the central channel, while the leg domain and the barrel domain are tilted by ~30° (Fig. 3e,f). As a result, in the dodecamer, each protomer spirals ~210° around the channel axis, to bury an overall surface of ~11,800 Å2 33, over a total length of 330 Å (Fig. 3e,f). The portal–portal monomer interface is stabilized by a complex network of salt bridges between a largely positive face of the portal monomer and the complementary largely negative face of its adjacent neighbor. A similar interface was previously observed in the phi29 portal protein 17,20, and is dramatically more extended in P22, where six Lys/Arg of one face interact with ten Glu/Asp of the neighboring face. This highly ionic interaction may explain previous observations about the portal oligomerization dynamics. First, the assembly of P22 portal monomers into oligomers was found to be strongly dependent on salt concentration 26,34, which is explained by the sensitivity of ionic interactions to ionic strength. Second, the high concentration of EDTA (60 mM or higher) required in vitro to assemble dodecameric portal protein 14 likely functions by chelating divalent cations nonspecifically trapped at the portal–portal interface, which would interfere with the correct assembly of dodecameric rings.
FIG. 3. The portal protein fold.
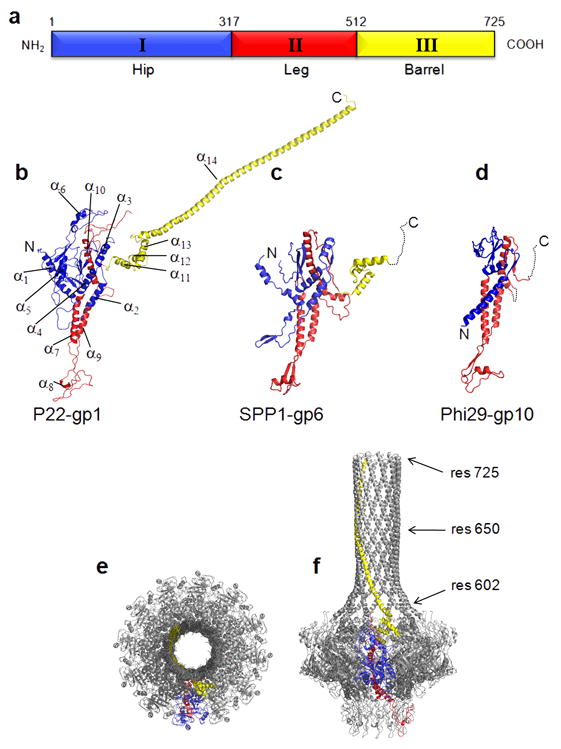
(a) Topology of the P22 portal protomer, colored by domains. Ribbon diagram of the portal protein monomers from the bacteriophages P22 (b), SPP1 (c) 23 and phi29 (d) 17,20 colored based on similarity to the P22 portal protein in (a). The three proteins have MW of ~83 kDa, ~55 kDa, and ~33 kDa, respectively. Despite the overall similarity in fold, the sequence similarity is below 20%. (e,f) Monomer organization in the portal dodecamer. Ribbon diagram of the full length portal protein, in (e) top and (f) side view, with only one protomer colored by domain as in Figs. 1 and 2, and the other eleven shown in gray. Each monomer of portal is nearly vertical in the hip domain (blue), but has an apparent tilt of ~30° relative to the 12-fold axis in both the leg domain (red) and the barrel (yellow).
The Barrel Domain
The barrel domain (Fig. 1c, 2b, 3b, 3f) is undoubtedly the most striking structural feature of the P22 portal protein. It is built by a largely α-helical stretch spanning ~120 residues. A leucine zipper motif is predicted between residues 600–650 (Supplementary Fig. 6), but was not confirmed in the structure of the full length portal protein due to the lack of visible side chains in the barrel domain. Almost one fifth of the residues in the barrel domain are Glns (Supplementary Fig. 3, 6), which is about four times higher than the average number of Glns in the remainder of the P22 portal protein. Interestingly, the Gln-rich domain of histone deacetylase 4 HDAC4 also folds into a straight α-helical homo tetramer 35. This helical oligomer is held together by multiple hydrophobic residues mixed with arrays of Glns that engage in extensive intra- and inter-helical stabilizing interactions. Similarly, it is possible that the arrays of Glns in the barrel domain of portal protein also contribute to the stabilization of this dodecameric α-helical structure. In vitro, deletion of the barrel domain does not prevent oligomerization 14, suggesting that this domain is not essential for assembly. Spontaneous loss of the last 50 C-terminal residues of the P22 portal protein in vivo (dif1 mutant) (Fig. 3f) are dispensable for phage viability 36. However, P22 virions expressing only the portal protein core (res. 1–602) are completely noninfectious (P.E. Prevelige, University of Alabama, personal communication). Interestingly, both the region 600–650 of the barrel domain and the putative coiled coil structure are highly conserved across the Podoviridae family of short tailed dsDNA bacteriophages, which includes members of the P22-like subgroup such as Sf6, CUS-3, epsilon34, APSE-1 (Supplementary Fig. 6). Interestingly, Podoviridae of the phi29-like genus, which encode a smaller portal protein and use a pRNA for DNA-packaging 21 do not have a detectable barrel domain. This suggests that an alternative mechanism to the portal barrel is possible in different members of the Podoviridae family.
Structure, Assembly and Conservation of gp4
On the opposite end of the portal assembly, gp4 is the first of three late gene products including gp10 and gp26 9 that add to the portal vertex marking the end of capsid morphogenesis. In solution gp4 exists as a monomer, even at millimolar concentrations, which readily oligomerizes upon binding to portal protein 13. The crystallographic structure of gp4 reveals a roughly globular protein consisting of four α-helices and an extended C-terminal tail (Fig. 4a, Supplementary Fig. 4). Helices α1-α3 pack loosely together to generate a small hydrophobic core, while the longer C-terminal helix α4 docks onto the outer surface of the portal protein (Fig. 4b). In the structure of the portal protein core–gp4 complex, a protomer of gp4 sits at the interface of two portal subunits (Fig. 4b,c) and extends its C-terminal tail into the interface of the neighboring two subunits, interacting in total with 4 portal monomers. The interface between a single portal subunit and a single gp4 subunit buries only ~500–650 Å2, while a protomer of gp4 buries as much as ~2250 Å2 of surface area in the dodecameric portal ring. Of the 166 residues of gp4 (146 of which have been modeled), 60 residues interact with portal protein, generating as many as 8 salt bridges and 34 hydrogen bonds. The small buried surface of a single gp4 with a single portal monomer explains why monomeric gp4 has no detectable binding affinity for unoligomerized portal protein 13. Likewise, the only contacts between gp4 subunits are mediated by helices α1-α2 of neighboring subunits (Fig. 4d). This modest binding interface buries ~900 Å2 with a free binding energy of ~4.0 kcal/mol 33, which explains the lack of oligomeric gp4 in the absence of portal protein 13. Despite an undetectable sequence similarity, the structure of gp4 superimposes with an rmsd of 3.0 Å and 2.9 Å to the middle tail factors gp16 and gp15 of bacteriophages HK97 and SPP1, respectively (Fig. 5b,c). These factors all share a similar helical fold (Fig. 5d), exist as monomers in solution, and readily oligomerize upon binding to their respective portal vertexes 13,37,38. We speculate that the portal protein-induced assembly of gp4-like middle tail factors is essential to displace the terminase complex and initiates portal protein closure after packaging is completed. This hypothesis is corroborated by the structural similarity between gp4 and the N-terminal domain of the bacteriophage Sf6 small terminase subunit 39.
FIG. 4. Three-dimensional structure of the middle ring factor gp4.

(a) Ribbon diagram of monomeric gp4. (b) Surface representation of portal protein core (in gray) in complex with 12 copies of gp4, shown as green ribbon. (c) Blow-up view of the portal protein core–gp4 binding interface. Portal protein and gp4 are shown as coils in gray and green, respectively. A semitransparent surface for neighboring gp4 protomers is highlighted in different tones of green. (d) Oligomeric conformation of gp4 extracted from (b), with one protomer of gp4 colored in green and the other eleven in gray. See also Supplementary Fig. 4.
FIG. 5. Structural conservation of the middle ring factor gp4.

Ribbon diagrams of (a) P22-gp4, (b) SPP1-gp15 37 and (c) HK97-gp6 38. (d) Superimposition of all three middle ring factors reveals a common, shared protein fold.
The DNA Translocation Channel
The overall length of the P22 DNA translocation channel is ~300 Å, with a lumen long enough to protect ~89 bases of double stranded linear B-DNA (Fig. 6a). The average internal diameter of the DNA translocating channel varies throughout the structure, between 35–75 Å. The diameter at the center of the portal channel between the loops connecting helices α9-α10 (Supplementary Fig. 3) is ~45 Å, considerably larger than predicted in the pseudo-atomic model of dodecameric SPP1 portal protein 23. Immediately above and below these loops, the portal channel presents two vestibules ~75 Å in diameter. In the barrel domain, the average interior diameter is ~35 Å, excluding side chains that are not visible in this region of our model (Supplementary Fig. 2). This is large enough to accommodate dsDNA, which has a diameter of ~20 Å in its hydrated form 40 (Fig. 6a–c). The large diameter of the DNA-translocation channel in our structure is therefore incompatible with a model where the tunnel loops tightly grip DNA and undergo sequential conformational changes coupled to portal rotation and DNA translocation 23. Likewise, the barrel domain has a left-handed twist, opposite to that of B-DNA (Fig. 6b), which is a right-handed helix. The lack of obvious “threads” in the portal channel, complementary to the double helical groove of DNA, is inconsistent with the nut and bolt model of DNA rotation inside the portal 41. Finally, the interior channel of the portal protein core is partially negatively charged with five notable rings of negative charge corresponding to residues Glu70/423 (which are clustered together), Glu414, Glu406, Glu393 and Glu396. This charge likely repulses the negatively charged backbone of DNA during genome ejection and packaging and may serve to keep the DNA centered in the channel.
FIG. 6. Architecture of the DNA translocating channel.
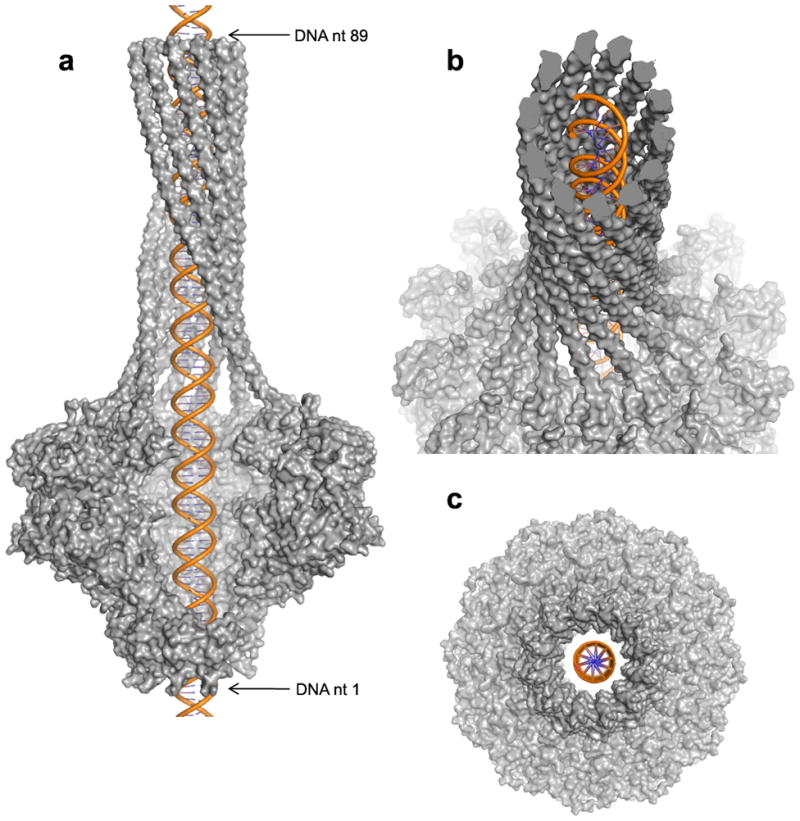
(a) Side view surface representation of P22 portal protein (in gray) with a ~100 nucleotide long dsB-DNA (in orange) modeled inside the DNA translocating channel. Four portal protein protomers have been omitted from the structure to visualize the interior of the DNA translocating channel. (b) Tilted view of the model in (a) revealing an opposite twist between the left-handed portal helices and the right-handed B-DNA helix. (c) Bottom view showing the lack of obvious contacts between the portal protein leg domains and modeled DNA.
Casjens et al. identified two point mutations in portal protein responsible for packaging ~2000 extra base pairs of chromosomal DNA 42. One of these mutations (V64M) is located on top of helix α2 (Supplementary Fig. 3), directly facing the DNA translocating channel. The longer side chain of Met at position 64 is expected to rigidify the hydrophobic environment between helix α2 and α4, thus reducing the mobility of the portal channel. Along the same lines, in phage SPP1, immobilization of the α-helices forming the DNA translocating channel by intersubunit cross-linking disrupts DNA packaging 43. However neither the mutation in P22 nor the cross-linking in SPP1 affect DNA ejection, emphasizing the idea that DNA translocation and ejection are not symmetric processes. Although a rotation of the entire portal vertex structure is not likely to accompany DNA translocation 44,45, our structure and previous mutational work provide clues that portal proteins are intrinsically dynamic macromolecular assemblies.
DISCUSSIONS
In this paper, we have described the 24-fold averaged structure of the portal protein core in complex with twelve copies of the tail factor gp4 to 3.25 Å resolution and the structure of the full length P22 portal protein to 7.5 Å resolution. Together these two structures reveal unique insights into the structure of a prototypical Podoviridae portal protein, similar in size (~1 MDa) and composition to Herpesvirus portal proteins. As suggested by our crystal structure and illustrated in the asymmetric cryo-EM reconstruction of the mature virion 1, in P22 and likely other Podoviridae, the viral genome does not enter the empty lumen of the capsid from a vertex as previously believed, but actually enters from the barrel of the portal, nearly in the center of the capsid shell. We propose distinct functions of the barrel domain in the lifecycle of Podoviridae. During DNA packaging, the barrel undulates in a circular motion while spooling the phage DNA outwards, onto the capsid shell, in a way reminiscent of a water sprinkler. This allows for filling the capsid with ordered, circumcentric layers of DNA 2. As the pressure of packaged DNA builds up inside the capsid, the barrel may function as a valve that senses the pressure and regulates the length and packaged density of DNA 42. In addition to its role in packaging, there likely exists a dual function of the barrel domain as it relates to ejection. First, the barrel would hold the trailing end of the packaged genome, keeping the DNA primed for ejection, and ready for a new cycle of infection. Since the diameter of the barrel domain is large enough to accommodate dsDNA, but too small to thread DNA as a loop, keeping the linear end of the viral genome in the portal channel is a prerequisite for pressure-driven ejection into the host. Second, the barrel domain likely stabilizes the linear momentum of the genome as it exits the capsid during infection, in much the same way as a longer barrel on a rifle allows for greater range and accuracy. In ballistics, the LeDuc formula 46 predicts that for a bullet travelling down a barrel, the velocity exponentially increases while the amount of pressure behind it exponentially decreases with the length of the barrel. While long-tailed phages use the length of the tail tube to balance pressure and velocity of genome ejection, we propose short-tailed bacteriophages of the Podoviridae family use an internal barrel domain to compensate for short tails. This ensures accurate genome delivery during infection and avoids dissipation of DNA into the periplasm due to the excessive pressure at the portal protein vertex. In summary, regardless of its location, be it interior in the barrel domain or exterior to the capsid in the tail, the viral genome will always be ejected through a tubular structure, which ensures accurate and efficient delivery into the host.
Accession codes
The atomic coordinates and structure factors for portal protein core–gp4 complex and full length portal protein have been deposited in the protein Data Bank with accession codes 3LJ4 and 3LJ5, respectively.
METHODS
Biochemical techniques
Purification of full length P22 portal protein (residues 1–725), portal protein core (residues 1–602), gp4 and portal protein core–gp4 complex were previously described 14,29. For crystallization experiments, a mutant of gp4 carrying the point mutation (A141P) was used. This mutant is less susceptible to C-terminal degradation as compared to wild-type gp4 14. For selenomethionine incorporation, E. coli BL21 cells were grown in M9 minimal medium and supplemented with selenomethionine and other essential amino acids before induction with 0.5 mM isopropyl β-D-1-thiogalactopyranoside. Purification of selenomethionine derivatized portal protein was identical to that of wild type protein.
Crystallization and X-ray data collection
Crystallization of full length portal protein was previously described 26. Crystals of portal protein core bound to gp4 grew in two interchangeable conditions, which resulted in the three distinct crystal forms presented in Supplementary Table 1. The first condition was 16 % (w/v) PEG 8,000, 0.1 M NaMoO4, 0.1 M MOPS at pH 7.0 and gave rise to the large P21 and small P21 crystal forms (indicated as L-P21 and S-P21 in Supplementary Table 1). The second crystallization condition was 20 % (w/v) PEG 8,000, 0.1 M (NH4)2HPO4, 0.1 M MES at pH 6.0, which gave predominantly primitive triclinic crystals (L-P1 in Supplementary Table 1). Hundreds of crystals were screened for diffraction between the years 2005–2009, resulting in ~20 complete datasets belonging to each of the three crystal forms. Complete diffraction data between 4.0 Å and 3.25 Å was obtained from each of the three different crystal forms (Supplementary Table 1), which all contain selenomethionine derivatized portal protein core bound to native gp4. The highest resolution dataset was measured for the L-P21 crystal form at CHESS beamline A1. All data were indexed, integrated and scaled using HKL-2000 47.
Structure determination and refinement of portal protein core–gp4 complex
The structure of portal protein core–gp4 complex was phased by molecular replacement using the cryo-EM structure of the P22 tail 7 as an initial phasing model. The density for portal protein–gp4 was computationally extracted from the whole tail and used as a 9.5 Å molecular replacement model in PHASER 48. Initial low resolution EM-phases were dramatically improved and extended to 3.25 Å resolution by a combination of intra- and inter-crystal symmetry averaging in MULTI–DM 49. A complete model for portal protein core–gp4 complex was built over ~20 rounds of manual model building in COOT 50 followed by positional and isotropic B-factor refinement of in Phenix 51. To increase the observation/parameter ratio, non-crystallographic symmetry restraints were included in refinement and found to significantly decrease the Rfree. Additionally, from the early stages of model building, the model was divided into 48 Translation/Libration/Screw (TLS)52 groups, with each monomer of portal protein core and gp4 being designated as a single TLS group (24 + 24 = 48 TLS groups). Sequence register for all 18,432 residues in the asymmetric unit was confirmed using the 432 selenomethionine sites present in the asymmetric unit. With the exception of a disordered loop in the portal protein between residues 464–492 and the N-terminus from residue 1–4, as well as the termini of gp4 (residues 1–13 and 160–166), all residues in the structure have been assigned in the structure. Finally, 528 water molecules were modeled in 3.5σ peaks of Fo–Fc density. Several larger blobs of density, likely ascribed to ions are clearly visible in the final electron density but have not been modeled. The portal protein core–gp4 complex model has been refined to an Rwork / Rfree ~ 22.2 / 23.7 %, using all reflections between 20–3.25 Å resolution (Supplementary Table 1) (reflections for calculating Rfree were selected in thin resolution shells). The final model has good geometry (r.m.s.d.bond = 0.006 Å, r.m.s.d.angle = 0.97 O) and the Ramachandran plot shows 73.9 % of residues in the most favored regions, 21.7 % of residues in allowed regions and only 4.4 % in generously allowed regions, with no disallowed residues.
Structure determination and refinement of full length portal protein
The atomic model of the dodecameric portal protein core was used to determine the structure of the full length portal protein by molecular replacement in PHASER 48. Phases calculated from the obtained model were used to compute Fo–Fc difference maps, which at a contour of 3σ revealed a strikingly tubular electron density extending above the top of the portal protein core model. After 12-fold averaging, this density became perfectly continuous, spanning almost 200 Å from residue 602 of portal outward (Supplementary Fig. 2). Based on the features of the density, as well as the secondary structure predictions of the C-terminal of the portal protein, this density was modeled in COOT 50 as a continuous helix starting at residue 589 and ending at residue 719, followed by a short random coil segment of 6 residues. The register was also confirmed by computing low resolution anomalous difference maps, which revealed discrete peaks of density for selenomethionine residues at position 608, 657 and 702. The complete model for full length portal protein was refined by rigid body refinement in Phenix 51 using data between 60–7.5 Å. The dataset used to refine the atomic model contains approximately 25,572 individual reflections (Supplementary Table 1). Further cycles of grouped B-factor refinement with 12 individual TLS domains resulted in a final model that has Rwork / Rfree ~18.6 / 26.3 %, using all reflections between 60–7.5 Å resolution. The loop between residues 464–492 and side chains in the barrel domain (residues 603–725) have not been modeled.
Fitting of X-ray models in cryo-EM reconstructions, figures and illustrations
All ribbon diagrams were generated using programs PyMol53 or Chimera54. All EM maps were visualized using the program Chimera54. The atomic models of full length portal protein and portal core–gp4 complex as well as the P22 tail-spike protein gp9 (PDB #1TSP, 1LKT)31,32 and gp26 (PDB # 2POH)11 were fit in the cryo-EM reconstructions manually. The fit was improved by rigid-body refined using the command “Fit Models in Maps” in Chimera54.
Supplementary Material
Acknowledgments
We thank Vivian Stojanoff and staff at the NSLS beamlines X6A, X25, X29A and the macCHESS staff for beam time and assistance in data collection. This work was supported by NIH grants 1R56 AI076509-01A1 (to G.C.), RO1 AI40101 (to J.E.J).
Footnotes
AUTHOR CONTRIBUTIONS
A.O. and G.C. crystallized the full length portal and portal protein core:gp4 complex, collected the X-ray data and determined the structures. P.E.P. isolated the gene encoding P22 portal protein and helped with data analysis. J.E.J. supervised the crystallization and data collection of full length portal protein and helped with data analysis. G.C. coordinated the overall project and wrote the manuscript with A.O.
COMPETING INTERESTS STATMENT
The authors declare no competing financial interests.
Note: Supplementary information is available on the Nature Structural and Molecular Biology website.
References
- 1.Lander GC, et al. The Structure of an Infectious p22 Virion Shows the Signal for Headful DNA Packaging. Science. 2006 doi: 10.1126/science.1127981. [DOI] [PubMed] [Google Scholar]
- 2.Johnson JE, Chiu W. DNA packaging and delivery machines in tailed bacteriophages. Curr Opin Struct Biol. 2007;17:237–43. doi: 10.1016/j.sbi.2007.03.011. [DOI] [PubMed] [Google Scholar]
- 3.Casjens S, Weigele P. Headful DNA packaging by bacteriophage P22. In: Catalano C, editor. Viral Genome Packaging Machines: Genetics, Structure and Mechanism. Landes Publishing; Georgetown, TX: 2005. pp. 80–88. [Google Scholar]
- 4.Ackermann HW. Bacteriophage observations and evolution. Res Microbiol. 2003;154:245–51. doi: 10.1016/S0923-2508(03)00067-6. [DOI] [PubMed] [Google Scholar]
- 5.Teschke CM, Parent KN. ‘Let the phage do the work’: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology. 2010;401:119–30. doi: 10.1016/j.virol.2010.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang J, Weigele P, King J, Chiu W, Jiang W. Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure. 2006;14:1073–82. doi: 10.1016/j.str.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 7.Lander GC, et al. The P22 tail machine at subnanometer resolution reveals the architecture of an infection conduit. Structure. 2009;17:789–99. doi: 10.1016/j.str.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olia AS, Bhardwaj A, Joss L, Casjens S, Cingolani G. Role of gene 10 protein in the hierarchical assembly of the bacteriophage P22 portal vertex structure. Biochemistry. 2007;46:8776–84. doi: 10.1021/bi700186e. [DOI] [PubMed] [Google Scholar]
- 9.Strauss H, King J. Steps in the stabilization of newly packaged DNA during phage P22 morphogenesis. J Mol Biol. 1984;172:523–43. doi: 10.1016/s0022-2836(84)80021-2. [DOI] [PubMed] [Google Scholar]
- 10.Olia AS, Casjens S, Cingolani G. Structural plasticity of the phage P22 tail needle gp26 probed with xenon gas. Protein Sci. 2009;18:537–48. doi: 10.1002/pro.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olia AS, Casjens S, Cingolani G. Structure of phage P22 cell envelope-penetrating needle. Nat Struct Mol Biol. 2007 doi: 10.1038/nsmb1317. [DOI] [PubMed] [Google Scholar]
- 12.Bhardwaj A, Walker-Kopp N, Casjens SR, Cingolani G. An evolutionarily conserved family of virion tail needles related to bacteriophage P22 gp26: correlation between structural stability and length of the alpha-helical trimeric coiled coil. J Mol Biol. 2009;391:227–45. doi: 10.1016/j.jmb.2009.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olia AS, et al. Binding-induced stabilization and assembly of the phage P22 tail accessory factor gp4. J Mol Biol. 2006;363:558–76. doi: 10.1016/j.jmb.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 14.Lorenzen K, Olia AS, Uetrecht C, Cingolani G, Heck AJ. Determination of stoichiometry and conformational changes in the first step of the P22 tail assembly. J Mol Biol. 2008;379:385–96. doi: 10.1016/j.jmb.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Israel V. E proteins of bacteriophage P22. I. Identification and ejection from wild-type and defective particles. J Virol. 1977;23:91–7. doi: 10.1128/jvi.23.1.91-97.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lenk E, Casjens S, Weeks J, King J. Intracellular visualization of precursor capsids in phage P22 mutant infected cells. Virology. 1975;68:182–99. doi: 10.1016/0042-6822(75)90160-9. [DOI] [PubMed] [Google Scholar]
- 17.Simpson AA, et al. Structure of the bacteriophage phi29 DNA packaging motor. Nature. 2000;408:745–50. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bazinet C, Benbasat J, King J, Carazo JM, Carrascosa JL. Purification and organization of the gene 1 portal protein required for phage P22 DNA packaging. Biochemistry. 1988;27:1849–56. doi: 10.1021/bi00406a009. [DOI] [PubMed] [Google Scholar]
- 19.Simpson AA, et al. Structure determination of the head-tail connector of bacteriophage phi29. Acta Crystallogr D Biol Crystallogr. 2001;57:1260–9. doi: 10.1107/s0907444901010435. [DOI] [PubMed] [Google Scholar]
- 20.Guasch A, et al. Detailed architecture of a DNA translocating machine: the high-resolution structure of the bacteriophage phi29 connector particle. J Mol Biol. 2002;315:663–76. doi: 10.1006/jmbi.2001.5278. [DOI] [PubMed] [Google Scholar]
- 21.Guo PX, Erickson S, Anderson D. A small viral RNA is required for in vitro packaging of bacteriophage phi 29 DNA. Science. 1987;236:690–4. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- 22.Orlova EV, et al. Structure of the 13-fold symmetric portal protein of bacteriophage SPP1. Nat Struct Biol. 1999;6:842–6. doi: 10.1038/12303. [DOI] [PubMed] [Google Scholar]
- 23.Lebedev AA, et al. Structural framework for DNA translocation via the viral portal protein. Embo J. 2007;26:1984–94. doi: 10.1038/sj.emboj.7601643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lurz R, et al. Structural organisation of the head-to-tail interface of a bacterial virus. J Mol Biol. 2001;310:1027–37. doi: 10.1006/jmbi.2001.4800. [DOI] [PubMed] [Google Scholar]
- 25.Orlova EV, et al. Structure of a viral DNA gatekeeper at 10 A resolution by cryo-electron microscopy. Embo J. 2003;22:1255–62. doi: 10.1093/emboj/cdg123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cingolani G, Moore SD, Prevelige PE, Jr, Johnson JE. Preliminary crystallographic analysis of the bacteriophage P22 portal protein. J Struct Biol. 2002;139:46–54. doi: 10.1016/s1047-8477(02)00512-9. [DOI] [PubMed] [Google Scholar]
- 27.Trus BL, et al. Structure and polymorphism of the UL6 portal protein of herpes simplex virus type 1. J Virol. 2004;78:12668–71. doi: 10.1128/JVI.78.22.12668-12671.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Newcomb WW, et al. The UL6 gene product forms the portal for entry of DNA into the herpes simplex virus capsid. J Virol. 2001;75:10923–32. doi: 10.1128/JVI.75.22.10923-10932.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng H, et al. A conformational switch in bacteriophage p22 portal protein primes genome injection. Mol Cell. 2008;29:376–83. doi: 10.1016/j.molcel.2007.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koronakis V, Sharff A, Koronakis E, Luisi B, Hughes C. Crystal structure of the bacterial membrane protein TolC central to multidrug efflux and protein export. Nature. 2000;405:914–9. doi: 10.1038/35016007. [DOI] [PubMed] [Google Scholar]
- 31.Steinbacher S, et al. Crystal structure of P22 tailspike protein: interdigitated subunits in a thermostable trimer. Science. 1994;265:383–6. doi: 10.1126/science.8023158. [DOI] [PubMed] [Google Scholar]
- 32.Steinbacher S, et al. Phage P22 tailspike protein: crystal structure of the head-binding domain at 2.3 A, fully refined structure of the endorhamnosidase at 1.56 A resolution, and the molecular basis of O-antigen recognition and cleavage. J Mol Biol. 1997;267:865–80. doi: 10.1006/jmbi.1997.0922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–97. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 34.Moore SD, Prevelige PE., Jr Structural transformations accompanying the assembly of bacteriophage P22 portal protein rings in vitro. J Biol Chem. 2001;276:6779–88. doi: 10.1074/jbc.M007702200. [DOI] [PubMed] [Google Scholar]
- 35.Guo L, Han A, Bates DL, Cao J, Chen L. Crystal structure of a conserved N-terminal domain of histone deacetylase 4 reveals functional insights into glutamine-rich domains. Proc Natl Acad Sci U S A. 2007;104:4297–302. doi: 10.1073/pnas.0608041104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eppler K, Wyckoff E, Goates J, Parr R, Casjens S. Nucleotide sequence of the bacteriophage P22 genes required for DNA packaging. Virology. 1991;183:519–38. doi: 10.1016/0042-6822(91)90981-g. [DOI] [PubMed] [Google Scholar]
- 37.Lhuillier S, et al. Structure of bacteriophage SPP1 head-to-tail connection reveals mechanism for viral DNA gating. Proc Natl Acad Sci U S A. 2009;106:8507–12. doi: 10.1073/pnas.0812407106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cardarelli L, et al. The crystal structure of bacteriophage HK97 gp6: defining a large family of head-tail connector proteins. J Mol Biol. 395:754–68. doi: 10.1016/j.jmb.2009.10.067. [DOI] [PubMed] [Google Scholar]
- 39.Zhao H, et al. Crystal structure of the DNA-recognition component of the bacterial virus Sf6 genome-packaging machine. Proc Natl Acad Sci U S A. 2010;107:1971–6. doi: 10.1073/pnas.0908569107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vlieghe D, Turkenburg JP, Van Meervelt L. B-DNA at atomic resolution reveals extended hydration patterns. Acta Crystallogr D Biol Crystallogr. 1999;55:1495–502. doi: 10.1107/s0907444999007933. [DOI] [PubMed] [Google Scholar]
- 41.Hendrix RW. Symmetry mismatch and DNA packaging in large bacteriophages. Proc Natl Acad Sci U S A. 1978;75:4779–83. doi: 10.1073/pnas.75.10.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Casjens S, et al. Bacteriophage P22 portal protein is part of the gauge that regulates packing density of intravirion DNA. J Mol Biol. 1992;224:1055–74. doi: 10.1016/0022-2836(92)90469-z. [DOI] [PubMed] [Google Scholar]
- 43.Cuervo A, Vaney MC, Antson AA, Tavares P, Oliveira L. Structural Rearrangements between Portal Protein Subunits Are Essential for Viral DNA Translocation. J Biol Chem. 2007;282:18907–18913. doi: 10.1074/jbc.M701808200. [DOI] [PubMed] [Google Scholar]
- 44.Hugel T, et al. Experimental test of connector rotation during DNA packaging into bacteriophage phi29 capsids. PLoS Biol. 2007;5:e59. doi: 10.1371/journal.pbio.0050059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baumann RG, Mullaney J, Black LW. Portal fusion protein constraints on function in DNA packaging of bacteriophage T4. Mol Microbiol. 2006;61:16–32. doi: 10.1111/j.1365-2958.2006.05203.x. [DOI] [PubMed] [Google Scholar]
- 46.Guy AE. A practical method of calculating the powder pressure and the velocity of projectile along the bore of a cannon. Journal of the American Society for Naval Engineers. 1921;33:720–734. [Google Scholar]
- 47.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymology. Macromolecular Crystallography. 1997;276:307–26. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 48.McCoy AJ, et al. Phaser crystallographic software. J Appl Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Collaborative Computational Project N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Adams PD, et al. PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–54. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 52.Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. [Google Scholar]
- 53.DeLano WL. 2002 www.pymol.org.
- 54.Pettersen EF, et al. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.