

Abstract
Superantigens have been implicated in a number of diseases including Kawasaki disease (KD), a multi-system vasculitis resulting in coronary artery aneurysms. We have characterized a murine disease model in which coronary arteritis is induced by a novel superantigen found in Lactobacillus casei cell wall extract (LCWE). Using this animal model of KD, we have identified three pathogenic steps leading to coronary artery aneurysm formation. These steps include T cell activation and proliferation, production of the proinflammatory cytokine tumour necrosis factor (TNF)-α and up-regulation of matrix metalloproteinase 9 (MMP-9), an elastolytic protease. In addition to their cholesterol-lowering effects, 3-hydroxy-3-methylglutaryl (HMG) coenzyme A (CoA) reductase inhibitors (statins) have pleotropic immunomodulatory properties. Thus, we examined the effect of atorvastatin in modulating each of these three critical pathogenic processes leading to aneurysm formation in the disease model. Atorvastatin inhibited lymphocyte proliferation in response to superantigen stimulation in a dose-dependent manner. This inhibition was also observed for production of soluble mediators of inflammation including interleukin (IL)-2 and TNF-α. The inhibitory effect on proliferation was rescued completely by mevalonic acid, confirming that the mechanism responsible for this inhibitory activity on immune activation was inhibition of HMG-CoA reductase. Similarly, TNF-α-induced MMP-9 production was reduced in a dose-dependent manner in response to atorvastatin. Inhibition of extracellular-regulated kinase (ERK) phosphorylation appears to be the mechanism responsible for inhibition of MMP-9 production. In conclusion, atorvastatin is able to inhibit critical steps known to be important in the development of coronary aneurysms, suggesting that statins may have therapeutic benefit in patients with KD.
Keywords: inflammation, Kawasaki disease, MMP, statins
Introduction
Kawasaki disease (KD) is the leading cause of acquired heart disease of children in the industrialized world. This multi-system vasculitis is characterized by prolonged fever, polymorphous skin rash, non-purulent conjunctival infection, extremity changes, oral–mucosal changes and cervical lymphadenopathy [1]. These classic signs and symptoms of systemic inflammation are prominent during the acute phase of illness, although KD then becomes a localized phenomenon with inflammation focused primarily at the coronary artery (CA), resulting in the development of aneurysms. Although the exact aetiology of KD is still debated [2,3], evidence suggests that the initial infectious trigger of KD may possess superantigenic activity leading to stimulation of the immune system. Evidence of a superantigen (SAg)-mediated disease process in KD includes identification of SAg-producing organisms in, isolation of bacterial SAgs from, or finding the hallmarks of SAg activation in the immune system of affected children. Specifically, the footprint of a SAg, T cell receptor (TCR)-Vβ skewing, has been identified during some outbreaks of KD, with skewing of TCR-Vβ2 and Vβ8 observed in children with KD, reflecting the TCR-Vβ footprint of toxic shock syndrome toxin-1 (TSST-1) and streptococcal pyrogenic exotoxin (SPE)-B and SPE-C [4–8]. SAgs encompass a group of proteins that are able to elicit a dramatic T cell-dependent immune response [9] via interaction with the TCR-Vβ chain. Exposure to SAgs leads to production of massive amounts of proinflammatory cytokines, including interferon (IFN)-γ, tumour necrosis factor (TNF)-α, interleukin (IL)-1α and IL-2 [10]. The resultant inflammatory cytokine cascade leads to many downstream effector functions, including up-regulation of matrix degrading enzymes. The most studied prototypical bacterial SAg is staphylococcal enterotoxin B (SEB), and it has been shown to induce the rapid production of IL-2, IFN-γ, TNF-α and TNF-β by splenocytes as soon as 30 min after injection in mice [11].
SAgs have been implicated in many human diseases, most notably food poisoning and toxic shock syndrome, as well as a number of inflammatory/autoimmune diseases, including insulin-dependent diabetes mellitus (IDDM) [12], rheumatoid arthritis (RA) [13], multiple sclerosis (MS) [14] and KD [6,15]. Common to each of these inflammatory diseases is the production of TNF-α, which mediates a number of important events during the inflammatory immune response. TNF-α is a pleiotropic cytokine with multiple downstream effects, one of which is up-regulation of matrix degrading proteases, including members of the matrix-metalloproteinase (MMP) family. MMPs are capable of degrading extracellular matrix proteins, and have been found to play a role in tissue destruction in RA, KD and MS [16–18].
A murine model of KD was first developed by Lehman et al. [19]. Lactobacillus casei cell wall extract (LCWE) containing SAg activity induces coronary arteritis in mice, which mimics closely that which develops in children with KD [19,20]. The disease induced in mice resembles that in human in terms of its time–course, susceptibility in the young, pathology and response to treatment with intravenous immunoglobulin (IVIG), the therapeutic agent used in KD children. The ability of LCWE to induce disease is dependent on its supergenic activity, with stimulation and expansion of the T cell subset expressing TCR-Vβ2, 4 and 6 [20]. Using this animal model of KD, we identified three critical steps involved in disease progression and aneurysm formation: T cell proliferation, TNF-α cytokine production and TNF-α-mediated MMP-9 production. The localized production of MMP-9 at the coronary artery results in elastin breakdown and aneurysm formation [21,22].
The 3-hydroxy-3-methylgultaryl co-enzyme A (HMG-CoA) reductase inhibitors, also known as statins, are very powerful inhibitors of the mevalonate pathway, which directs the biosynthesis of isoprenoids and cholesterol. They are the leading therapeutic regimen for treating hypercholesterolaemia and reducing cardiovascular morbidity and mortality in the setting of atherosclerotic cardiovascular disease [23]. Interestingly, a pilot study has reported that statin therapy appeared to improve chronic vascular inflammation and endothelial dysfunction significantly in children complicated with coronary arterial abnormality late after KD [24]. Recent evidence suggests that statins have multiple effects and are able to modulate the immune response independent of their cholesterol attenuating ability [25]. The anti-inflammatory and immunomodulatory effects of statins stem from downstream effects of inhibiting the mevalonate pathway leading to decreased activity of the small guanosine triphosphate (GTPases) Rac, Ras and Rho [26], which are crucial for many cellular functions including proliferation and transcriptional regulation [27], key processes in inflammation. We hypothesize a beneficial therapeutic effect of statins in SAg-mediated diseases through the modulation of T cell activation and MMP-9 production.
In this study, we studied the role of atorvastatin in modulating three critical steps in the pathogenesis of coronary artery inflammation and aneurysm formation in a disease model of KD. These include T cell proliferation, TNF-α cytokine production and TNF-α-mediated MMP-9 production [28,29]. We show that atorvastatin inhibits each one of these critical processes leading to aneurysm formation, suggesting a potential beneficial effect of statins in the treatment of KD.
Materials and methods
Reagents
Atorvastatin calcium (Pfizer, Kirkland, Quebec, Canada) was dissolved in dimethyl sulphoxide (DMSO) (Sigma-Aldrich, St Louis, MO, USA). Mevalonic acid (MVA) (Sigma-Aldrich) was also dissolved in DMSO, and Staphylococcal enterotoxin B (SEB) (Toxin Technology Inc, Sarasota, FL, USA) was dissolved in phosphate-buffered saline (PBS).
Preparation of LCWE
LCWE was prepared as described previously [19]. Briefly, Lactobacillus casei (ATCC 11578) was harvested after ∼18 h and washed in PBS. Bacteria lysis by overnight sodium dodecyl sulphate (SDS) incubation was followed by incubation with DNAase I, RNAse and trypsin (Sigma Chemicals) to remove any adherent material from the cell wall. The cell wall was fragmented through sonication in a dry ice/ethanol bath for 2 h. Phenol-sulphuric colorimetric determination assay was used to determine the measurement of rhamnose concentration, which was expressed in mg/ml PBS. Total protein concentration was determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories, Mississauga, ON, Canada) following the manufacturer's instructions.
Experimental mice
Wild-type 6–12-week-old C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA, USA) and housed under specific pathogen-free conditions at the Hospital for Sick Children under an approved animal use protocol.
Lymphocyte proliferative assays
Splenocytes (5 × 105) from C57BL/6 mice were cultured in medium alone (Iscove's supplemented with 10% heat-inactivated fetal bovine serum (FBS), sodium pyruvate, non-essential amino acid, 50 µM 2-mercaptoethanol (ME), 2 mM l-glutamine and 10 mM HEPES), medium containing 0·03125 µg/ml highly purified SEB (Toxin Technology Inc., Sarasota, FL, USA), medium containing 0·1 µg/ml anti-mouse CD3ε chain (BD Biosciences, San Jose, CA, USA) plus 0·4 µg/ml anti-mouse CD28 (BioLegend, San Diego, CA, USA), or medium containing 6·25 µg/ml LCWE, together with atorvastatin (0–12·5 mM). Cells were incubated at 37°C in 5% CO2 for 72 h and pulsed with 1 µCi/well [3H]-thymidine (GE General Health, Mississauga, ON, Canada) during the last 16 h. Disintegrations per minute (dpm) from triplicate wells were analysed. Data are presented as mean dpm ± standard error of the mean (s.e.m.). The same experiment was performed three times using five to eight animals.
Enzyme-linked immunosorbent assays (ELISA)
Culture supernatant was collected from splenocytes 48 h after incubation with SEB and atorvastatin. IL-2 protein levels were quantified by the mouse IL-2 Duoset ELISA (R&D Systems, Minneapolis, MN, USA), as per the manufacturer's protocol, and read using a SpectraMAX 250 plate reader (Molecular Devices, Sunnyvale, CA, USA). Similarly, TNF-α concentration was assayed in culture supernatant at 24 h and quantified by the mouse TNF-α Ready-SET-Go Kit (eBioscience, San Diego, CA, USA), as per the manufacturer's protocol. In some experiments, MVA was also added to the SEB plus atorvastatin, and supernatants assayed for IL-2 and TNF-α as described. Results presented were representative of at least three independent experiments.
Quantitative real time reverse transcription–polymerase chain reaction (RT–PCR)
Mouse vascular smooth muscle cells (SMC) (MOVAS) (generously provided by Dr M. Husain, Toronto General Hospital Research Institute, Toronto, Ontario, Canada) were cultured [Dulbecco'smodified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), sodium pyruvate, non-essential amino acid, 2 mM l-glutamine and 10 mM HEPES] for 6 h with atorvastatin in addition to 25 ng/ml recombinant mouse TNF-α (eBioscience). In experiments to determine the effect of the mitogen-activated protein (MEK) 1/2 inhibitor U0126 (Cell Signaling, Beverly, MA, USA) on MMP-9 production, U0126 was used instead of atorvastatin. After the incubation period, the MOVAS cells were lysed with TRIzol reagent (Invitrogen Life Technologies, Carlsbad, CA, USA) and total RNA was isolated with a standard chloroform extraction method. Complementary DNA (cDNA) was synthesized using the GeneAmp RNA PCR kit and murine leukaemia virus reverse transcriptase (Applied Biosystems, Foster City, CA, USA). cDNA was then amplified by real-time RT–PCR following the manufacturer's protocol with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers and probe (Applied Biosystems) and the MMP-9 primers and probe set (Assays-on-Demand; Applied Biosystems) in an ABI PRISM 7900 Sequence Detection System (Applied Biosystems). Data were collected and analysed using GraphPad Prism 4 software (GraphPad Software Inc, La Jolla, CA, USA). Relative quantities of PCR products were determined off the standard curve generated in each run from cDNA known to contain MMP-9 and expressed as a ratio against the housekeeping gene GAPDH. Real-time RT–PCR was performed in at least three independent experiments.
Western blotting
MOVAS cells were serum-starved for 48 h and then preincubated with various concentrations of atorvastatin for 2 h prior to the addition of 25 ng/ml recombinant TNF (rTNF)-α for 15 min. Cells were exposed immediately to ice-cold lysis buffer [50 mM Tris (pH 7·6), 2% sodium dodecyl sulphate, 0·1 mM phenylmethylsulphonyl fluoride, 10 µg/ml leupeptin] and sonicated for 5 s. Standard immunoblotting with peroxidase-based detection was performed with equal amounts of total protein (10 µg). Blots were incubated with antibodies specific for the active p44/42 MAP kinase (Cell Signaling). Once the results were analysed, the blot was stripped with Restore Western Blot Stripping Buffer (Thermo Scientific, Rockford, IL, USA) and incubated with antibodies specific for p44/42 MAP kinase (Cell Signaling) in order to normalize the results. All bands were semi-quantified by reverse image scanning densitometry with Adobe Photoshop CS3 (Adobe Systems, San Jose, CA, USA). Four independent Western blotting experiments were performed.
Statistical analysis
All values are expressed as mean ± standard error (s.e.), unless specified otherwise. For the comparison of multiple groups, a one-way analysis of variance and a Tukey post-hoc test were performed. P-values less than 0·05 were considered significant.
Results
Atorvastatin inhibits superantigen-mediated T cell activation in a dose-dependent manner
To investigate the effect of atorvastatin on lymphocyte activation, we measured the proliferative response to different TCR agonists including: anti-CD3 + anti-CD28; SEB; and LCWE together with atorvastatin. Both pan-stimulation of T cells, using aCD3 + aCD28, and stimulation by superantigens, both SEB and LCWE [20], produced a marked proliferative response which maximized at day 3. Atorvastatin was able to inhibit this proliferative response to all three mitogens in a dose-dependent fashion (Fig. 1a–c). Interestingly, the dose–response to atorvastatin had a direct correlation with the strength of TCR activation. Anti-CD3 + anti-CD28 cultures exhibits the most robust proliferative response, and thus higher concentrations of atorvastatin (12·5–8·7 µM) were required to exert an inhibitory effect, whereas much lower atorvastatin amounts (0·16–2·5 µM) were effective at inhibition in LCWE cultures. Similarly, SEB cultures proliferated moderately to SEB, and thus correspondingly moderate atorvastatin concentrations (1·72–6·9 µM) were sufficient. T cell activation is also characterized by production of the growth/proliferative cytokine, IL-2. Production of IL-2 by superantigen-activated T cells was assayed by ELISA on the supernatant of splenocytes cultured with SEB. Atorvastatin decreased IL-2 production in a dose-dependent manner (Fig. 1d). The observed inhibitory effects were not due to the diluant (DMSO) used to deliver atorvastatin to the cell culture system. DMSO was assayed for potential toxic effects and was found to have no effect on cell proliferation at the concentrations used (data not shown).
Fig. 1.
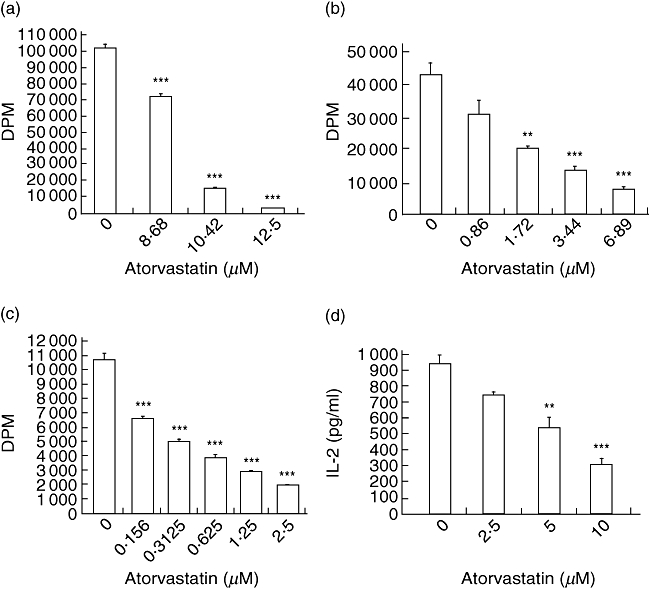
Atorvastatin inhibits T cell proliferation and interleukin (IL)-2 production in a dose-dependent manner. Splenocytes from C57BL/6 mice were cultured in the presence of either (a) anti-CD3 + anti-CD28 antibodies (b) staphylococcal enterotoxin B (SEB) or (c) Lactobacillus casei cell wall extract (LCWE) and atorvastatin for 72 h, with [3H]-thymidine added for the last 16 h of incubation. Cells were harvested and radioactivity measured in a liquid scintillation counter. Data representative of at least three independent experiments. ***P < 0·001, **P < 0·01 versus control (no atorvastatin). (d) Splenocytes from C57BL/6 mice were cultured in the presence of SEB and various concentrations of atorvastatin for 48 h, supernatant collected and concentration of IL-2 determined by enzyme-linked immunosorbent assay. Data represent at least three independent experiments. ***P < 0·001, **P < 0·01 versus control (no atorvastatin), by analysis of variance.
Atorvastatin inhibits T cell activation via the mevalonate pathway
To determine whether atorvastatin inhibits lymphocyte proliferation through the inhibition of the mevalonate pathway, mevalonic acid (MVA) was added to the culture conditions described above. The addition of MVA rescued the inhibitory effect on cell proliferation caused by atorvastatin in a dose-dependent manner (Fig. 2a). Similarly, the addition of MVA also abrogated the inhibitory effect of atorvastatin on IL-2 production in response to SEB in a dose-dependent manner (Fig. 2b), confirming that atorvastatin inhibits both superantigen-mediated lymphocyte proliferation and IL-2 production through inhibition of the mevalonate pathway acting at HMG-CoA reductase.
Fig. 2.
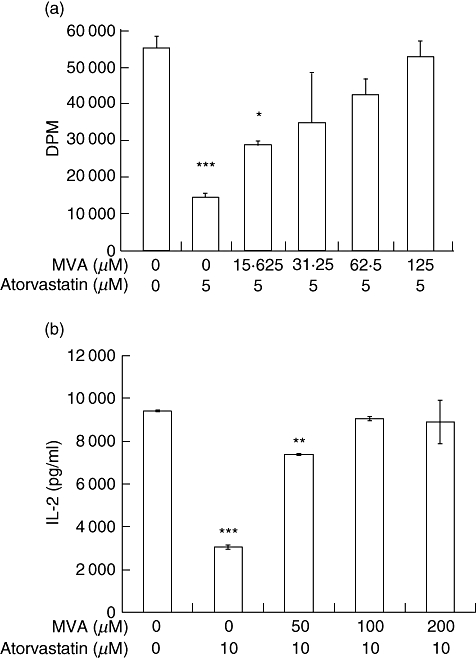
Mevalonic acid (MVA) rescues atorvastatin mediated inhibition of lymphocyte proliferation. (a) Splenocytes from C57BL/6 mice were cultured in the presence of staphylococcal enterotoxin B (SEB), 5 µM atorvastatin and MVA lactone for 72 h, with addition of [3H]-thymidine for the last 16 h of incubation. The cells were harvested and radioactivity was measured in a liquid scintillation counter. Data representative of at least three independent experiments. ***P < 0·001, *P < 0·05 versus control (no atorvastatin, no MVA). (b) Splenocytes from C57BL/6 mice were cultured in the presence of SEB, 10 µM atorvastatin and various concentrations of MVA lactone. Supernatants were collected 48 h later, and concentration of interleukin (IL)-2 determined by enzyme-linked immunosorbent assay. **P < 0·01.
Atorvastatin inhibits superantigen-mediated TNF-α production
The inflammatory response in acute KD is characterized by high levels of circulating TNF-α. TNF-α production is a key proinflammatory cytokine in the pathogenesis of coronary artery inflammation and elastin breakdown in the LCWE model of KD [21]. Local production of TNF-α at the coronary artery leads to up-regulation of MMP-9 production by vascular smooth muscle cells and localized elastolytic activity and matrix breakdown of affected coronary arteries [22,28]. To investigate the effect of atorvastatin on SAg-mediated TNF-α production, the supernatant of splenocytes co-cultured with SEB and atorvastatin was assayed by ELISA. Atorvastatin was able to inhibit TNF-α production dramatically (Fig. 3a). Furthermore, the addition of MVA abrogated the inhibitory effect of atorvastatin on TNF-α production in a dose-dependent manner (Fig. 3b) indicating that, as in the case of IL-2, atorvastatin inhibits TNF-α production in response to SAg by interfering with the mevalonic pathway.
Fig. 3.
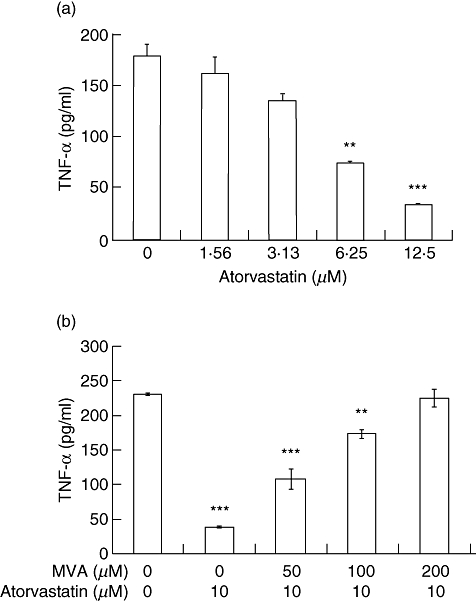
Atorvastatin inhibits tumour necrosis factor (TNF)-α production in a dose-dependent manner. Splenocytes from C57BL/6 mice were cultured with staphylococcal enterotoxin B (SEB) and atorvastatin (a) in the presence or (b) absence of various concentrations of mevalonic acid lactone. Twenty-four-h supernatants were collected, and concentration of TNF-α determined by enzyme-linked immunosorbent assay. Data representative of at least three independent experiments. ***P < 0·001, **P < 0·01 versus control (no atorvastatin).
Atorvastatin inhibits TNF-α-mediated MMP-9 production
In the LCWE disease model, MMP-9 production by vascular SMC at the coronary artery is directed by TNF-α. The production of MMP-9 leads to elastin breakdown and coronary vessel wall destruction [22,28]. To determine whether atorvastatin modulates TNF-α-induced MMP-9 production, MOVAS cells were stimulated with TNF-α and atorvastatin and quantitative RT–PCR assay was used to determine MMP-9 transcription. Atorvastatin inhibited MMP-9 production in a dose-dependent fashion (Fig. 4a). The higher concentrations of atorvastatin required to exert an inhibitory effect may reflect the differential sensitivity to statin of different cell types (i.e. SMC versus lymphocytes) and/or of different cellular pathways (i.e. proliferation and cytokine production versus MMP-9 production). The observed inhibitory effects were not due to the diluant (DMSO) used to deliver atorvastatin to the cell culture system. DMSO was assayed for potential toxic effects and was found to have no effect on cell proliferation at the concentrations used (Fig. S1; see Supporting information at end).
Fig. 4.
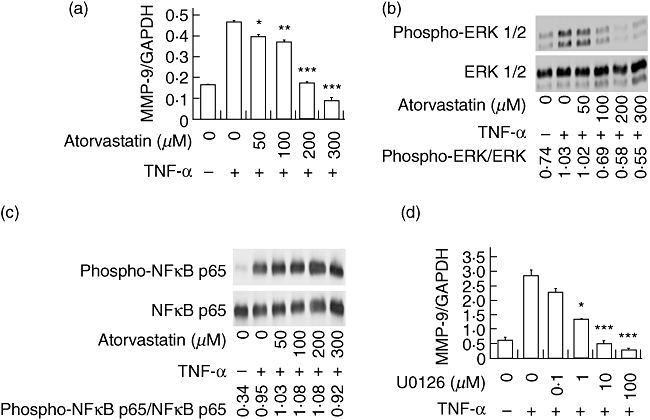
Atorvastatin inhibits tumour necrosis factor (TNF)-α-mediated matrix metalloproteinase 9 (MMP-9) production via the mitogen-activated protein/extracellular-regulated kinase (MEK/ERK) pathway. (a) Mouse vascular smooth muscle cells (MOVAS) cells were cultured with TNF-α and atorvastatin for 6 h, total RNA isolated, and cDNA synthesized. Real-time reverse transcription–polymerase chain reaction was performed and relative quantities of MMP-9 were expressed as a ratio against the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). (b) MOVAS cells were incubated with atorvastatin and TNF-α. Total protein was extracted and Western blot analysis was performed using antibodies specific for phospho-ERK 1/2 (in panel b) or antibodies specific for phospho-nuclear factor (NF)-κB (c). (d) Similar conditions to those in (a) but U0126, a MEK 1/2 inhibitor, was used instead of atorvastatin. All data presented represent at least three independent experiments. ***P < 0·001, **P < 0·01, *P < 0·05 versus control (TNF-α, but no chemical inhibitor).
Atorvastatin inhibits signalling via the MEK/ERK pathway
To determine whether the MEK/extracellular-regulated kinase (ERK) signalling pathway was responsible for atorvastatin-mediated inhibition of MMP-9 production, the effects of atorvastatin on ERK phosphorylation was determined by phospho-Western blots on MOVAS cells stimulated with TNF-α and given atorvastatin. Atorvastatin inhibited the phosphorylation of ERK 1/2 in a dose-dependent manner, at the same concentrations in which MMP-9 transcription is inhibited by atorvastatin (Fig. 4b). Nuclear factor (NF)-κB signalling is also involved in TNF-α-mediated MMP-9 production, but interestingly, this pathway was not affected by atorvastatin (Fig. 4c).
The MEK/ERK signalling pathway mediates TNF-α-induced MMP-9 production
To determine whether the MEK/ERK signalling pathway mediates TNF-α-induced MMP-9 production by MOVAS cells, cultures were co-incubated with a MEK inhibitor, U0126 and MMP-9 message levels assayed by quantitative RT–PCR. U0126 effectively inhibited MMP-9 production in a dose-dependent manner (Fig. 4d), indicating that the signalling via the MEK/ERK pathway is necessary for TNF-α-mediated MMP-9 production by MOVAS cells.
Discussion
We have identified previously three key steps in the development of coronary artery damage in a disease model of KD [30]. These pathogenic steps include T cell activation and proliferation, production of TNF-α and TNF-α-mediated MMP-9 production. In the mouse model of KD, T cell activation triggers a massive inflammatory response characterized by marked lymphocyte proliferation and cytokine production. Local inflammation and production of TNF-α at the coronary arteries stimulates the production of MMP-9 by SMC, resulting in elastin breakdown and aneurysm formation. All three steps in concert lead to coronary artery damage and aneurysm formation in the animal model of KD.
Atorvastatin inhibited lymphocyte proliferation in response to superantigen stimulation in a dose-dependent manner. This inhibition was also observed for production of soluble mediators of inflammation including IL-2 and TNF-α. The inhibitory effect on both proliferation and cytokine production was rescued completely by mevalonic acid, confirming that the mechanism responsible for this inhibitory activity on immune activation was at HMG-CoA reductase, a similar mechanism of action in inhibiting cholesterol metabolism. Similarly, TNF-α-induced MMP-9 production was reduced in a dose-dependent manner in response to atorvastatin. Inhibition of ERK phosphorylation appears to be the mechanism responsible for inhibition of MMP-9 production.
The ability of atorvastatin to modulate these key pathogenic steps stems from its ability to inhibit the conversion of HMG-CoA to l-mevalonate. Consistent with previous findings, our data confirm that the inhibition of T cell proliferation is dependent on the mevalonate pathway, as the addition of mevalonic acid to statin-treated cells rescued the inhibitory effect observed [31]. The inhibition of the mevalonate pathway by statins leads to the loss of isoprenoid intermediates, such as geranyl pyrophosphate and farnesyl pyrophosphate. These isoprenoid intermediates act as essential lipid attachments for the post-translational modification of several small GTP-binding proteins, one of which is Ras [32]. The Ras/Raf/Mek/Erk pathway has been demonstrated previously to be a key element involved in T cell activation, as it is involved in production of the activator protein 1 (AP1) transcription factor. AP1 activity is necessary for the up-regulation of proinflammatory genes such as IL-2 and TNF-α[33–35], but also necessary for TNF-α-mediated MMP-9 production by SMC [36]. Our previous study suggested that IVIG, the therapeutic agent of choice in acute KD, may prevent aneurysm formation through its ability to reduce TNF-α production and, thus, inhibit MMP-9 production indirectly. However, IVIG has no direct effect on MMP-9 production mediated by TNF-α[37]. Thus, the ability of atorvastatin to mitigate MMP-9 production both indirectly through inhibition of TNF-α production and directly via inhibition of TNF-α-mediated ERK phosphorylation in SMC is very noteworthy and has important clinical implications.
Our earlier studies in the animal model of KD revealed that whereas T cell proliferation and TNF-α production in the periphery occurred early following LCWE stimulation, TNF-α and MMP-9 production at the coronary arteries were detected days later, corresponding to the late stage of the acute or subacute phase of KD in children indicating ongoing inflammation leading to elastin breakdown and end-organ damage [21,22]. Our results demonstrate a modulatory effect of atorvastatin at early (e.g. T cell activation and/or TNF-α production) as well as later (e.g. TNF-α-mediated MMP-9 production by SMC) events during disease progression, thus pointing to a potential therapeutic role of this agent even after immunological activation has taken place. This is relevant clinically, as systemic inflammation is well under way at diagnosis of KD, and atorvastatin, with its ability to interfere with both early and late pathogenic events, may be of added therapeutic value. There remain many factors to consider prior to clinical use of statin therapy in children with KD, especially in the acute phase. The potential benefits of statin therapy during the acute inflammation of KD include its role in reducing both the cellular proliferative response and production of proinflammatory soluble mediators. Additionally, statin treatment can inhibit elastin degradation and matrix breakdown via down-regulation of MMP-9 production. Potential contraindications include hepatic toxicity evidenced by raised liver-derived enzymes. Liver dysfunction evidenced by elevation of transaminases is already common during acute KD, and in fact is one of the supportive laboratory criteria to help identify children with incomplete KD [1]. Additionally, limited toxicity data are available on statin use in young children, and young children comprise the at-risk population for KD. In children and adolescents with familial hypercholesterolaemia who are more than 8 years old, current evidence suggests that statin treatment is well tolerated without significant adverse concerns [38–41]; however, no data are available for those less than 5 years old, corresponding to the majority of children with KD. Before statin treatment can be initiated in very young children, additional pharmacokinetic and toxicity data are needed. Interestingly, a pilot study reported that short-term statin therapy improved chronic vascular inflammation and endothelial dysfunction with no detectable adverse effects in a small population of children with ages ranging from 9·25 to 16·67 years, and complicated with coronary arterial abnormality late after KD [24].
Another important consideration in translating the in vitro murine data to the bedside is the dosing regimen. Serum concentrations of atorvastatin, for lipid lowering, are in the nanomolar range [42], while inhibition of T cell activation and MMP-9 production occurs only at micromolar concentrations in tissue culture. Direct comparisons of human serum concentrations to in vitro experiments are not appropriate, especially for a lipophilic drug such as atorvastatin. Additionally, previous work has shown that statin treatment inhibits MMP-9 production indirectly in the vessel wall of abdominal aortic aneurysms [43,44], thus serum levels may not reflect accurately local tissue concentrations at work, similar to the disconnect between serum and tissue levels of MMP-9 in KD [45].
An altered lipid profile has been reported in children with KD. During the acute phase of disease a pro-atherogenic lipid profile [46,47], with a decrease in total cholesterol, high-density lipoprotein-cholesterol (HDL-C), apoA1 and apoA2 and an increase in triglycerides and apoB, is observed [46,48,49]. Total cholesterol returns quickly to normal, but HDL-C recovery is slow and remains significantly lower than expected up to years later [46]. In addition to the observed mild dyslipidaemia in patients with KD, arterial function may be abnormal with abnormal measures of endothelial dysfunction even in those without aneurysms [50–53]. Carotid artery intima-media thickness among KD patients is greater, and endothelial dysfunction has been reported both in children with persistent coronary lesions as well as in those without detectable early coronary artery involvement, indicated by decreased brachial artery flow-mediated dilatation [40,48,49,53]. Thus, all patients with KD, even in the absence of echocardiographic evidence of coronary artery involvement, may be at risk for premature atherosclerosis even if managed appropriately during the acute phase of the illness. The potential benefits of statin therapy are recognized outside the acute phase of illness.
Recognizing the limitations of the in-vitro results reported, confirmation of the immunomodulatory effects of atorvastatin are needed in vivo using the LCWE-induced coronary arteritis animal model of KD. This model, which mimics accurately the histopathological changes seen in the coronary arteries of KD patients [19,20,54], provides a unique opportunity to study treatment protocols and potential side effects of statin therapy in young animals, providing important insight prior to human studies.
In conclusion, our results show that atorvastatin is able to inhibit critical pathogenic steps in the development of coronary artery damage in KD. The use of statins in treating KD may be beneficial due to its observed immunomodulatory properties, including the inhibition of T cell proliferation and cytokine production as well as inhibiting MMP-9 production, suggesting that statins may have benefit beyond that of cholesterol-lowering in Kawasaki disease. More study is needed to determine the safety and efficacy of this class of therapeutic agents in young children.
Acknowledgments
This study was funded by operating grants from the Canadian Institutes of Health Research (MOP-81378) and the Heart and Stroke Foundation of Canada (T6365). R.S.M.Y. is a recipient of an Investigator Award from the Arthritis Society of Canada and BWM is the holder of the CIBC World Markets Children's Miracle research chair.
Disclosure
All authors have no conflicts of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Cytotoxicity assay. Mouse vascular smooth muscle cells (MOVAS) cells were cultured [Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS), sodium pyruvate, non-essential amino acid, 2 mM L-glutamine and 10 mM HEPES] for 6 h in a 96-well culture plate with 25 ng/ml recombinant mouse tumour necrosis factor (TNF)-α (eBioscience, San Diego, CA, USA), and with either various atorvastatin concentrations or of the drug vehicle, dimethyl sulphoxide (DMSO). After the incubation period, cytotoxicity was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) using a commercial kit, following the manufacturer's protocol (Roche Applied Science, Mannheim, Germany). Open and solids bars represent cultures in the presence of atorvastatin and corresponding concentrations of DMSO, respectively.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis, and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Pediatrics. 2004;114:1708–33. doi: 10.1542/peds.2004-2182. [DOI] [PubMed] [Google Scholar]
- 2.Rowley AH, Shulman ST. New developments in the search for the etiologic agent of Kawasaki disease. Curr Opin Pediatr. 2007;19:71–4. doi: 10.1097/MOP.0b013e328012720f. [DOI] [PubMed] [Google Scholar]
- 3.Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. 2001;166:1334–43. doi: 10.4049/jimmunol.166.2.1334. [DOI] [PubMed] [Google Scholar]
- 4.Abe J, Kotzin BL, Meissner C, et al. Characterization of T cell repertoire changes in acute Kawasaki disease. J Exp Med. 1993;177:791–6. doi: 10.1084/jem.177.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yamashiro Y, Nagata S, Oguchi S, Shimizu T. Selective increase of Vbeta2+ T cells in the small intestinal mucosa in Kawasaki disease. Pediatr Res. 1996;39:264–6. doi: 10.1203/00006450-199602000-00013. [DOI] [PubMed] [Google Scholar]
- 6.Leung D, Meissner HC, Fulton DR, Murray DL, Kotzin BL, Schlievert PM. Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. 1993;342:1385–8. doi: 10.1016/0140-6736(93)92752-f. [DOI] [PubMed] [Google Scholar]
- 7.Curtis N, Levin M. Evidence for a superantigen mediated process in Kawasaki disease – comment. Arch Dis Child. 1995;73:275–6. doi: 10.1136/adc.72.4.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung DY, Giorno RC, Kazemi LV, Flynn PA, Busse JB. Evidence for superantigen involvement in cardiovascular injury due to Kawasaki syndrome. J Immunol. 1995;155:5018–21. [PubMed] [Google Scholar]
- 9.White J, Herman A, Pullen AM, Kubo R, Kappler JW, Marrack P. The Vβ-specific superantigen straphylococcal enterotoxin B: stimulation of mature T cells and clonal deletion in neonatal mice. Cell. 1989;56:27–35. doi: 10.1016/0092-8674(89)90980-x. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalo JA, de Alboran IM, Kroemer G. Dissociation of autoaggression and self-superantigen reactivity [Editorial] Scand J Immunol. 1993;37:1–6. doi: 10.1111/j.1365-3083.1993.tb01657.x. [DOI] [PubMed] [Google Scholar]
- 11.Bette M, Schafer MK, van Rooijen N, Weihe E, Fleischer B. Distribution and kinetics of superantigen-induced cytokine gene expression in mouse spleen. J Exp Med. 1993;178:1531–9. doi: 10.1084/jem.178.5.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conrad B, Weidmann E, Trucco G, et al. Evidence for superantigen involvement in insulin-dependent diabetes mellitus aetiology. Nature. 1994;371:351–5. doi: 10.1038/371351a0. [DOI] [PubMed] [Google Scholar]
- 13.Paliard X, West SG, Lafferty JA, et al. Evidence for the effects of a superantigen in rheumatoid arthritis. Science. 1991;253:325–9. doi: 10.1126/science.1857971. [DOI] [PubMed] [Google Scholar]
- 14.Brocke S, Gaur A, Piercy C, et al. Induction of relapsing paralysis in experimental autoimmune encephalomyelitis by bacterial superantigen. Nature. 1993;365:642–4. doi: 10.1038/365642a0. [DOI] [PubMed] [Google Scholar]
- 15.Yeung RSM. The etiology of Kawasaki disease – a superantigen-mediated process. Prog Pediatr Cardiol. 2004;19:109–13. [Google Scholar]
- 16.Gruber BL, Sorbi D, French DL, et al. Markedly elevated serum MMP-9 (gelatinase B) levels in rheumatoid arthritis: a potentially useful laboratory marker. Clin Immunol Immunopathol. 1996;78:161–71. doi: 10.1006/clin.1996.0025. [DOI] [PubMed] [Google Scholar]
- 17.Gavin PJ, Crawford SE, Shulman ST, Garcia FL, Rowley AH. Systemic arterial expression of matrix metalloproteinases 2 and 9 in acute Kawasaki disease. Arterioscler Thromb Vasc Biol. 2003;23:576–81. doi: 10.1161/01.ATV.0000065385.47152.FD. [DOI] [PubMed] [Google Scholar]
- 18.Cossins JA, Clements JM, Ford J, et al. Enhanced expression of MMP-7 and MMP-9 in demyelinating multiple sclerosis lesions. Acta Neuropathol. 1997;94:590–8. doi: 10.1007/s004010050754. [DOI] [PubMed] [Google Scholar]
- 19.Lehman TJ, Walker SM, Mahnovski V, McCurdy D. Coronary arteritis in mice following the systemic injection of group B Lactobacillus casei cell walls in aqueous suspension. Arthritis Rheum. 1985;28:652–9. doi: 10.1002/art.1780280609. [DOI] [PubMed] [Google Scholar]
- 20.Duong TT, Silverman ED, Bissessar MV, Yeung RSM. Superantigenic activity is responsible for induction of coronary arteritis in mice: an animal model of Kawasaki disease. Int Immunol. 2003;15:79–89. doi: 10.1093/intimm/dxg007. [DOI] [PubMed] [Google Scholar]
- 21.Hui-Yuen JS, Duong TT, Yeung RS. TNF-{alpha} is necessary for induction of coronary artery inflammation and aneurysm formation in an animal model of Kawasaki disease. J Immunol. 2006;176:6294–301. doi: 10.4049/jimmunol.176.10.6294. [DOI] [PubMed] [Google Scholar]
- 22.Lau AC, Duong TT, Ito S, Yeung RS. Matrix metalloproteinase 9 activity leads to elastin breakdown in an animal model of Kawasaki disease. Arthritis Rheum. 2008;58:854–63. doi: 10.1002/art.23225. [DOI] [PubMed] [Google Scholar]
- 23.LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. JAMA. 1999;282:2340–6. doi: 10.1001/jama.282.24.2340. [DOI] [PubMed] [Google Scholar]
- 24.Huang SM, Weng KP, Chang JS, Lee WY, Huang SH, Hsieh KS. Effects of statin therapy in children complicated with coronary arterial abnormality late after Kawasaki disease. Circ J. 2008;72:1583–7. doi: 10.1253/circj.cj-08-0121. [DOI] [PubMed] [Google Scholar]
- 25.Wick G, Schett G, Amberger A, Kleindienst R, Xu Q. Is atherosclerosis an immunologically mediated disease? Immunol Today. 1995;16:27–33. doi: 10.1016/0167-5699(95)80067-0. [DOI] [PubMed] [Google Scholar]
- 26.Liao JK. Isoprenoids as mediators of the biological effects of statins. J Clin Invest. 2002;110:285–8. doi: 10.1172/JCI16421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mackay DJ, Hall A. Rho GTPases. J Biol Chem. 1998;273:20685–8. doi: 10.1074/jbc.273.33.20685. [DOI] [PubMed] [Google Scholar]
- 28.Lau AC, Duong TT, Ito S, Wilson GJ, Yeung RSM. Inhibition of matrix metalloproteinase-9 activity improves coronary outcome in an animal model of Kawasaki disease. Clin Exp Immunol. 2009;157:300–9. doi: 10.1111/j.1365-2249.2009.03949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeung RS. Kawasaki disease: update on pathogenesis. Curr Opin Rheumatol. 2010;22:551–60. doi: 10.1097/BOR.0b013e32833cf051. [DOI] [PubMed] [Google Scholar]
- 30.Yeung RS. Pathogenesis and treatment of Kawasaki's disease. Curr Opin Rheumatol. 2005;17:617–23. doi: 10.1097/01.bor.0000174184.15901.ee. [DOI] [PubMed] [Google Scholar]
- 31.Blank N, Schiller M, Krienke S, et al. Atorvastatin inhibits T cell activation through 3-hydroxy-3-methylglutaryl coenzyme A reductase without decreasing cholesterol synthesis. J Immunol. 2007;179:3613–21. doi: 10.4049/jimmunol.179.6.3613. [DOI] [PubMed] [Google Scholar]
- 32.Kuipers HF, van den Elsen PJ. Immunomodulation by statins: inhibition of cholesterol vs. isoprenoid biosynthesis. Biomed Pharmacother. 2007;61:400–7. doi: 10.1016/j.biopha.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Jain J, Valge-Archer VE, Rao A. Analysis of the AP-1 sites in the IL-2 promoter. J Immunol. 1992;148:1240–50. [PubMed] [Google Scholar]
- 34.Koike T, Yamagishi H, Hatanaka Y, et al. A novel ERK-dependent signaling process that regulates interleukin-2 expression in a late phase of T cell activation. J Biol Chem. 2003;278:15685–92. doi: 10.1074/jbc.M210829200. [DOI] [PubMed] [Google Scholar]
- 35.Schafer PH, Wang L, Wadsworth SA, Davis JE, Siekierka JJ. T cell activation signals up-regulate p38 mitogen-activated protein kinase activity and induce TNF-alpha production in a manner distinct from LPS activation of monocytes. J Immunol. 1999;162:659–68. [PubMed] [Google Scholar]
- 36.Moon SK, Cha BY, Kim CH. ERK1/2 mediates TNF-alpha-induced matrix metalloproteinase-9 expression in human vascular smooth muscle cells via the regulation of NF-kappaB and AP-1: involvement of the ras dependent pathway. J Cell Physiol. 2004;198:417–27. doi: 10.1002/jcp.10435. [DOI] [PubMed] [Google Scholar]
- 37.Lau AC, Duong TT, Ito S, Yeung RSM. Intravenous immunoglobulin and salicylate differentially modulate pathogenic processes leading to vascular damage in an animal model of Kawasaki disease. Arthritis Rheum. 2009;60:2131–41. doi: 10.1002/art.24660. [DOI] [PubMed] [Google Scholar]
- 38.de Jongh S, Ose L, Szamosi T, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized, double-blind, placebo-controlled trial with simvastatin. Circulation. 2002;106:2231–7. doi: 10.1161/01.cir.0000035247.42888.82. [DOI] [PubMed] [Google Scholar]
- 39.Knipscheer HC, Boelen CC, Kastelein JJ, et al. Short-term efficacy and safety of pravastatin in 72 children with familial hypercholesterolemia. Pediatr Res. 1996;39:867–71. doi: 10.1203/00006450-199605000-00021. [DOI] [PubMed] [Google Scholar]
- 40.Wiegman A, Hutten BA, de Groot E, et al. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. JAMA. 2004;292:331–7. doi: 10.1001/jama.292.3.331. [DOI] [PubMed] [Google Scholar]
- 41.McCrindle BW, Ose L, Marais AD. Efficacy and safety of atorvastatin in children and adolescents with familial hypercholesterolemia or severe hyperlipidemia: a multicenter, randomized, placebo-controlled trial. J Pediatr. 2003;143:74–80. doi: 10.1016/S0022-3476(03)00186-0. [DOI] [PubMed] [Google Scholar]
- 42.Stern RH, Yang BB, Hounslow NJ, MacMahon M, Abel RB, Olson SC. Pharmacodynamics and pharmacokinetic–pharmacodynamic relationships of atorvastatin, an HMG-CoA reductase inhibitor. J Clin Pharmacol. 2000;40:616–23. [PubMed] [Google Scholar]
- 43.Nagashima H, Aoka Y, Sakomura Y, et al. A 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor, cerivastatin, suppresses production of matrix metalloproteinase-9 in human abdominal aortic aneurysm wall. J Vasc Surg. 2002;36:158–63. doi: 10.1067/mva.2002.123680. [DOI] [PubMed] [Google Scholar]
- 44.Evans J, Powell JT, Schwalbe E, Loftus IM, Thompson MM. Simvastatin attenuates the activity of matrix metalloprotease-9 in aneurysmal aortic tissue. Eur J Vasc Endovasc Surg. 2007;34:302–3. doi: 10.1016/j.ejvs.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 45.Lau A, Rosenberg H, Duong T, McCrindle B, Yeung R. Matrix metalloproteinases and coronary outcome in children with Kawasaki disease. Pediatr Res. 2007;61:710–15. doi: 10.1203/pdr.0b013e318053418b. [DOI] [PubMed] [Google Scholar]
- 46.Newburger JW, Burns JC, Beiser AS, Loscalzo J. Altered lipid profile after Kawasaki syndrome. Circulation. 1991;84:625–31. doi: 10.1161/01.cir.84.2.625. [DOI] [PubMed] [Google Scholar]
- 47.Ikemoto Y, Ogino H, Teraguchi M, Kobayashi Y. Evaluation of preclinical atherosclerosis by flow-mediated dilatation of the brachial artery and carotid artery analysis in patients with a history of Kawasaki disease. Pediatr Cardiol. 2005;26:782–6. doi: 10.1007/s00246-005-0921-8. [DOI] [PubMed] [Google Scholar]
- 48.Salo E, Pesonen E, Viikari J. Serum cholesterol levels during and after Kawasaki disease. J Pediatr. 1991;119:557–61. doi: 10.1016/s0022-3476(05)82404-7. [DOI] [PubMed] [Google Scholar]
- 49.Cabana VG, Gidding SS, Getz GS, Chapman J, Shulman ST. Serum amyloid A and high density lipoprotein participate in the acute phase response of Kawasaki disease. Pediatr Res. 1997;42:651–5. doi: 10.1203/00006450-199711000-00017. [DOI] [PubMed] [Google Scholar]
- 50.Niboshi A, Hamaoka K, Sakata K, Yamaguchi N. Endothelial dysfunction in adult patients with a history of Kawasaki disease. Eur J Pediatr. 2008;167:189–96. doi: 10.1007/s00431-007-0452-9. [DOI] [PubMed] [Google Scholar]
- 51.McCrindle B. A childhood disease with important consequences into adulthood. Circulation. 2009;120:6–8. doi: 10.1161/CIRCULATIONAHA.109.874800. [DOI] [PubMed] [Google Scholar]
- 52.Cheung YF, Wong SJ, Ho MHK. Relationship between carotid intima-media thickness and arterial stiffness in children after Kawasaki disease. Arch Dis Child. 2007;92:43–7. doi: 10.1136/adc.2006.096628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dalla Pozza R, Bechtold S, Urschel S, Kozlik-Feldmann R, Netz H. Subclinical atherosclerosis, but normal autonomic function after Kawasaki disease. J Pediatr. 2007;151:239–43. doi: 10.1016/j.jpeds.2007.03.057. [DOI] [PubMed] [Google Scholar]
- 54.Lehman TJ, Warren R, Gietl D, Mahnovski V, Prescott M. Variable expression of Lactobacillus casei cell wall-induced artertis: an animal model of Kawasaki's disease in selected inbred mouse strains. Clin Immunol Immunopathol. 1988;48:108–18. doi: 10.1016/0090-1229(88)90161-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.