

Abstract
Oncogenic activation of the RAS-ERK MAP kinase signaling pathway can lead to uncontrolled proliferation but can also result in apoptosis or premature cellular senescence, both regarded as natural protective barriers to cell immortalization and transformation. In FGFR3-related skeletal dyplasias, oncogenic mutations in the FGFR3 receptor tyrosine kinase cause profound inhibition of cartilage growth resulting in severe dwarfism, although many of the precise mechanisms of FGFR3 action remain unclear. Mutated FGFR3 induces constitutive activation of the ERK pathway in chondrocytes and, remarkably, can also cause both increased proliferation and apoptosis in growing cartilage, depending on the gestational age. Here, we demonstrate that FGFR3 signaling is also capable of inducing premature senescence in chondrocytes, manifested as reversible, ERK-dependent growth arrest accompanied by alteration of cellular shape, loss of the extracellular matrix, upregulation of senescence markers (α-GLUCOSIDASE, FIBRONECTIN, CAVEOLIN 1, LAMIN A, SM22α and TIMP 1), and induction of senescence-associated β-GALACTOSIDASE activity. Our data support a model whereby FGFR3 signaling inhibits cartilage growth via exploiting cellular responses originally designed to eliminate cells harbouring activated oncogenes.
Introduction
Activating mutations in FGFR3 result in several human skeletal dysplasias such as thanatophoric dysplasia (TD) and achondroplasia (ACH), the most common genetic forms of lethal and non-lethal dwarfism, respectively [1]. Despite recent progress in unraveling the mechanisms of FGFR3 signaling in cartilage, many aspects of this signaling still remain elusive. It is clear that FGFR3 activation leads to inhibition of chondrocyte proliferation and alteration of their differentiation, together contributing to marked shortening and disorganization of the endochondral growth plate cartilage [2]. This phenotype appears paradoxical when compared to the general action of FGFR tyrosine kinases as well as the role of FGFR3 in other diseases. Apart from skeletal dysplasias, the same activating FGFR3 mutations are associated with at least seven other conditions affecting skin (epidermal nevi, seborrheic keratosis, acanthosis nigricans) and cancer development (multiple myeloma, prostate, bladder cancer, seminoma) [1, 3, 4], which are all caused by excessive cell proliferation. The ability of FGFR3 to transform cells is perhaps best documented in multiple myeloma, where expression of FGFR3 harbouring the TD activating mutations enhances cell proliferation and survival, and reduces cell-growth dependency on interleukin 6. Furthermore, mice with transplanted TD-FGFR3 expressing bone marrow cells or NIH3T3 fibroblasts develop lethal leukemia or soft tissue tumors, altogether demonstrating the oncogenic capacity of FGFR3 [5-7].
The growth-inhibitory outcome of FGFR3 signaling in cartilage has not been satisfactorily explained to date, and it may stem from yet unknown, unique feature(s) of FGFR3 signaling in chondrocytes. Mutations associated with FGFR3-related skeletal dysplasias activate the FGFR3 oncogene in cartilage that, in turn, uses another oncogenic pathway, the ERK pathway, to cause many of its effects therein [8-13].
Aside from induction of proliferation, oncogene activation may also result in premature senescence, which represents a specific form of growth arrest and acts as a natural barrier to tumor development [14]. This study was undertaken to test the hypothesis that FGFR3 signaling inhibits chondrocyte proliferation via induction of premature senescence.
Materials and Methods
Cell studies, senescence-associated (SA) β-GALACTOSIDASE staining, FACS analysis and cell sorting
Rat chondrosarcoma chondrocytes (RCS) were propagated in DMEM media (Gibco, Gaithersburg, MD), containing 10% FBS (Atlanta Biological, Nordcross, GA) and antibiotics. Cells were analyzed for SA-β-GALACTOSIDASE activity using the SA-β-GALACTOSIDASE staining kit (Cell Signaling, Beverly, MA), according to the manufacturer's protocol. For the growth arrest assays, 1×104 cells were seeded per well in 24-well plates (Corning, Cambridge, MA), treated with FGF2 (RnD Systems, Minneapolis, MN) for the indicated period, and counted. SU5402, U0126, camptothecin and etoposide were purchased from Calbiochem (San Diego, CA), Sigma-Aldrich (St. Louis, MO), and Pharmacia-Upjohn (Bridgewater, NJ), respectively. For flow-cytometry analyses, cells were either transfected with FGFR3 vectors or treated with FGF2 for 48 or 24 hours, and stained for apoptosis and viability using combined propidium iodide (PI) and Annexin-FITC labelling kit according to manufacturer's protocol (Roche Applied Science, Indianapolis, IN). Both signals (2×104 cells analyzed per sample) were measured by Cyan Adp FACS instrument (Beckman Coulter, Brea, CA). For cell sorting, RCS chondrocytes were co-transfected with FGFR3 and GFP expression plasmids, grown for 48 hours, and GFP-positive cells were isolated using MoFlo XDP cell sorter (Beckman).
Extracellular matrix studies, phalloidin staining and confocal microscopy
For Alcian blue staining, growing RCS cultures were treated with FGF2 for 72 hours, fixed with 4% paraformaldehyde and stained with Alcian blue as described before [15]. For quantification of the extracellular matrix loss, cells were treated with FGF2 for 72 hours in the presence of 10 μCi/ml of [35S]sulfate (Perkin Elmer, Boston, MA). Following the cultivation period, cells were harvested and the incorporated radioactivity was determined by liquid scintillation. For immunocytochemistry, cells were grown on glass coverslips inserted into 6-well tissue culture plates (Corning), treated with FGF2 for 72 hours and fixed in 4% formaldehyde. Cells were stained overnight with vinculin-FITC antibody (Sigma-Aldrich) followed by one hour staining with Alexa Fluor 594-conjugated phalloidin (Invitrogen, Carlsbad, CA), and mounted in a solution containing DAPI for nuclear staining (Vector Laboratories, Burlingame, CA). The confocal microscopy was described in detail elsewhere [16].
Preparation of cell extracts, western blotting (WB), cell transfection, p53 luciferase reporter assay and real-time RT-PCR analysis
Cells were lysed in ice-cold lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM EDTA, 25 mM NaF) supplemented with proteinase inhibitors. Protein samples were resolved by SDS-PAGE and transferred onto a PVDF membrane (Bio-Rad, Hercules, CA). The following antibodies were used: ERK1/2, P-ERK1/2(T202/Y204), p53, P-p53(S15), CAVEOLIN 1, LAMIN A/C, ID2 (Cell Signaling); FGFR3, ACTIN (Santa Cruz Biotechnology, Santa Cruz, CA); α-GLUCOSIDASE and SM22α (Abcam, Cambridge, MA). NORE 1 antibodies were from Sigma and Cell Signaling, and TIMP-1 antibodies were from Santa Cruz and RnD Systems. WB signal was quantified by determining the integrated optical density (I.O.D.) of a given band using Scion Image software (Scion Corporation, Frederick, MA). Transfections were carried-out using the Fugene6 reagent according to the manufacturer's protocol (Roche). The pRK7 vectors encoding C-terminally FLAG-tagged both wild-type and mutant FGFR3 were described elsewhere [17]. The GFP-encoding plasmid was pCCEY and the empty plasmid was pcDNA3. In a standard transfection protocol, 6 μg of plasmid DNA was mixed with Fugene6 in a 1:3.2 (μg/μl) ratio, and transfected into 1.5×105 cells. For cell sorting experiments, 1×106 cells were co-transfected with FGFR3 and pCCEY in a 3:1 ratio, with the total amount of plasmid being 12 μg. For p53 luciferase reporter assays, RCS chondrocytes were co-transfected with a plasmid encoding firefly luciferase driven by a minimal CMV promoter containing tandem repeats of the p53 transcriptional response element and a constitutively expressing Renilla reniformis luciferase control vector (SABiosciences, Frederick, MD). Cells were grown for 24 hours after transfection, treated with FGF2 for 20 hours and analyzed for firefly and Renilla luciferase using a Dual Luciferase Reporter Assay kit according to manufacturer's protocol (Promega, Madison, WI). For real-time RT-PCR analyses, total RNA was isolated using the RNeasy kit (Qiagen, Valencia, CA) and treated with DNase I. cDNA was synthesized from 2 μg of total RNA using the Omniscript RT kit (Qiagen). Real-time RT-PCR conditions are described in detail elsewhere [13]. Table 1 lists the PCR primers used.
TABLE 1.
PCR primers used for real-time RT-PCR analyses
| gene | forward (5′ to 3′) | reverse (5′ to 3′) | product (bp) |
|---|---|---|---|
| α-Glucosidase | CCTGTTCTTCGCTGACCAGTTC | CGCCTGTTGACCTCCAAGTTAG | 290 |
| Apa1 | TGGCGATGGGCAACTCAAAG | CCGTAGTGAACTGCTTACCACACC | 275 |
| Caveolin1 | GAACCAGAAGGGACACAGTTTC | CGTAGATGGAATAGACACGGCTG | 235 |
| Caveolin2 | AGCTAGGCTTCGAGGATCTGATCG | GCTAGAAACACCGTCAGGAACTTG | 130 |
| Fkbp12 | TTGACTCCTCTCGGGACAGAAAC | ATGTGGTGGGATGATGCCTG | 176 |
| Gapdh | ACCACAGTCCATGCCATCAC | TCCACCACCCTGTTGCTGTA | 452 |
| Id1 | TTGGTCTGTCGGAGCAAAGC | GTCCCTGATGTAGTCGATAACATGC | 206 |
| Id2 | GACCCGATGAGTCTGCTCTACAAC | GATGCTGATGTCCGTGTTCAGG | 246 |
| Lamin A | CCACCGAAGTTCACCCTAAAGG | CCTCATCGTCATTGTCCTCAACC | 220 |
| Nore1 | CAAGAATGTCTGTAAGGCTGTGGAG | AAGTGGCAGGTAGAAGGATGTCCG | 277 |
| Ral-A | TCCCATTTCTCCTGGTTGGTAAC | TCTTTCTGGCTCGTATTTCCCTC | 180 |
| Sm22α | CAAGTTGGTGAACAGCCTGTACC | TCCATCGTTCTTGGTCACGG | 241 |
| Timp1 | ATGTCCACAAGTCCCAGAACCGCAG | ACCCCAAGGTATTGCCAGGTG | 328 |
Results
FGFR3 activation inhibits chondrocyte cell proliferation and extracellular matrix production, and alters the cellular shape of chondrocytes in vitro and in vivo
Inhibition of chondrocyte proliferation represents one of the most obvious cellular phenotypes of aberrant FGFR3 activation in vivo. When compared with a normal controls, the growth plate cartilage of an individuals with TD show marked shortening and disorganization of the proliferative zone due to the loss of colonies of proliferating cells. Apart from inhibition of proliferation, an increased cell-to-extracellular matrix ratio is evident in TD cartilage, suggesting a negative influence of FGFR3 activation on production of chondrocyte extracellular matrix [18]. Finally, the alteration of chondrocyte cellular shape appears in TD cartilage, manifested as an increased number of spindle-shaped, fibroblast-like chondrocytes (W. R. Wilcox, unpublished).
In vitro, all three phenotypes of FGFR3 signaling in cartilage can be reproduced in RCS chondrocytes, a FGFR3-expressing chondrocytic cell line that represents the best characterized cellular model for FGFR3-related skeletal dysplasias [19]. RCS chondrocytes respond to FGF2-mediated activation of endogenous FGFR3 via potent growth arrest in the G1-phase of the cell cycle (Fig. 1A)[20], that is dependent on signaling through the ERK MAP kinase pathway [13, 16]. Upon FGFR3 activation, RCS chondrocytes lose their cartilaginous extracellular matrix as evidenced by both Alcian blue staining and metabolic labelling by [35S]sulphate (Fig. 1B, C). This matrix loss is mediated by FGF2-mediated inhibition of matrix production and matrix metalloproteinase (MMP2, 3, 9, 10 and 13)-induced degradation [15]. Interestingly, FGFR3 activation induces also a shape change in RCS chondrocytes, manifested as cellular enlargement, flattening and elongation as compared to the round or cubical morphology of untreated cells (Fig. 1D). These changes are likely due to the cytoskeletal remodelling and increased substrate adhesion, as demonstrated by phalloidin staining of polymerized actin fibers and visualization of focal adhesions by vinculin immunostaining (Fig. 1E).
FIGURE 1.
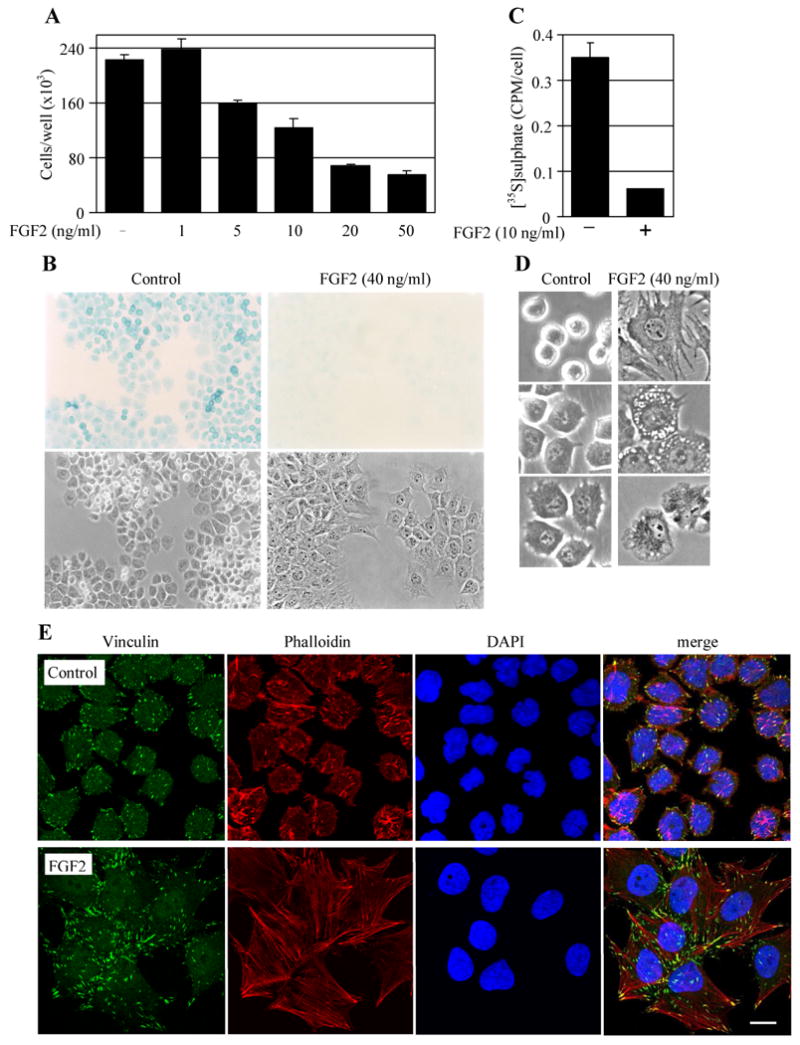
FGFR3 effects on proliferation, extracellular matrix production and cellular shape in RCS chondrocytes. (A) Cells were treated with FGF2 for 72 hours and counted. Note the potent FGF2-mediated inhibition of RCS growth. Data represent an average from four wells with the indicated standard deviation. (B) RCS chondrocytes were treated with FGF2 for 72 hours and stained with Alcian blue to visualize the cartilaginous extracellular matrix (upper panel). The corresponding bright field with contrast photograph is also shown (lower panel, 100×). Note the significant loss of extracellular matrix in FGF2-treated cells. (C) RCS chondrocytes were treated with FGF2 for 72 hours in the presence of [35S]sulfate and the amount of incorporated radioactivity was determined by liquid scintillation. Note the potent downregulation of [35S]sulfate incorporation by FGF2. Data represent an average from four wells with the indicated standard deviation. (D) RCS chondrocytes were treated with FGF2 for 72 hours and photographed. The left panel shows the round to cubical shape of control cells. Note the cellular shape alteration that accompanies the FGF2-mediated growth arrest (right panel, 200×). (E) RCS chondrocytes were treated with FGF2 (10 ng/ml) for 72 hours, co-stained for polymerized actin (Alexa Fluor 594-phalloidin) and focal adhesions (vincullin-FITC) and photographed. Note the significant cytoskeleton remodelling accompanied by increased substrate-adhesion that is induced by FGFR3 signaling. Scale bar: 20 μm.
FGFR3 activation induces premature senescence in RCS chondrocytes
To test whether FGFR3 signaling causes cellular senescence in RCS chondrocytes, we first evaluated the FGF2-mediated effect on activation of SA-β-GALACTOSIDASE, one of the most universally accepted markers of senescence [21]. Figure 2 shows that prolonged FGF2 treatment lead to activation of SA-β-GALACTOSIDASE in nearly 100% of RCS cells (Fig. 2A, B). We next focused on other markers known to be upregulated (α-GLUCOSIDASE, APA1, CAVEOLIN 1/2, FKBP12, LAMIN A, NORE 1, RAL-A, SM22α and TIMP 1) or downregulated (ID 1/2) in senescent cells [22-27]. As determined by real-time RT-PCR, FGF2 upregulated α-Glucosidase, Caveolin1, Lamin A, SM22α, Timp1 and Nore1 transcripts while the levels of Id2 decreased (Fig. 2C). Similar FGF2-mediated up- or down-regulation was found for Caveolin2 and Id1 transcripts, respectively (not shown). No FGF2-mediated changes were found in the quantities of Apa1, Ral-A and Fkbp12 transcripts (not shown). At the protein level, FGF2 caused upregulation of α-GLUCOSIDASE, CAVEOLIN 1, LAMIN A/C, SM22α and TIMP 1 and downregulation of ID 2 (Fig. 2D). We were unable to detect NORE 1 with two different commercially available antibodies (not shown).
FIGURE 2.

FGFR3-mediated senescence in RCS chondrocytes. (A) Cells were treated with FGF2 for 72 hours and the activity of SA-β-GALACTOSIDASE was determined as described in the Materials and Methods. Note a potent, FGF2-mediated induction of SA-β-GALACTOSIDASE activity that appears as blue cytoplasmic staining (upper panels). The corresponding bright field with contrast photograph is also shown (lower panels, 100×). (B) The experiment presented in (A) was quantified by counting the SA-β-GALACTOSIDASE positive cells under the light microscope. The data represent an average from 10 counts (50 cells each) with the indicated standard deviation. The results are representative for three experiments. (C) Cells were treated with FGF2 (20 ng/ml) for the indicated times and expression of a panel of senescence markers was determined by real-time RT-PCR. Quantities of the transcript are relative to untreated cells. The data represent an average from technical duplicates with the indicated range. Results are representative for five experiments. (D) RCS chondrocytes were treated with FGF2 (20 ng/ml) for the indicated times and the quantities of given proteins were determined by WB. The α-GLUCOSIDASE signal was quantified by densitometry and graphed. ACTIN serves as a loading control. Data are representative for four experiments.
We have previously shown that FGFR3 signaling in RCS chondrocytes results in a potent upregulation of FIBRONECTIN and MMP3 [15], which are also considered markers of cellular senescence [26]. Since both FIBRONECTIN and MMP3 induction was dependent on the ERK MAP kinase pathway [15], we asked whether the FGFR3-mediated changes in the expression of senescence markers observed here (Fig. 2C, D) were also induced via ERK. RCS chondrocytes were treated with FGF2 in the presence of the MEK inhibitor U0126 and the quantities of α-Glucosidase, Caveolin1, Lamin A, Id2, SM22α, Timp1 and Nore1 transcripts were determined by real-time RT-PCR. FGF2-mediated modulation of expression of all tested senescence markers was at least partially rescued by U0126 (Fig. 3A). Similar results were obtained at the protein level, where U0126 antagonized FGF2-mediated modulation of all but one (α-GLUCOSIDASE) of the tested proteins (Fig. 3B).
FIGURE 3.

Reversal of FGF2-mediated senescence by chemical inhibition of the ERK pathway. (A) Cells were treated with FGF2 for 24 hours in the presence of the MEK inhibitor U0126, and analyzed for expression of a panel of senescence markers by real-time RT-PCR. Quantities of the transcript are relative to untreated cells. Note that U0126 partially antagonizes FGF2-mediated induction of all tested markers. The data represent an average from technical duplicates with the indicated range. Results are representative for three experiments. A similar experiment was performed in (B), with the quantities of the given proteins determined by WB. Note the FGF2-mediated induction or downregulation of senescence markers, which was rescued by U0126 in all but one case (α-GLUCOSIDASE, evidenced by densitometry). ACTIN serves as a loading control. Results are representative for four experiments. SM22α was not analyzed by WB since its protein induction by FGF2 required at least 48 hours of treatment (Fig. 2D).
FGFR3-mediated premature senescence in RCS chondrocytes is a reversible phenotype
As activation of p53 tumor suppressor pathway is typically one of the major events underlying premature senescence [28], we next asked whether FGFR3 utilizes p53 to cause senescence in RCS chondrocytes. Treatment with known p53 activators such as etoposide, camptothecin or UV irradiation lead to activatory p53 phosphorylation at Serine 15 [29-31], in contrast to treatment with FGF2 that produced no such phosphorylation (Fig. S1A). Similarly, FGF2 did not cause transcriptional activation of p53 in RCS chondrocytes, as determined by a p53-dependent luciferase reporter assay (Fig. S1B). Altogether, these data suggest that although the p53 pathway is functional in RCS chondrocytes, FGFR3 signaling induces senescence independent of p53. As p53-mediated senescence is often irreversible [32], the lack of p53 activation by FGF2 opened the possibility that the senescence phenotype observed here could be alleviated upon removal of the FGF2 stimulus. Indeed, when FGF2 was washed-off during the course of prolonged treatment, the cells responded with reversal of both CAVEOLIN 1 and LAMIN A/C induction, and resumption of proliferation (Fig. 4A, C). Similar results were obtained in cells treated with the FGFR inhibitor SU5402 (Fig. 4B, cell growth data not shown).
FIGURE 4.
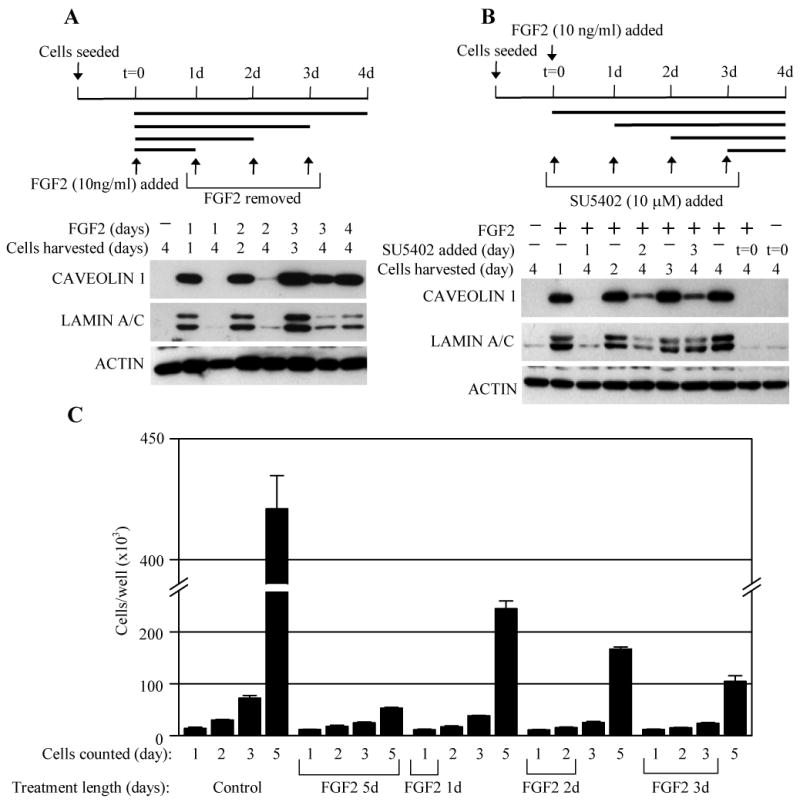
FGFR3-mediated senescence is a reversible phenotype. (A) RCS chondrocytes were seeded and treated with FGF2 for four days as indicated. In some samples, the length of FGF2 treatment was restricted by washing away after one, two and three days of cultivation. Cell lysates were analyzed by WB for CAVEOLIN 1 and LAMIN A/E expression at the indicated times. ACTIN serves as a loading control. Note the FGF2-mediated induction of CAVEOLIN 1 and LAMIN A/E which was reversed when FGF2/FGFR3 stimulus diminished as a result of FGF2 removal. (B) The experiment was carried-out similarly to (A) except that the all cell cultures were treated with FGF2 for the whole duration of the experiment, and FGFR3 signaling was inhibited by addition of the FGFR inhibitor SU5402. Note the reversal of the FGF2/FGFR3-mediated effect on senescence markers by SU5402. The results are representative for three experiments. (C) RCS chondrocytes were grown for up to five days, treated with FGF2 (10 ng/ml) for a variable length of time and counted at the indicated times. Note that despite the complete growth arrest caused by the initial FGF2 treatment, the cells resumed normal growth upon FGF2 removal. The results are representative for three experiments.
FGFR3 overexpression causes apoptosis in RCS chondrocytes
We next evaluated whether overexpression of activating FGFR3 mutants causes senescence similar to FGF2-mediated activation of endogenous FGFR3. For this purpose, we transfected RCS chondrocytes with FGFR3 mutants associated with hypochondroplasia (N540K), ACH (G380R), TDI (R248C, Y373C, K650E) and TDII (K650M). Figure 5A shows that all of the mutants cause ERK activation, with the amount roughly correlating with both the level of FGFR3 activation and severity of the dwarfing phenotype caused by a given mutation [1, 17]. As documented previously, the expression of the FGFR3 mutants causes growth-arrest in RCS chondrocytes, with the intensity of the phenotype correlating with the level of FGFR3 activation [17]. We however found no SA-β-GALACTOSIDASE activation induced by expression of any of the FGFR3 mutants used (not shown). Furthermore, no other senescence markers were induced by the expression of the FGFR3 mutants with exception of CAVEOLIN 1, which was upregulated to the levels similar to those induced by FGF2-mediated activation of endogenous FGFR3 (Figs 2D, 5A). Based on these findings it is unlikely that FGFR3 mutants inhibit RCS proliferation via premature senescence. Despite the relatively low efficiency of the standard transfection protocol (∼40%), we noticed that FGFR3 transfection lead to cell detachment and shrinkage, as opposed to enlargement and increased adhesion in FGF2-treated cells (Figs 1D, S2), suggesting that FGFR3 overexpression might be toxic for RCS chondrocytes. In order to look closely at the morphology of the FGFR3-transfected cells, we co-transfected RCS chondrocytes with FGFR3 together with a GFP-encoding plasmid and isolated GFP-positive cells by flow-cytometry based cell sorting. Figure 5B shows poor growth and adhesion of cells expressing activating FGFR3 mutants with many putative dead or dying cells, thus suggesting that ectopic expression of active FGFR3 causes apoptosis in RCS chondrocytes. This was confirmed by flow-cytometric analysis of cell viability and annexin binding to the cellular surface, which serves as a marker for apoptosis [33] (Fig. 5C).
FIGURE 5.

Apoptosis induced by FGFR3 overexpression. (A) RCS chondrocytes were transfected with wild-type FGFR3 as well as its activating mutants associated with hypochondroplasia (N540K), ACH (G380R), and TD (R248C, Y373C, K650M, K650E), and analyzed for the indicated molecules by WB 24 hours later. Note the typical migration pattern of FGFR3 in both K650 mutants that form only an immature, intracellular variant of FGFR3 (lower band). Also note the different levels of ERK activation (P-ERK blot) that correspond to the severity of the disease. KD is an inactive, kinase-dead FGFR3 mutant (K508M), whereas cells transfected with a GFP or empty plasmid serve as transfection controls. ACTIN serves as loading control. (B) Cells were co-transfected with indicated FGFR3 mutants together with a GFP plasmid. 48 hours later, GFP-positive cells were isolated by cell-sorting as described in Material and Methods, plated and grown for additional 48 or 72 hours, and photographed (left panel, bright field with contrast, 100×), or counted (right graph). Note the poor attachment of cells transfected with FGFR3 carrying highly activating K650M and K650E mutants, with many small, dying (white arrows) or dead (black arrows) cells. Also note the poor growth in GFP-positive cells expressing activating FGFR3 mutants when compared to GFP-positive cells expressing KD-FGFR3 or an empty plasmid. Also note the growth of GFP-negative cells (right graph, open bars), which was normal in all the transfections. Data represent an average from three wells with the indicated standard deviation. (C) Cells were transfected as indicated, grown for 48 hours, stained with propidium iodide (PI) and annexin-FITC, and analyzed by flow-cytometry to visualize the apoptotic (annexin positive) and necrotic (PI/annexin positive) cells. The data represent an average from three independent experiments with the indicated standard deviation. Cells transfected with an empty plasmid serve as a transfection control. Control – untransfected cells, FGF2 – untransfected cells treated with FGF2 (25 ng/ml) for 24 hours.
Discussion
FGFR3 promotes cell transformation when activated ectopically, as demonstrated in multiple myeloma where FGFR3, activated by mutation or overexpression, acts as an oncogene [6, 7]. In the growing bones, however, the cancer-associated activating mutations in FGFR3 suppress chondrocyte growth and differentiation, resulting in marked shortening and disorganization of epiphyseal growth plates, which leads to severe dwarfism [34]. Probably the most striking effect of aberrant FGFR3 signaling in cartilage is the profound inhibition of chondrocyte proliferation [35-37], although the nature of the cellular processes underlying this phenotype is not clear. Increased apoptosis was found in the cartilage of mice overexpressing FGF2 [38], however other studies failed to confirm this effect [39, 40] (P. Krejci and W. R. Wilcox, unpublished). Ectopic expression of activating FGFR3 mutants induces apoptosis in ATDC5 chondrogenic cells and RCS chondrocytes [41] (Figs 5, S2), however this effect may represent an artifact of FGFR3 overexpression rather then a true physiological response.
In addition to apoptosis, FGFR3 signaling is known to increase chondrocyte proliferation. The supporting evidence comes from the analysis of the cartilage of mice harbouring a K644M mutation in FGFR3 [42]. Such animals display expected inhibition of chondrocyte proliferation similar to human TD cases carrying the homologous K650M mutation [1]. However, this inhibition is preceded by potent but transient increase of chondrocyte proliferation, observed between embryonic day 15.5 and 18.5 [42].
In this article, we used RCS chondrocytes to test the possibility that FGFR3 signaling inhibits chondrocyte growth via induction of premature senescence. RCS cells represent the best characterized in vitro cell model for FGFR3-related skeletal dysplasias to date, as demonstrated by numerous studies on the effect of FGFR3 signaling on chondrocyte proliferation, differentiation, extracellular matrix homeostasis and others [15, 19, 20, 43, 44]. RCS chondrocytes respond to FGFR3 activation with potent G1 growth arrest due to upregulation of cyclin D1, which leads to stabilization of p21WAF1 and p27Kip1 proteins and, ultimately, to inhibition of the cdk2/4-cyclin activity necessary for cell cycle progression [13]. Data from various experimental systems implicate signaling via the RAS-ERK MAP kinase pathway as important for FGFR3-mediated inhibition of chondrocyte growth both in vitro and in vivo [9-12]. Constitutive activation of ERK MAP kinase is necessary for FGFR3-mediated RCS chondrocyte growth arrest, as demonstrated by targeting ERK via chemical or C-natriuretic peptide-mediated inhibition, over-expression of a dominant-negative RAS mutant or RNAi-mediated knock-down [13, 15, 16, 43]. The analyses of the senescence markers performed here clearly demonstrate that FGFR3 inhibits RCS chondrocyte proliferation via induction of premature senescence (Fig. 2). Moreover, this phenotype depends on the ERK pathway and is fully reversible upon removal of the FGF ligand or chemical inhibition of FGFR3 (Figs 3, 4). Thus the in vitro evidence, presented in this article, opens the possibility that premature senescence might also be involved in the pathological effects of FGFR3 in skeletal dysplasias. Although the effects of FGFR3 signaling in RCS chondrocytes appear identical to many effects of pathological FGFR3 signaling in cartilage in vivo [13, 15, 19, 20, 43-47], this cell line by no means fully models the complexity of growing cartilage where chondrocyte proliferation and differentiation take place simultaneously, being influenced by many signaling pathways. Thus the lack of in vivo evidence represents one limitation to the conclusions of this study. Experiments are now ongoing to evaluate the effect of FGFR3 signaling on induction of chondrocyte senescence in vivo.
Taken together, FGFR3 appears to induce diverse cellular phenotypes in chondrocytes such as apoptosis, senescence or unscheduled proliferation. It is unclear what mechanisms underlie such differential responses to the same stimulus. It is important to note that many aspects of chondrocyte function are likely to change with advancing gestational and postnatal age, most likely altering the way how cells respond to FGFR3. However, as demonstrated here, FGFR3 activation may trigger senescence or apoptosis in the same cell type (Figs 2-5, S2), likely depending on the intensity and duration of the signal as well as on other yet unknown factors, which are presently under investigation. Mutations associated with FGFR3-skeletal dysplasias activate the FGFR3 oncogene in cartilage, which signals via constitutive activation of another oncogenic pathway, i.e. RAS-ERK pathway [9-13]. Moreover, the chondrocyte responses to the FGFR3 activation, i.e. unscheduled proliferation, apoptosis or premature senescence, all resemble typical cellular responses to oncogene activation. Oncogene activation may result in uncontrolled cell proliferation but more frequently leads to induction of apoptosis or premature senescence, both acting as natural barriers to tumor development [14]. We speculate that the growth-inhibitory outcome of FGFR3 signaling in cartilage might be explained on the basis of a normal cellular response to an activated oncogene.
Supplementary Material
Acknowledgments
We are grateful to M. Rock for assistance with phalloidin staining, M. Schibler for assistance with confocal microscopy, and P. Mekikian and K. Pejchalova for technical assistance. This work was supported by National Institutes of Health (5P01-HD22657), Yang Sheng Tang USA company, the Ministry of Education, Youth and Sports of the Czech Republic (MSM0021622430), Grant Agency of the Czech Republic (301/09/0587), Cedars-Sinai Medical Genetics Institute fellowship (PK), and a Winnick Family Scholars Award (WRW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Passos-Bueno MR, Wilcox WR, Jabs EW, Sertie AL, Alonso LG, Kitoh H. Clinical spectrum of fibroblast growth factor receptor mutations. Hum Mutat. 1999;14:115–25. doi: 10.1002/(SICI)1098-1004(1999)14:2<115::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 2.Ornoy A, Adomian GE, Eteson DJ, Burgeson RE, Rimoin DL. The role of mesenchyme-like tissue in the pathogenesis of thanatophoric dysplasia. Am J Med Genet. 1985;21:613–30. doi: 10.1002/ajmg.1320210402. [DOI] [PubMed] [Google Scholar]
- 3.Goriely A, Hansen RM, Taylor IB, Olesen IA, Jacobsen GK, McGowan SJ, Pfeifer SP, McVean GA, Meyts ER, Wilkie AO. Activating mutations in FGFR3 and HRAS reveal a shared genetic origin for congenital disorders and testicular tumors. Nat Genet. 2009;41:1247–52. doi: 10.1038/ng.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hafner C, Stoehr R, van Oers JM, Zwarthoff EC, Hofstaedter F, Landthaler M, Hartmann A, Vogt T. FGFR3 and PIK3CA mutations are involved in the molecular pathogenesis of solar lentigo. Br J Dermatol. 2009;160:546–51. doi: 10.1111/j.1365-2133.2008.08963.x. [DOI] [PubMed] [Google Scholar]
- 5.Hernández S, de Muga S, Agell L, Juanpere N, Esgueva R, Lorente JA, Mojal S, Serrano S, Lloreta J. FGFR3 mutations in prostate cancer: association with low-grade tumors. Mod Pathol. 2009;22:848–56. doi: 10.1038/modpathol.2009.46. [DOI] [PubMed] [Google Scholar]
- 6.Plowright EE, Li Z, Bergsagel PL, Chesi M, Barber DL, Branch DR, Hawley RG, Stewart AK. Ectopic expression of fibroblast growth factor receptor 3 promotes myeloma cell proliferation and prevents apoptosis. Blood. 2000;95:992–8. [PubMed] [Google Scholar]
- 7.Li Z, Zhu YX, Plowright EE, Bergsagel PL, Chesi M, Patterson B, Hawley TS, Hawley RG, Steward AK. The myeloma-associated oncogene fibroblast growth factor receptor 3 is transforming in hematopoietic cells. Blood. 2001;97:2413–9. doi: 10.1182/blood.v97.8.2413. [DOI] [PubMed] [Google Scholar]
- 8.Chesi M, Brents LA, Ely SA, Bais C, Robbiani DF, Mesri EA, Kuehl WM, Bergsagel PL. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Blood. 2001;97:729–36. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 9.Yasoda A, Komatsu Y, Chusho H, Miyazawa T, Ozasa A, Miura M, Kurihara T, Rogi T, Tanaka S, Suda M, Tamura N, Ogawa Y, Nakao K. Overexpression of CNP in chondrocytes rescues achondroplasia through a MAPK-dependent pathway. Nat Med. 2004;10:80–6. doi: 10.1038/nm971. [DOI] [PubMed] [Google Scholar]
- 10.Nowroozi N, Raffioni S, Wang T, Apostol BL, Bradshaw RA, Michels Thompson L. Sustained Erk1/2 but not STAT1 or 3 activation is required for thanatophoric dysplasia phenotypes in PC12 cells. Hum Mol Genet. 2005;14:1529–38. doi: 10.1093/hmg/ddi161. [DOI] [PubMed] [Google Scholar]
- 11.Murakami S, Balmes G, McKinney S, Zhang Z, Givol D, de Crombrugghe B. Constitutive activation of MEK1 in chondrocytes causes Stat1-independent achondroplasia-like dwarfism and rescues the Fgfr3-deficient mouse phenotype. Genes Dev. 2004;18:290–305. doi: 10.1101/gad.1179104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matsushita T, Wilcox WR, Chan YY, Kawanami A, Bukulmez H, Balmes G, Krejci P, Mekikian PB, Otani K, Yamaura I, Warman ML, Givol D, Murakami S. FGFR3 promotes synchondrosis closure and fusion of ossification centers through the MAPK pathway. Hum Mol Genet. 2008;18:227–40. doi: 10.1093/hmg/ddn339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krejci P, Bryja V, Pachernik J, Hampl A, Pogue R, Mekikian P, Wilcox WR. FGF2 inhibits proliferation and alters the cartilage-like phenotype of RCS cells. Exp Cell Res. 2004;297:152–64. doi: 10.1016/j.yexcr.2004.03.011. [DOI] [PubMed] [Google Scholar]
- 14.Yaswen P, Campisi J. Oncogene-induced senescence pathways weave an intricate tapestry. Cell. 2007;128:233–4. doi: 10.1016/j.cell.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 15.Krejci P, Masri B, Fontaine V, Mekikian PB, Weis M, Prats H, Wilcox WR. Interaction of fibroblast growth factor and C-natriuretic peptide signaling in regulation of chondrocyte proliferation and cartilage matrix homeostasis. J Cell Sci. 2005;118:5089–100. doi: 10.1242/jcs.02618. [DOI] [PubMed] [Google Scholar]
- 16.Krejci P, Salazar L, Goodridge HS, Kashiwada TA, Schibler MJ, Jelinkova P, Thompson LM, Wilcox WR. STAT1 and STAT3 do not participate in FGF-mediated growth arrest in chondrocytes. J Cell Sci. 2008;121:272–81. doi: 10.1242/jcs.017160. [DOI] [PubMed] [Google Scholar]
- 17.Krejci P, Salazar L, Kashiwada TA, Chlebova K, Salasova A, Thompson LM, Bryja V, Kozubik A, Wilcox WR. Analysis of STAT1 activation by six FGFR3 mutants associated with skeletal dysplasia undermines dominant role of STAT1 in FGFR3 signaling in cartilage. PLoS One. 2008;3:e3961. doi: 10.1371/journal.pone.0003961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilcox WR, Tavormina PL, Krakow D, Kitoh H, Lachman RS, Wasmuth JJ, Thompson LM, Rimoin DL. Molecular, radiologic, and histopathologic correlations in thanatophoric dysplasia. Am J Med Genet. 1998;78:274–81. doi: 10.1002/(sici)1096-8628(19980707)78:3<274::aid-ajmg14>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 19.Dailey L, Laplantine E, Priore R, Basilico C. A network of transcriptional and signaling events is activated by FGF to induce chondrocyte growth arrest and differentiation. J Cell Biol. 2003;161:1053–66. doi: 10.1083/jcb.200302075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aikawa T, Sagre GV, Lee K. Fibroblast growth factor inhibits chondrocytic growth through induction of p21 and subsequent inactivation of cyclin E-Cdk2. J Biol Chem. 2001;276:29347–52. doi: 10.1074/jbc.M101859200. [DOI] [PubMed] [Google Scholar]
- 21.Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007;371:21–31. doi: 10.1007/978-1-59745-361-5_3. [DOI] [PubMed] [Google Scholar]
- 22.Benvenuti S, Cramer R, Bruce J, Waterfield MD, Jat PS. Identification of novel candidates for replicative senescence by functional proteomics. Oncogene. 2002;21:4403–13. doi: 10.1038/sj.onc.1205525. [DOI] [PubMed] [Google Scholar]
- 23.Benanti JA, Williams DK, Robinson KL, Ozer HL, Galloway DA. Induction of extracellular matrix-remodeling genes by the senescence-assosiated protein Apa-1. Mol Cell Biol. 2002;22:7385–97. doi: 10.1128/MCB.22.21.7385-7397.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park WY, Park JS, Cho KA, Kim DI, Ko YG, Seo JS, Park SC. Up-regulation of caveolin attenuates epidermal growth factor signaling in senescent cells. J Biol Chem. 2000;275:20847–52. doi: 10.1074/jbc.M908162199. [DOI] [PubMed] [Google Scholar]
- 25.Barradas M, Gonos ES, Zebedee Z, Kolettas E, Petropoulou C, Delgado MD, Leon J, Hara E, Serrano M. Identification of a candidate tumor-suppressor gene specifically activated during ras-induced senescence. Exp Cell Res. 2002;273:127–37. doi: 10.1006/excr.2001.5434. [DOI] [PubMed] [Google Scholar]
- 26.Gonos ES, Derventzi A, Kveiborg M, Agiostratidou G, Kassem M, Clark BFC, Jat PS, Rattan SIS. Cloning and identification of genes that associate with mammalian replicative senescence. Exp Cell Res. 1998;240:66–74. doi: 10.1006/excr.1998.3948. [DOI] [PubMed] [Google Scholar]
- 27.Dimri GP, Testori A, Acosta M, Campisi J. Replicative senescence, aging and growth-regulatory transcription factors. Biol Signals. 1996;5:154–62. doi: 10.1159/000109185. [DOI] [PubMed] [Google Scholar]
- 28.Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J Pathol. 2009;19:3–15. doi: 10.1002/path.2584. [DOI] [PubMed] [Google Scholar]
- 29.Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol. 2000;20:3224–33. doi: 10.1128/mcb.20.9.3224-3233.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ljungman M, O'Hagan HM, Paulsen MT. Induction of ser15 and lys382 modifications of p53 by blockage of transcription elongation. Oncogene. 2001;20:5964–71. doi: 10.1038/sj.onc.1204734. [DOI] [PubMed] [Google Scholar]
- 31.Karpinich NO, Tafani M, Rothman RJ, Russo MA, Farber JL. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J Biol Chem. 2002;277:16547–52. doi: 10.1074/jbc.M110629200. [DOI] [PubMed] [Google Scholar]
- 32.Nakagawa H, Opitz OG. Inducing cellular senescence using defined genetic elements. Methods Mol Biol. 2007;371:167–78. doi: 10.1007/978-1-59745-361-5_13. [DOI] [PubMed] [Google Scholar]
- 33.Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–20. [PubMed] [Google Scholar]
- 34.Horton WA, Hall JG, Hecht JT. Achondroplasia. Lancet. 2007;14:162–72. doi: 10.1016/S0140-6736(07)61090-3. [DOI] [PubMed] [Google Scholar]
- 35.Segev O, Chumakov I, Nevo Z, Givol D, Madar-Shapiro L, Sheinin Y, Weinreb M, Yayon A. Restrained chondrocyte proliferation and maturation with abnormal growth plate vascularization and ossification in human FGFR-3(G380R) transgenic mice. Hum Mol Genet. 2000;9:249–58. doi: 10.1093/hmg/9.2.249. [DOI] [PubMed] [Google Scholar]
- 36.Chen L, Li C, Qiao W, Xu X, Deng C. A Ser(365)-->Cys mutation of fibroblast growth factor receptor 3 in mouse downregulates Ihh/PTHrP signals and causes severe achondroplasia. Hum Mol Genet. 2001;10:457–65. doi: 10.1093/hmg/10.5.457. [DOI] [PubMed] [Google Scholar]
- 37.Li C, Chen L, Iwata T, Kitagawa M, Fu XY, Deng CX. A lys644glu substitution in fibroblast growth factor receptor 3 (FGFR3) causes dwarfism in mice by activation of STATs and ink4 cell cycle inhibitors. Hum Mol Genet. 1999;8:35–44. doi: 10.1093/hmg/8.1.35. [DOI] [PubMed] [Google Scholar]
- 38.Sahni M, Raz R, Coffin JD, Levy D, Basilico C. STAT1 mediates the increased apoptosis and reduced chondrocyte proliferation in mice overexpressing FGF2. Development. 2001;128:2119–29. doi: 10.1242/dev.128.11.2119. [DOI] [PubMed] [Google Scholar]
- 39.Naski MC, Colvin JS, Coffin JD, Ornitz DM. Repression of hedgehog signaling and BMP4 expression in growth plate cartilage by fibroblast growth factor receptor 3. Development. 1998;125:4977–88. doi: 10.1242/dev.125.24.4977. [DOI] [PubMed] [Google Scholar]
- 40.Legeai-Mallet L, Benoist-Lasselin C, Delezoide AL, Munnich A, Bonaventure J. Fibroblast growth factor receptor 3 mutations promote apoptosis but do not alter chondrocyte proliferation in thanatophoric dysplasia. J Biol Chem. 1998;273:13007–14. doi: 10.1074/jbc.273.21.13007. [DOI] [PubMed] [Google Scholar]
- 41.Harada D, Yamanaka Y, Ueda K, Nishimura R, Morishima T, Seino Y, Tanaka H. Sustained phosphorylation of mutated FGFR3 is a crucial feature of genetic dwarfism and induces apoptosis in the ATDC5 chondrogenic cell line via PLCgamma-activated STAT1. Bone. 2007;41:273–81. doi: 10.1016/j.bone.2006.11.030. [DOI] [PubMed] [Google Scholar]
- 42.Iwata T, Li CL, Deng CX, Francomano CA. Highly activated Fgfr3 with the K644M mutation causes prolonged survival in severe dwarf mice. Hum Mol Genet. 2001;10:1255–64. doi: 10.1093/hmg/10.12.1255. [DOI] [PubMed] [Google Scholar]
- 43.Raucci A, Laplantine E, Mansukhani A, Basilico C. Activation of the Erk1/2 and p38 mitogen-activated protein kinase pathways mediates fibroblast growth factor-induced growth arrest of chondrocytes. J Biol Chem. 2004;279:1747–56. doi: 10.1074/jbc.M310384200. [DOI] [PubMed] [Google Scholar]
- 44.Ben-Zvi T, Yayon A, Gertler A, Monsonego-Ornan E. Suppressors of cytokine signaling (SOCS) 1 and SOCS3 interact with and modulate fibroblast growth factor receptor signaling. J Cell Sci. 2006;119:380–7. doi: 10.1242/jcs.02740. [DOI] [PubMed] [Google Scholar]
- 45.Zhang R, Murakami S, Coustry F, Wang Y, de Crombrugghe B. Constitutive activation of MKK6 in chondrocytes of transgenic mice inhibits proliferation and delays endochondral bone formation. Proc Natl Acad Sci USA. 2006;103:365–70. doi: 10.1073/pnas.0507979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laplantine E, Rossi F, Sahni M, Basilico C, Cobrinik D. FGF signaling targets the pRb-related p107 and p130 proteins to induce chondrocyte growth arrest. J Cell Biol. 2002;158:741–50. doi: 10.1083/jcb.200205025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rozenblatt-Rosen O, Mosonego-Ornan E, Sadot E, Madar-Shapiro L, Sheinin Y, Ginsberg D, Yayon A. Induction of chondrocyte growth arrest by FGF: transcriptional and cytoskeletal alterations. J Cell Sci. 2002;115:553–62. doi: 10.1242/jcs.115.3.553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.