

Abstract
The elucidation of the genes leading to selected immune defects has accelerated our understanding of the molecular basis of tolerance in autoimmunity disorders. Mutations in genes of the immune system are known to lead to a catalogue of functional deficits, including loss of activation-induced Fas-mediated apoptosis, an inability to remove self-reactive T and/or B cells and insufficient numbers or functions of regulatory T cells. In most cases, microbial antigen stimulation occurs simultaneously, leading to further inflammatory responses. In each case, probing the molecular pathways involved in these primary immune defects has led to a better understanding of autoimmune diseases in general. While subjects with X-linked agammaglobulinaemia are almost devoid of autoimmune diseases, B cells which are present, but dysfunctional in other defects, lead to a significant incidence of autoimmune disease. Autoimmunity is also particularly common in the antibody deficiency states. Although organ-based autoimmunity also occurs, for unclear reasons the main conditions are immune thrombocytopenia purpura and autoimmune haemolytic anaemia. The common variable immune deficiency subjects most afflicted by these cytopenias are those with specific peripheral blood memory B cell phenotypes. B cells of these subjects have a retained autoimmune potential, lack of somatic hypermutation, profound loss of proliferative potential, accelerated apoptosis and loss of normal Toll-like receptor signalling. Treatment with high-dose immunoglobulin and/or steroids can be helpful, while rituximab provides benefits in the treatment of refractory cytopenias with apparently little risk, even with repeated use, due to ongoing immune globulin therapy.
Keywords: autoimmune diseases, autoimmune haemolytic anaemia, common variable immune deficiency, immune globulin, immune thrombocytopenia purpura, X-linked agammaglobulinaemia
Introduction
For many years the association between the presence of autoimmunity in subjects with primary immune deficiency has been examined as a puzzling and yet potentially revealing biological phenomenon. While these immune defects are usually understood as leading to infections, the truth is that most of these inborn errors also lead to greater or lesser degrees of immune dysregulation. Autoimmunity is certainly one of the most important of these manifestations. The autoimmune complications in primary immune deficiency are common in defects of both the adaptive and innate immune system, demonstrating that all these immune components must be required for the appropriate development of tolerance in humans. It may not be surprising that so many unique pathways to exclude autoimmunity are the norm in humans; what is not clear is the role that each component plays. However, careful dissection of these molecular pathways has proved fruitful in immune deficiency, and has led to enhanced understanding of autoimmunity in general. All immune defects have characteristic general clinical manifestations, based on the specific immune component that is defective. Similarly, primary immune deficiencies that lead to autoimmunity also have a characteristic autoimmune phenotype, often overlapping with each other, but only in few cases are these well understood. Some of the more common autoimmune manifestations of primary immune deficiency are shown in Table 1.
Table 1.
Overview of autoimmune manifestations of primary immune deficiency
| Disease | Genes | Autoimmunity |
|---|---|---|
| Severe combined immunodeficiency (SCID) | Many | Alopecia, dermatitis, thrombocytopenia |
| X-linked agammaglobulinaemia (XLA) | BTK | Juvenile rheumatoid arthritis and rheumatoid arthritis/dermatomyosisits |
| Autoimmune polyendocrinopathy | AIRE | Endocrine |
| Immune dysregulation, polyendocrinopathy and enteropathy, X-linked (IPEX) | FoxP3 | Juvenile diabetes, cytopenias, dermatitis, enteropathy |
| Wiskott–Aldrich | WASP | Haemolytic anaemia, Henoch–Schonlein inflammatory bowel disease, vasculitis |
| Chronic granulomatous disease (CGD) | NADPH oxidase | Inflammatory bowel disease, mothers with systemic lupus erythmatosus |
| Hyper-IgM | CD40L and others | Autoimmune hepatitis, rheumatoid arthritis, inflammatory bowel disease, uveitis, diabetes |
| Common variable immune deficiency (CVID) | TACI/ICOS/CD19 CD81, CD20, BAFFr | Immune thrombocytopenia purpura, haemolytic anemia, alopecia pernicious anaemia, systemic lupus erythmatosus, inflammatory bowel disease |
NADPH: nicotinamide adenine dinucleotide phosphate-oxidase; IPEX: X-linked inheritance; FoxP3: forkhead box P2; WASP: Wiskott–Aldrich syndrome protein; BAFF: B cell activating factor; TACI: transmembrane activator and calcium-modulating ligand interactor; AIRE: autoimmune regulator gene; BTK: Bruton agammaglobulinaemia tyrosine kinase.
Why does autoimmunity occur in primary immune defects?
Turning first to the control of self-reactive T cells, the great majority of these cells are deleted in the thymus, leading to central tolerance. These events depend upon the assembly of an effective T cell receptor that can display self-antigens, as these cells are best targeted for elimination. How a vast number of self-antigens can actually be arrayed in the thymus is unclear, but the crucial role of the autoimmune regulator gene (AIRE) in their expression is illustrated by the autoimmune polyendocrinopathy–candidasis–ectodermal dystrophy (APECED) syndrome, an autosomal recessive disease due to mutations in AIRE. The clinical condition includes hypoparathyroidism, mucocutaneous candidiasis, adrenal insufficiency, gonad failure, malabsorption and other tissue damages due to autoimmune attack. Loss of the AIRE gene, a thymic transcription factor that up-regulates the expression of tissue-specific genes in thymic epithelial cells, results in loss of tissue tolerance [1]. The expression of AIRE in the thymus is related closely to thymocyte differentiation; as this requires cooperation between early thymocytes and medullary thymic epithelial cells, loss of AIRE expression leading to retention of autoreactive T cells in the thymus might be expected in defects of T cell development such as Omen syndrome [2]. Here, the leaky severe combined immunodeficiency (SCID) phenotype and relative loss of AIRE expression permits the survival of a few T cells with autoimmune potential.
However, some self-reactive T cells escape thymic selection and must be removed in the periphery. The mechanisms for removal of these cells are different than for central tolerance, and probably involve a number of different pathways, including the development of regulatory T cells (Tregs), among others. Here, the study of mutations in the X-chromosome gene for the forkheard box P3 (FoxP3) transcription factor has led to a clearer understanding of the essential role of Tregs in tolerance. FoxP3 is essential for the development of CD25+ Tregs. Its loss leads to the clinical condition called immune dysregulation, polyendocrinopathy and enteropathy, X-linked (IPEX) manifested by early-onset type 1 diabetes mellitus, severe enteropathy, eczema, anaemia, thrombocytopenia, hypothyroidism and other organ-specific tissue damage [3–5]. The lack of Tregs in this syndrome explains many facets of the immune-mediated tissue destruction which occurs.
Loss of counter-selection of self-reactive B cells
Normal B cell development also includes stages in which potentially autoimmune B cell clones can be eliminated; these steps include the bone marrow and peripheral tissues. B cell receptors of naive B cells do not contain somatic hypermutations, and any diversity that is present is due to random immunoglobulin (Ig) V(D)J gene recombination events. However, early immature B cells in the bone marrow are often both autoreactive and polyreactive, having the capacity to bind to many antigens. Thus random recombination normally leads to the production of numerous deleterious B cells, unless these are eliminated. As autoreactive cells are much less common in the peripheral blood, it is clear that mechanisms for their removal are generally successful [6]. However, with regard to T cell clonal elimination, both central and peripheral checkpoints appear to be operative to remove autoimmune B cells in blood. If new emigrant B cells in peripheral blood do express autoimmune potential, a failure of central tolerance is suggested; if mature naive B cells in this compartment contain autoimmune potential, peripheral checkpoints have failed. Again, using selected defects in primary immune deficiency, it has been possible to analyse the molecular requirements for these checkpoints in humans. The first surprise was that subjects with X-linked agammaglobulinaemia (XLA), who have no circulating mature B cells, still have a small population of immature B cells in peripheral blood, the majority of which expressed self-reactivity, suggesting the failure of both central and peripheral tolerance mechanisms. In fact, one of the first autoimmune complications to be described in primary immune deficiency was the rheumatological disease that occurs in XLA [7].
While subjects with the hyper-IgM syndromes have recurrent opportunistic infections with Pneumocystis jiroveci and Cryptosporidium and for the X-linked version, a tendency to develop biliary tract disease [8,9], autoimmune complications are also common. These occur in both the X-linked and autosomal forms and include joint, bowel, liver and haematological disease. Table 2 outlines the most common autoimmune conditions in groups of subjects with the X-linked and the autosomal form of hyper-IgM syndrome due to mutations in the activation-induced cytidine deaminase gene (AID). Characteristics of these defects are the development of IgM antibodies but not IgG or IgA, lack of response to T dependent antigens and an inability to develop memory B cells. For the X-linked form, loss of CD40L signals on dendritic cells and thymic epithelial cells also occurs, and potentially a loss of development of Tregs. Some or all of these molecular defects leads to an increased number of mature naive B cells, which express a high proportion of autoreactive antibodies.
Table 2.
Autoimmunity in the hyper-immunoglobulin M (IgM) syndromes
As for subjects with XLA, subjects with hyper-IgM syndromes have circulating B cells with autoimmune potential; however, these are not new emigrant B cells but naive B cells, suggesting loss of peripheral tolerance. Alterations in B cell receptor signalling pathways are also found in patients with defects in Toll-like receptor (TLR) signalling, such as interleukin-1 receptor-associated kinase 4 (IRAK-4), myeloid differentiation primary response gene 88 (MyD88) and unc-93 homologue B1 (UNC-93B) [10–12] for less clear reasons. These observations demonstrate that B cell tolerance in humans normally relies upon a number of pathways working as an interactive network to exclude B cell autoimmunity.
Mechanisms of autoimmunity in common variable immune deficiency (CVID)
In CVID, B cells secrete immune globulins poorly, and fail to differentiate into plasma cells. About 25–30% of these subjects develop autoimmune complications; for unclear reasons, more than 50% of these involve the haematological system, with immune thrombocytopenia and haemolytic anaemia being foremost [13–17] (Table 3). While defects of single genes have helped to elucidate autoimmunity in selected primary immune defects, the cause of autoimmunity in CVID has proved more complex and a number of mechanisms are likely. Similar to the hyper-IgM syndrome, CVID B cells exhibit impaired somatic hypermutation [18], and there are reduced numbers of CD27+ memory B cells and an even greater losses of isotype-switched (IgD– IgM– CD27+) memory B cells [19]. Loss of these cells is associated with both the development of autoimmunity, lymphoid hyperplasia, splenomegaly and granulomatous disease [19–22] (Fig. 1 shows data for a Mount Sinai cohort). Another peripheral blood B cell marker of CVID subjects with propensity towards autoimmunity concerns those with low numbers of CD19, CD21–/lo B cells in peripheral blood [23]. These cells also lack somatic hypermutations, contain germline autoreactive antibodies and have an unusual phenotype on gene array.
Table 3.
Autoimmune conditions in common variable immune deficiency (CVID)
| Condition | % |
|---|---|
| Immune thrombocytopenia | 34 |
| Evans syndrome | 12 |
| Autoimmune haemolytic anaemia | 10 |
| Rheumatoid arthritis | 7 |
| Anti-IgA | 5 |
| Systemic lupus erythematosus | 4 |
| Alopecia | 3 |
| Diabetes mellitus | 3 |
| Inflammatory bowel disease | 3 |
| Pernicious anaemia | 3 |
| Myasthenia gravid | 3 |
| Neutropenia | 3 |
| Primary biliary cirrhosis | 3 |
| Immune urticaria | 3 |
| Anti-cardiolipin | 2 |
| Juvenile rheumatoid arthritis | 2 |
| Uveitis | 2 |
| Vasculitis | 2 |
| Lichen planus | 1 |
| Thyroiditis | 1 |
| Vitiligo | 1 |
IgA: immunoglobulin A.
Fig. 1.
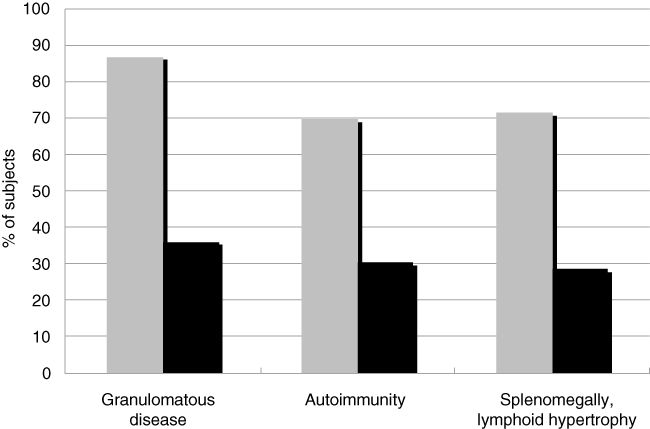
Isotype-switched memory B cells in common variable immune deficiency (CVID). For 105 subjects with CVID, the number of isotype-switched memory B cells in peripheral blood was determined based on the percentge of the total lymphocyte numbers. Those with more than 0·55% of these cells (grey bars) had significantly fewer of each of the noted complications compared to those with less than this number (black bars) This breakpoint was determined by receiver operating characteristic (ROC) curves. [21]
Turning to potential genetic reasons, 7–10% of CVID subjects have a mutation in the gene encoding the related receptor, transmembrane activator and calcium-modulating ligand interactor (TACI), which is expressed mainly on mature B cells [24,25]. While mutations in TACI are associated clearly with CVID, the same mutations are found in non-immunodeficient family members and some normal controls [26,27]. However, CVID patients with mutations in TACI have an increased incidence of autoimmunity. In a study of 199 patients, 14 (7%) had mutations in TACI; six of these had marked splenomegaly and one or more episodes of immune thrombocytopenia purpura (ITP) or autoimmune haemolytic anaemia (AIHA); five had undergone splenectomy. Significant differences were found when compared to the 163 CVID patients without TACI mutations; 20 had a history of ITP (P = 0·012), 17 had splenomegaly (P = 0·012), eight had splenectomy (P = 0·001) and six had AIHA [27]. A review of the European data showed that heterozygous inheritance of the C104R mutation was associated particularly with both autoimmunity and lymphoid hyperplasia in this cohort [28]. As TACI–/– mice develop splenomegaly, lymphadenopathy, lymphoma and a fatal autoimmune syndrome similar to human systemic lupus erythematosus (SLE) [29], it seems probable that this receptor exerts selected inhibitory effects, impaired in subjects with CVID who have mutations.
Another factor potentially important in autoimmunity in CVID is that both B cell activating factor (BAFF) and acidic protein rich in leucines (APRIL), cytokines important for survival and maturation of B cells [30], are found in excessive amounts in serum [31]. Over-expression of BAFF in mice leads to B cell hyperplasia, hyperglobulinaemia, splenomegaly and autoimmunity [32]. Both BAFF (and APRIL) are present in excess amounts in the sera of patients with systemic autoimmune disease such as rheumatoid arthritis, systemic lupus erythematosus and systemic sclerosis [32–34].
It is entirely probable that autoimmunity in CVID is also due to many other factors, including the known dysregulation of many cytokines and cellular factors, as reviewed recently [17]. Several groups have pointed out that the relative loss of Tregs in CVID is related to autoimmunity, splenomegaly and other inflammatory markers [35–37].
Conclusions
Primary immune defects are associated commonly with autoimmune manifestations. These may be organ- or tissue-based, and from the medical perspective are difficult to treat, as prolonged immune suppression, undesirable in these patients, may be required. The pathogenesis of autoimmunity in immune deficiency is unclear for the most part, but careful dissection of immune mechanisms in some have led to greater understanding of autoimmunity in general. The best examples of these include the mutations in CD95 in autoimmunity lymphoproliferative disorder, which lead to defective apoptosis and autoimmune cytopenias, and mutations in the interleukin (IL)-2 receptor, signal transducer and activator of transcription 5 (STAT 5) and FoxP3, that lead to loss of functional Tregs. For other genetic immune defects, including CVID, the pathogenesis of autoimmunity remains more obscure, although recently genetic studies have provided some illumination. However, the heterogeneity in both pathogenesis and clinical complications in CVID makes these investigations challenging.
Acknowledgments
This work was supported by grants from the National Institutes of Health, AI 101093, AI-467320, and AI-48693.
Disclosure
This paper is part of a supplement supported by an unrestricted grant from Grifols. The author received payment for the preparation of this article and attendance at the symposium in which it was presented.
References
- 1.Gardner JM, Fletcher AL, Anderson MS, Turley SJ. AIRE in the thymus and beyond. Curr Opin Immunol. 2009;21:582–9. doi: 10.1016/j.coi.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Poliani PL, Facchetti F, Ravanini M, et al. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood. 2009;114:105–8. doi: 10.1182/blood-2009-03-211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gambineri E, Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, and X-linked inheritance (IPEX), a syndrome of systemic autoimmunity caused by mutations of FOXP3, a critical regulator of T-cell homeostasis. Curr Opin Rheumatol. 2003;15:430–5. doi: 10.1097/00002281-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 4.Och HD, Gambineri E, Torgerson TR, et al. IPEX, FOXP3 and regulatory T-cells: a model for autoimmunity. Immunol Res. 2007;38:112–21. doi: 10.1007/s12026-007-0022-2. [DOI] [PubMed] [Google Scholar]
- 5.Moraes-Vasconcelos D, Costa-Carvalho BT, Togerson TR, Ochs HD. Primary immune deficiency disorders presenting as autoimmune diseases: IPEX and APECED. J Clin Immunol. 2008;28(Suppl. 1):S11–19. doi: 10.1007/s10875-008-9176-5. [DOI] [PubMed] [Google Scholar]
- 6.Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. 2008;20:632–8. doi: 10.1016/j.coi.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 7.Rotstein J, Good RA. Significance of the simultaneous occurrence of connective tissue disease and agammaglobulinaemia. Ann Rheum Dis. 1962;21:202–6. doi: 10.1136/ard.21.2.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levy J, Espanol-Boren T, Thomas C, et al. Clinical spectrum of X-linked hyper-IgM syndrome. J Pediatr. 1997;131:47–54. doi: 10.1016/s0022-3476(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 9.Winkelstein JA, Marino MC, Ochs HD, et al. The X-linked hyper-IgM syndrome: clinical and immunologic features of 79 patients. Medicine (Balt) 2003;82:373–84. doi: 10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]
- 10.Ng YS, Wardemann H, Chelnis J, Cunningham-Rundles C, Meffre E. Bruton's tyrosine kinase is essential for human B cell tolerance. J Exp Med. 2004;200:927–34. doi: 10.1084/jem.20040920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herve M, Isnardi I, Ng YS, et al. CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J Exp Med. 2007;204:1583–93. doi: 10.1084/jem.20062287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Isnardi I, Ng YS, Srdanovic I, et al. IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity. 2008;29:746–57. doi: 10.1016/j.immuni.2008.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cunningham-Rundles C. Common variable immunodeficiency. Curr Allergy Asthma Rep. 2001;1:421–9. doi: 10.1007/s11882-001-0027-1. [DOI] [PubMed] [Google Scholar]
- 14.Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709–27. doi: 10.1111/j.1365-2141.2009.07669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 16.Cunningham-Rundles C. Autoimmune manifestations in common variable immunodeficiency. J Clin Immunol. 2008;28(Suppl. 1):S42–5. doi: 10.1007/s10875-008-9182-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agarwal S, Cunningham-Rundles C. Autoimmunity in common variable immunodeficiency. Curr Allergy Asthma Rep. 2009;9:347–52. doi: 10.1007/s11882-009-0051-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonhomme D, Hammarstrom L, Webster D, et al. Impaired antibody affinity maturation process characterizes a subset of patients with common variable immunodeficiency. J Immunol. 2000;165:4725–30. doi: 10.4049/jimmunol.165.8.4725. [DOI] [PubMed] [Google Scholar]
- 19.Warnatz K, Denz A, Dräger R, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(–)IgD(–)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–51. doi: 10.1182/blood.v99.5.1544. [DOI] [PubMed] [Google Scholar]
- 20.Piqueras B, Lavenu-Bombled C, Galicier L, et al. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23:385–400. doi: 10.1023/a:1025373601374. [DOI] [PubMed] [Google Scholar]
- 21.Sanchez-Ramon S, Radigan L, Yu JE, et al. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128:314–21. doi: 10.1016/j.clim.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 23.Isnardi I, Ng YS, Menard L, et al. Complement receptor 2/CD21– human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115:5026–36. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–34. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 25.Salzer U, Chapel HM, Webster AD, et al. Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet. 2005;37:820–8. doi: 10.1038/ng1600. [DOI] [PubMed] [Google Scholar]
- 26.Pan-Hammarstrom Q, Salzer U, Du L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429–30. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Radigan L, Salzer U, et al. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol. 2007;120:1178–85. doi: 10.1016/j.jaci.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salzer U, Bacchelli C, Buckridge S, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967–76. doi: 10.1182/blood-2008-02-141937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seshasayee D, Valdez P, Yan M, Dixit VM, Tumas D, Grewal IS. Loss of TACI causes fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLyS receptor. Immunity. 2003;18:279–88. doi: 10.1016/s1074-7613(03)00025-6. [DOI] [PubMed] [Google Scholar]
- 30.Khare SD, Hsu H. The role of TALL-1 and APRIL in immune regulation. Trends Immunol. 2001;22:61–3. doi: 10.1016/s1471-4906(00)01843-3. [DOI] [PubMed] [Google Scholar]
- 31.Knight AK, Radigan L, Marron T, Langs A, Zhang L. High serum levels of BAFF, APRIL, and TACI in common variable immunodeficiency. Clin Immunol. 2007;124:182–9. doi: 10.1016/j.clim.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stohl W, Xu D, Kim KS, et al. BAFF overexpression and accelerated glomerular disease in mice with an incomplete genetic predisposition to systemic lupus erythematosus. Arthritis Rheum. 2005;52:2080–91. doi: 10.1002/art.21138. [DOI] [PubMed] [Google Scholar]
- 33.Mackay F, Sierro F, Grey ST, Gordon TP. The BAFF/APRIL system: an important player in systemic rheumatic diseases. Curr Dir Autoimmun. 2005;8:243–65. doi: 10.1159/000082106. [DOI] [PubMed] [Google Scholar]
- 34.Matsushita T, Hasegawa M, Yanaba K, Kodera M, Takehara K, Sato S. Elevated serum BAFF levels in patients with systemic sclerosis: enhanced BAFF signaling in systemic sclerosis B lymphocytes. Arthritis Rheum. 2006;54:192–201. doi: 10.1002/art.21526. [DOI] [PubMed] [Google Scholar]
- 35.Fevang B, Yndestad A, Sandberg WJ, et al. Low numbers of regulatory T cells in common variable immunodeficiency: association with chronic inflammation in vivo. Clin Exp Immunol. 2007;147:521–5. doi: 10.1111/j.1365-2249.2006.03314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Genre J, Errante PR, Kokron CM, Toledo-Barros M, Câmara NO, Rizzo LV. Reduced frequency of CD4(+)CD25(HIGH)FOXP3(+) cells and diminished FOXP3 expression in patients with common variable immunodeficiency: a link to autoimmunity? Clin Immunol. 2009;132:215–21. doi: 10.1016/j.clim.2009.03.519. [DOI] [PubMed] [Google Scholar]
- 37.Yu GP, Chiang D, Song SJ, et al. Regulatory T cell dysfunction in subjects with common variable immunodeficiency complicated by autoimmune disease. Clin Immunol. 2009;131:240–53. doi: 10.1016/j.clim.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quartier P, Bustamante J, Sanal O, et al. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to activation-induced cytidine deaminase deficiency. Clin Immunol. 2004;110:22–9. doi: 10.1016/j.clim.2003.10.007. [DOI] [PubMed] [Google Scholar]