

Abstract
Acute lung injury is characterized by a diffuse inflammatory parenchymal process, implicated in the context of significant morbidity and mortality. Previously, we have reported that soluble ST2 (sST2), a member of the Toll-interleukin (IL)-1 receptor (TIR) superfamily, represses proinflammatory cytokine production of macrophage exposed to lipopolysaccharide (LPS). In this study, we examined the possibility of modulating LPS-induced murine inflammatory pulmonary damage by recombinant adenovirus-mediated sST2-Fc (Ad-sST2-Fc) gene transfer. Single intranasal administration of Ad-sST2-Fc led to a profound decrease in LPS-induced bronchoalveolar lavage leucocyte exudation and lung tissue myeloperoxidase activity (reflecting phagocyte infiltration). Histological examination revealed alveolitis with inflammatory cell infiltration and alveolar haemorrhage in the alveolar airspace was less severe in Ad-sST2-Fc-treated mice when compared with control groups. In addition, high levels of sST2-Fc in vivo reduced the transcription of tumour necrosis factor-α, IL-6 and Toll-like receptor-4 gene remarkably, and suppressed the nuclear translocation of nuclear factor-κB in lung tissues in response to LPS challenge. Taken together, these results suggested that administration of Ad-sST2-Fc gene transfer may have therapeutic potential for the immunomodulatory treatment of LPS-mediated inflammatory lung injury.
Keywords: acute lung injury, adenoviral vector, inflammation, NF-κB, soluble ST2
Introduction
Acute lung injury (ALI) and its severest form, acute respiratory distress syndrome (ARDS), are related closely to the development of multiple organ dysfunction syndrome (MODS), which plays an essential role in the death of patients with sepsis, shock, trauma, ischaemia/reperfusion and pancreatitis [1–4]. The pathogenesis of ALI/ARDS involves the presence of significant vascular leakage, with movement of fluid and macromolecules into the airspaces and lung parenchyma, events that are directly responsible for the serious physiological derangements characteristic of this disorder. Moreover, lung microvascular barrier dysfunction increases the transendothelial diapedesis of inflammatory cells into lung tissues, further exacerbating microvascular injury and diffuse alveolar damage with intrapulmonary haemorrhage, oedema and fibrin deposition. Despite intense research and diverse therapeutic trials, there are still no effective therapeutic strategies in preventing or reversing the severe pulmonary inflammation [3–5].
The ST2/T1 gene was identified originally as an oncogene and serum-responsive gene expressed in mouse fibroblasts [6]. Four distinct types of ST2 molecules, the soluble form (sST2), the transmembrane form (ST2L) and two variant forms (ST2V and ST2LV), are recognized to be generated by alterative splicing [7–9]. Recently the natural ligand for ST2L was discovered and named interleukin (IL)-33 by Schmitz et al. [10]. Among ST2 gene products, soluble ST2 has been known to be correlated with various immune disorders. Elevated levels of soluble ST2 have been observed in patients with such conditions as autoimmune diseases [11], trauma [12], dengue virus infection [13] and myocardial infarction, especially on acute exacerbation [14]. The functional role of soluble ST2 in vivo has not been elucidated fully. Several experimental studies have revealed that administration of a recombinant soluble ST2-Fc fusion protein or a soluble ST2 vector attenuates inflammatory responses in allergic airway inflammation [15] and decreases inflammation and lethality in intestinal and hepatic ischaemia/reperfusion injury [16,17]. In addition, soluble ST2 pretreatment was shown to suppress the production of inflammatory cytokines and mortality in a mouse endotoxin shock model [18].
Given that ST2 possibly contributes to the modulation of immune response during severe inflammation, we hypothesized that high levels of soluble ST2 in vivo could improve LPS-induced inflammatory pulmonary injury. Therefore, we constructed a recombinant replication-deficient adenovirus encoding soluble ST2-human immunoglobulin (Ig)G1 Fc (sST2-Fc) fusion protein, and investigated its beneficial effect in a murine model of lipopolysaccharide (LPS)-induced ALI. We report here that the expression of the adenoviral vector encoding sST2-Fc in vivo prevents lung tissue damage and suppresses the activation of inflammatory cascades. Furthermore, the protective effect of sST2-Fc in ALI was correlated with down-regulation of the Toll-like receptor (TLR)-4 signalling pathway.
Materials and methods
Animals
Male BALB/c mice, 8–12 weeks old, were obtained from the Center of Experimental Animals of Chinese Academy of Medical Science and Center of Medical Experimental Animals of Hubei Province, and maintained in an animal facility under pathogen-free conditions. All studies involving mice were approved by the Guangdong Pharmaceutical University Animal Care and Use Committee.
Construction and preparation of E1/E3-defective adenovirus vectors
The E1/E3-defective adenoviral vector encoding murine soluble ST2-human IgG1 Fc (Ad-sST2-Fc) was prepared using HEK293 cells and the AdEasy system, as described elsewhere [19]. The cDNA fragment coding for the mouse soluble ST2 gene was cloned by reverse transcription polymerase chain reaction (RT-PCR) from mouse spleen, and coupled to the Fc portion of human IgG1. The two sequences were subcloned into the shuttle plasmid [pAdTrack-cytomegalovirus (CMV)] to produce the adenovirus vectors. After homologous recombination between the shuttle plasmids and an adenoviral backbone plasmid (pAdEasy-1) in Escherichia coli BJ5183 cells, E1/E3-defective adenoviral vectors encoding sST2-Fc were prepared in HEK293 cells. After several rounds of passage in HEK293 cells, the adenovirus vectors were purified using two rounds of caesium chloride (CsCl) density gradient centrifugation. An adenovirus expressing green fluorescence protein (Ad-EGFP) was also constructed and used as a control vector. The concentrated viruses were dialysed in phosphate-buffered saline (PBS) and the titres of the purified adenovirus were determined using green fluorescent protein (GFP) assay in the HEK293 cells. The prepared adenovirus was stored at −80°C.
LPS-induced ALI model
After BALB/c mice were dieghylether-anaesthetized, 50 µg LPS (Escherichia coli O111:B4; Sigma, St Louis, MO, USA) was administrated intranasally (i.n.) in 50 µl PBS to induce lung injury. Control mice were given 50 µl PBS i.n. without LPS, as described previously [20]. Ad-sST2-Fc [5 × 108 plaque-forming units (pfu)/mice] or vehicle (Ad-EGFP) was administered i.n. 48 h prior to LPS administration, bronchoalveolar lavage fluid (BALF) was performed by intratracheal instillation, and then the lungs were lavaged five times with 0·8 ml of sterile PBS. Recovered fluid from each sample was centrifuged (800 g, 5 min at 4°C) to pellet cells. The supernatant was stored at −80°C for cytokine analysis, and the cell pellet was resuspended in PBS and counted with a haemocytometer.
ST2-Fc protein measurement
The levels of ST2-Fc in the lung tissue were measured by enzyme-linked immunosorbent assay (ELISA) as described previously [17]. Briefly, microtitre wells were coated with horse anti-human IgG (25 µg/ml; SIBP, Shanghai, China) in 50 mM carbonate buffer pH 9·6 overnight at 4°C and rinsed three times with washing buffer (PBS containing 0·05% Tween-20). Microtitre wells were blocked with 10% foetal calf serum (FCS) for 3 h at 37°C. After supernatant of BALF was added and incubated for 1 h at 37°C, the plates were washed five times with the washing buffer and incubated for 1 h with a 1:3000 horseradish peroxidase (HRP)-conjugated anti-human IgG (ZhongShan Biotechnology, Beijing, China) in the blocking buffer. After washing, plates were incubated with 3,3′,5,5′,-tetramethylbenzidine (TMB) subtracted for 15 min and the reaction was stopped with the addition of 2 N H2SO4. Optical densities (OD) at 450 nm wavelengths were determined by using a Microplate Reader (Labsystems, Helsinki, Finland).
Enzyme-linked immunosorbent assay (ELISA)
Concentrations of tumour necrosis factor (TNF)-α and interleukin (IL)-6 in mouse BALF were determined with ELISA kits from eBioscience (San Diego, CA, USA), performed according to the manufacturer's instructions.
Myeloperoxidase assay
Measurement of myeloperoxidase (MPO) activity of the lung was performed as described previously [21]. Briefly, the lung tissues were homogenized and centrifuged (30 000 g, 30 min at 4°C). Pellets were resuspended in extraction buffer (0·05 M potassium phosphate buffer dissolved in 0·5% hexadecyltrimethylammonium bromide) and then freeze–thawed three times. The supernatants collected (13 000 g, 15 min at 4°C) were assayed for MPO activity by measuring the change in optical density at 460 nm using kinetic readings over 3 min (200 µl sample with 800 µl reaction buffer containing 0·05 M potassium phosphate buffer, 0·167 mg/ml of O-dianisidine dihydrochloride, and 0·0006% H2O2). Sample protein concentrations were determined [bicinchoninic acid (BCA) assay], and the results are presented as MPO units per milligram of protein.
Histochemistry
Histopathological evaluation was performed on animals that were not subjected to BAL. Lungs were inflated and fixed with 10% buffered formalin. Samples were embedded in paraffin, and then tissue sections (4 µm thick) were stained with haematoxylin and eosin (H&E) and examined under light microscope.
Real-time quantitative RT-PCR (qRT-PCR)
Total RNA was extracted from the lung tissues using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. After removal of potentially contaminating DNA with DNase I (Invitrogen), cDNA was synthesized from 2 µg total RNA using a first-strand cDNA synthesis kit (MBI Fermentas Inc., Burlington, ON, USA). PCR mixture was prepared using SYBR Green qPCR kit (Invitrogen) using the primers as follows: TNF-α (forward 5′-CATCTTCTCAAAATTCGAGTGACAA-3′, reverse 5′-TGGGAGTAG ACAAGGTACAACCC-3′), IL-6 (forward 5′-GAGACTTCCATCCAGTTGCC-3′, reverse 5′-AAGTGCATCATCGTTGTTCATACA-3′), TLR-4 (forward 5′-CTGTATTCCCTCAGCACTCTTG-3′, reverse 5′-CCCACTGCAGGAATTTCTGATG-3′) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward 5′-TTCACCACCATGGAGAAGGC-3′, reverse 5′-GGCATGGACTGTGGTCATGA-3′). All qRT–PCR reactions were performed with an ABI PRISM® 7000 Sequence Detector Systems (Applied Biosystems, Foster City, CA, USA), and each gene expression was normalized with GAPDH mRNA content.
Electrophoretic mobility shift assay (EMSA)
Nuclear factor (NF)-κB DNA-binding activity was measured by EMSA using nuclear extracts from lung tissues. The biotin 5′ end-labelled duplex NF-κB oligonucleotide (5′-AGTTGAGGGGACTTTCCCAGGC-3′) was used as the probe, according to the LightShift Chemiluminescent EMSA (Pierce Biotechnology Inc., Rockford, IL, USA). Incubations were performed in a buffer containing 10 mM Tris (pH 7·6), 2·5% glycerol, 0·05% Nonidet P-40 and 50 ng/µl of polydeoxyinosinic-deoxycytidylic acid [poly(dI·dC)]. This reaction is then subjected to gel electrophoresis on a native (6%) polyacrylamide gel and transferred to a nylon membrane. The biotin end-labelled DNA is detected using the peroxidase-conjugated streptavidin system. For the cold competition assay, unlabelled probe was added in a 500-fold molar excess.
Statistical analysis
Results were analysed by using Student's t-test or one-way analysis of variance (anova). All data are expressed as the mean ± standard error of the mean (s.e.m.). Differences were considered to be statistically significant when P < 0·05.
Results
Expression of sST2-Fc after Ad-sST2-Fc administration in the mouse lung
In order to elucidate the immunomodulatory effect of soluble ST2 in LPS-induced pulmonary injury, we first examined whether exogenous administration of sST2-Fc by transgene delivery could induce sST2-Fc expression in the mouse lung. We performed initial experiments with recombinant adenovirus encoding sST2-Fc (Ad-sST2-Fc) to determine the timing of sST2-Fc expression after intranasal instillation. sST2-Fc protein expression levels were measured by a specific ELISA of BALF from day 1 to day 6 after in vivo Ad-sST2-Fc administration (5 × 108 pfu/mice). As shown in Fig. 1a, the levels of sST2-Fc protein increased as early as day 1, with peak level at day 2 after sST2-Fc gene delivery. However, there was no detectable sST2-Fc protein in the non-transfected mice, nor was any sST2-Fc detected in the Ad-EGFP group.
Fig. 1.
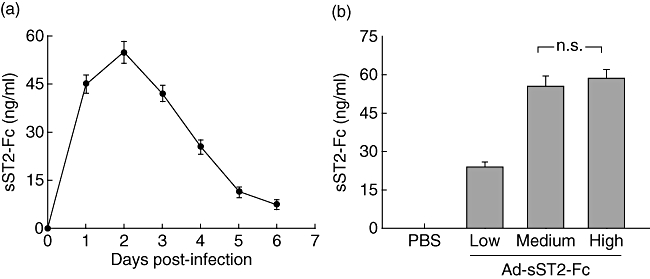
Expression of recombinant adenovirus-mediated soluble ST2 (Ad-sST2-Fc) in vivo. (a) Ad-sST2-Fc was administered intranasally to BALB/c mice at a dose of 5 × 108 plaque-forming units (pfu)/mice. Time–course of sST2-Fc expression in bronchoalveolar lavage fluid (BALF) was measured by enzyme-linked immunosorbent assay (ELISA). (b) Animals were treated with different virus doses (high: 1 × 109 pfu/mice; medium: 5 × 108 pfu/mice; low: 1 × 108 pfu/mice). Bronchoalveolar lavage was performed 2 days later after viral administration and sST2-Fc expression in BALF was determined by ELISA. Data are presented as the mean ± standard error of the mean (n = 4–6 in each group); n.s.: not significant.
Because high doses of adenoviral vectors are known to cause pulmonary inflammation, we performed dose–range experiments with a high dose of Ad-sST2-Fc (1 × 109 pfu/mice), a medium dose of Ad-sST2-Fc (5 × 108 pfu/mice) or a low dose of Ad-sST2-Fc (1 × 108 pfu/mice) by intranasal delivery. The expression levels of sST2-Fc protein were assessed on day 2 later after adenoviral administration. sST2-Fc concentrations in BALF did not show significant differences between high- and medium-dose groups (Fig. 1b). However, there was more evidence of vascular congestion and hypersecretion of mucous in bronchioles in the high-dose group compared with that of the medium-dose group, as shown by lung H&E staining (data not shown). Therefore, all subsequent studies were performed with 5 × 108 pfu, a dose that is able to induce strong sST2-Fc expression without triggering an inflammatory response.
sST2-Fc overexpression attenuates LPS-induced inflammatory lung injury
Subsequently, we identified further the protective effect of sST2-Fc on severe inflammatory response by using a murine model of LPS-induced acute lung injury. Consistent with previous reports [4,22], LPS treatment resulted in marked proinflammatory alterations characterized by neutrophil infiltration, oedema of the alveolar septa and alveolar haemorrhage at 24 h (Fig. 2a). These findings were determined clearly by blinded pathological examination and were supported by measuring the number of neutrophils in the BALF (Fig. 2b) and assessing the MPO activity of the lung (Fig. 2c) from mice exposed to LPS when compared with PBS-challenged control mice. In contrast, pretreatment of mice with Ad-sST2-Fc dramatically reduced the lung histopathological damage, BAL neutrophilia and lung MPO activity that followed LPS exposure (Fig. 2). As expected, Ad-EGFP pretreatment did not alleviate these pathological changes after LPS challenge.
Fig. 2.
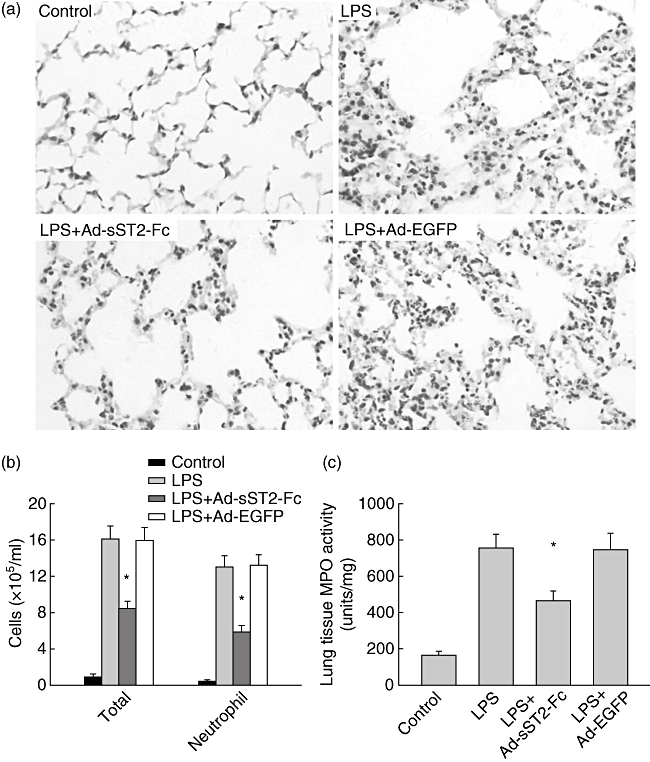
Pretreatment with adenovirus-mediated soluble ST2 (Ad-sST2-Fc) attenuates lung tissue damage in mice challenged with lipopolysaccharide (LPS). (a) Lungs from each experimental group were processed for histological examination after haematoxylin and eosin staining (400×). Compared with control mice, mice exposed to intranasal LPS alone or LPS + adenovirus expressing green fluorescence protein (Ad-EGFP) for 24 h led to profound neutrophil infiltration and alveolar haemorrhage. These features were decreased dramatically in mice pretreated with Ad-sST2-Fc prior to LPS challenge. (b) Bronchoalveolar lavage fluid (BALF) was collected and cell differentiation was determined at 24 h after LPS challenge. Exposure to intranasal LPS markedly enhanced the total cells and percentage of neutrophils that was attenuated in mice pretreated with Ad-sST2-Fc. (c) Myeloperoxidase (MPO) content was also assessed in lung tissue homogenates, as described in Materials and methods. Exposure to LPS resulted in a significant increase in lung MPO activity assessed at 24 h after intranasal LPS administration. The level of tissue MPO activity was reduced markedly in mice pretreated with Ad-sST2-Fc. Data are presented as the mean ± standard error of the mean (n = 4–6 in each group). *P < 0·05 versus the LPS group.
sST2-Fc decreases the production of proinflammatory cytokines
Next, we continued to determine the effect of sST2-Fc overexpression on the production of proinflammatory cytokines such as TNF-α and IL-6, which is known to be involved in the pathophysiology of LPS-mediated pulmonary inflammation. The transcription of TNF-α and IL-6 in lung tissues was determined by real-time PCR. As shown in Fig. 3a, animals exposed to LPS resulted in an enhanced expression of TNF-α and IL-6 mRNA at 3 h compared with a control group. In contrast, animals that pretreated with Ad-sST2-Fc drastically reduced TNF-α and IL-6 mRNA levels following LPS challenge. Similarly, overexpression of sST2-Fc also decreased significantly LPS-induced TNF-α and IL-6 protein in BALF at 6 h compared with control mice (Fig. 3b). Again, pretreatment with Ad-EGFP had no inhibitory effect on the production of both of the proinflammatory cytokines.
Fig. 3.
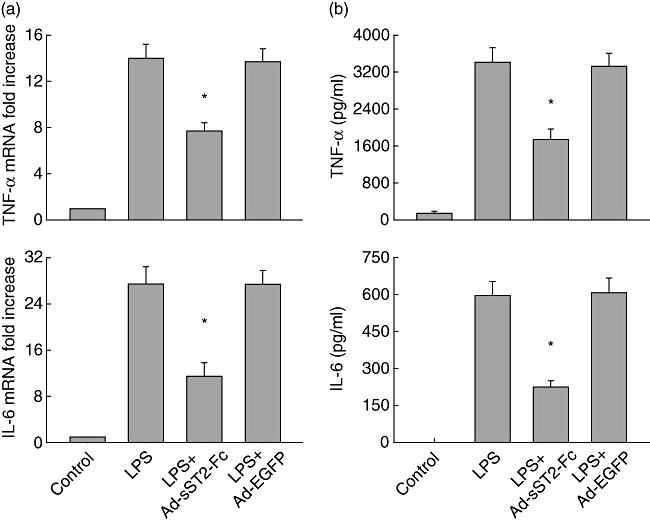
Overexpression of soluble ST2 (sST2-Fc) decreases the production of proinflammatory cytokines. (a) Lung tumour necrosis factor (TNF)-α and interleukin (IL)-6 mRNA levels were determined in mice pretreated with adenovirus-mediated sST2-Fc (Ad-sST2-Fc) following lipopolysaccharide (LPS) challenge at 3 h. Results were obtained using real-time reverse transcription–polymerase chain reaction and expressed as relative increase of mRNA expression compared with control animals. (b) The levels of TNF-α and IL-6 protein in bronchoalveolar lavage fluid (BALF) were also evaluated by enzyme-linked immunosorbent assay in mice after LPS stimulation at 6 h. Data are presented as the mean ± standard error of the mean (n = 4–6 in each group). *P < 0·05 versus the LPS group.
sST2-Fc suppresses TLR-4 levels in LPS-inflamed mouse lungs
Increasing evidence shows that the TLR-4-associated pathway plays a crucial role in the LPS-stimulated inflammatory response in vivo and in vitro[23]. Therefore, we sought to observe whether the protection of sST2-Fc on LPS-induced acute lung injury was correlated with TLR-4 signalling. TLR-4 mRNA levels were examined using real-time PCR in lung tissues after LPS challenge. The results showed that animals exposed to LPS led to an increased expression of TLR-4 mRNA at 3 h compared with the negative control group (Fig. 4). In contrast, animals that were pretreated with Ad-sST2-Fc decreased TLR-4 mRNA levels remarkably following LPS stimulation. Also, pretreatment with Ad-EGFP did not show an inhibitory effect on the expression of TLR-4 mRNA.
Fig. 4.
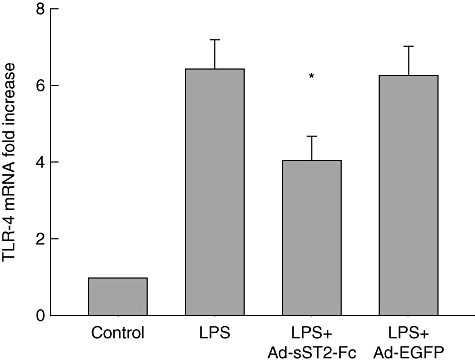
Overexpression of soluble ST2 (sST2-Fc) suppresses the transcription of Toll-like receptor (TLR)-4. Lung TLR-4 mRNA expression was measured in mice pretreated with adenovirus-mediated sST2-Fc (Ad-sST2-Fc) followed by lipopolysaccharide (LPS) exposure at 3 h. Results were obtained using real-time reverse transcription–polymerase chain reaction and expressed as relative increase of mRNA expression compared with control animals. Data are presented as the mean ± standard error of the mean (n = 4–6 in each group). *P < 0·05 versus the LPS group.
sST2-Fc regulates the inflammatory signalling pathway
NF-κB is an essential transcription factor implicated in the signal transduction of a variety of extracellular stress stimuli [24]. It has been proved that NF-κB is activated following LPS-exposed acute pulmonary injury and modulates both inflammatory and protective responses in the lung [25]. To investigate the potential molecular mechanism of sST2-Fc suppressing the gene expression of proinflammatory cytokines, the activation of NF-κB was measured by EMSA. We found that NF-κB DNA binding activity was increased significantly in the lung tissue at 3 h in LPS-challenged mice compared with control mice (Fig. 5). However, mice pre-administered with Ad-sST2-Fc exhibited less NF-κB DNA binding activity. The specificity of the NF-κB bands was identified by cold competition analysis in the presence of excess unlabelled NF-κB consensus motif (Fig. 5).
Fig. 5.
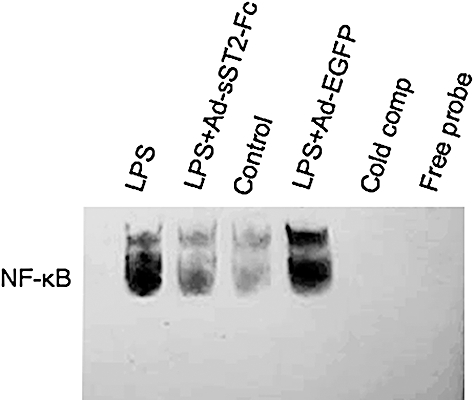
High levels of soluble ST2 (sST2-Fc) in vivo inhibit nuclear factor-κB activation. Mice werepretreated with adenovirus-mediated sST2-Fc (Ad-sST2-Fc) following lipopolysaccharide (LPS) challenge. Nuclear extracts were prepared at 3 h from the lung tissues and subjected to electrophoretic mobility shift assay. Assay shown is representative of three experiments with similar results.
Discussion
The accumulated evidence has shown that enhanced levels of soluble ST2 protein provide a protective effect in idiopathic pulmonary fibrosis, septic shock, asthma and ischaemia/reperfusion [15,16,18,26]. As is already known, the pathogenesis of ALI is complex and the molecular mechanisms have been investigated extensively using distinct animal models. Intranasal instillation of LPS induces a more reasonable experimental model of ALI, as it results in lung injury without causing systemic inflammation and multiple organ failure [20]. Meanwhile, the precise mechanisms by which soluble ST2 affords protection in inflammatory pulmonry damage need to be illustrated further.
The results of this study have demonstrated that overexpression of sST2-Fc fusion protein transfused with adenoviral vector in vivo suppressed dramatically the pulmonary vascular leakage and MPO activity, as well as leucocyte influx in BALF. The histopathological examination indicated that inflammatory cell infiltration and alveolar haemorrhage were less severe in Ad-sST2-Fc-treated mice than control groups after LPS challenge, accompanied by the immediate down-modulation of proinflammatory cytokines TNF-α and IL-6 expression. Furthermore, we found that high levels of sST2-Fc protein repressed considerably the expression of the TLR-4 gene and the translocation of transcription factor NF-κB to the nucleus, and this may be part of the mechanisms whereby sST2-Fc elicits its salutary effects.
Several studies have shown that inflammatory cell sequestration into the pulmonary microvasculature and migration into the lung parenchyma is critical for host defence, but also contributes to the development of ALI [27]. The characteristic pulmonary hypertension and increased vascular permeability to endotoxin are abrogated if the animals are subjected to intravascular macrophage depletion or administration of detergent disposal [28,29]. Intriguingly, Fagundes and colleagues [16] found recently that treatment with soluble ST2 suppressed remarkably the accumulation of neutrophils into lung tissues after reperfusion through inhibiting the production of CCL2 and CXCL1, which are well-characterized mediators of leucocyte migration. Consistent with earlier reports [22,30], our results indicated that significant recruitment of neutrophils was observed in lung tissue by pathological examination in the murine model of LPS-induced ALI. LPS also caused an increase in the number of inflammatory cell extravasation in BALF. Moreover, MPO activity, which reflects neutrophil and macrophage diapedesis, in pulmonary tissue was elevated markedly followed by LPS challenge. Conversely, pretreatment with Ad-sST2-Fc remarkably reversed the inflammatory histological changes in lung tissues produced by LPS stimulation, as well as suppressed LPS-induced BALF neutrophilia and MPO activity (Fig. 2). These findings confirmed that the protective effect of sST2-Fc protein in ALI is related to attenuation of leucocyte sequestration and migration into the lung tissue.
Proinflammatory cytokines appear rapidly in the early phase of inflammatory response and play a pivotal role in ALI and acute respiratory distress syndrome (ARDS) [4,31]. The increased levels of TNF-α, Il-1β and IL-6 in BALF are noted in ARDS patients, and the persistent elevation of proinflammatory cytokines in humans with ALI or sepsis has been linked to a worse outcome [32]. Some evidence indicates that soluble ST2 could act directly as an anti-inflammatory mediator through a mechanism that involves the inhibition of TLRs, especially TLR-4 and TLR-2 signalling by sequestration of MyD88 and Mal adapter proteins [18,33]. In vitro and in vivo experiments have demonstrated that sST2-Fc protein decreases the production of proinflammatory cytokines in septic shock and hepatic ischaemia/reperfusion [17,18]. Other evidence shows that soluble ST2 could also be acting as a decoy receptor for IL-33 regulating its biological function. The interaction of IL-33 with its receptor ST2L leads to the induction of the Th2 cytokines IL-4, IL-5 and IL-13 through a signalling mechanism that involves the activation of NF-κB and mitogen-activated protein (MAP) kinases [34]. Hence, the inhibitory effect of soluble ST2 may be due to its ability to trap IL-33 and prevent it from binding to ST2L on the surface of Th2 cells and mast cells. In the present study, we found that enhanced levels of sST2-Fc protein in vivo profoundly suppresses the gene expression of TNF-α, IL-6 and TLR-4 in lung tissues in response to LPS challenge. Meanwhile, the potential regulatory effect of sST2-Fc on blocking IL-33 signalling could not be negligible in lung inflammatory injury.
To illuminate further the molecular mechanisms of soluble ST2, which exerts anti-inflammatory signals leading to down-regulation of TLR-4 expression, we investigated the transcription factor NF-κB signalling pathway in lung tissue following LPS stimulation. NF-κB is known as a critical nuclear factor required for the production of a mass of proinflammatory cytokines [24,35]. Furthermore, it has been demonstrated that the network of proinflammatory cytokines is regulated by the transcription factor NF-κB in lung inflammation challenged by LPS [36]. Using EMSA, we found that high levels of sST2-Fc protein remarkably suppressed the NF-κB DNA binding activity. Subsequently, this effect led to considerable suppression of TNF-α and IL-6 genes, the expression of which is modulated by NF-κB. Given that a large variety of proinflammatory cytokines regulated by NF-κB are produced primarily by inflammatory cells in the lung, our results consequently suggest that inactivation of inflammatory cells, especially alveolar macrophages and neutrophils, through inhibiting translocation of NF-κB into the nucleus is possibly the potential mechanism of sST2-Fc observed in this study.
In conclusion, these results revealed that soluble ST2 exerts anti-inflammatory and protective function in acute lung injury followed by LPS challenge. Treatment with recombinant adenovirus encoding sST2-Fc in vivo leads to a remarkable reduction in inflammatory cell infiltration and proinflammatory cytokine production in lung tissues. Moreover, the mechanism of action of sST2-Fc is associated closely with negative regulation of TLR-4 signalling cascades followed by inhibition of NF-κB activity.
Acknowledgments
This work was supported by Medical and Scientific Research Foundation of Guangdong Province (A2008314), PhD Research Foundation of Guangdong Pharmaceutical University (2007JCX06) and the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry.
Disclosure
No conflicts of interest to declare.
References
- 1.Steinberg KP, Hudson LD. Acute lung injury and acute respiratory distress syndrome. The clinical syndrome. Clin Chest Med. 2000;21:402–17. doi: 10.1016/s0272-5231(05)70156-8. [DOI] [PubMed] [Google Scholar]
- 2.Piantadosi CA, Schwartz DA. The acute respiratory distress syndrome. Ann Intern Med. 2004;141:460–70. doi: 10.7326/0003-4819-141-6-200409210-00012. [DOI] [PubMed] [Google Scholar]
- 3.Tomashefski JF., Jr Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med. 2000;21:435–66. doi: 10.1016/s0272-5231(05)70158-1. [DOI] [PubMed] [Google Scholar]
- 4.Bhatia M, Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol. 2004;202:145–56. doi: 10.1002/path.1491. [DOI] [PubMed] [Google Scholar]
- 5.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–27. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Werenskiold AK, Hoffmann S, Klemenz R. Induction of a mitogen-responsive gene after expression of the Ha-ras oncogene in NIH 3T3 fibroblasts. Mol Cell Biol. 1989;9:5207–14. doi: 10.1128/mcb.9.11.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yanagisawa K, Takagi T, Tsukamoto T, Tetsuka T, Tominaga S. Presence of a novel primary response gene ST2L, encoding a product highly similar to the interleukin 1 receptor type 1. FEBS Lett. 1993;318:83–7. doi: 10.1016/0014-5793(93)81333-u. [DOI] [PubMed] [Google Scholar]
- 8.Trajkovic V, Sweet MJ, Xu D. T1/ST2–an IL-1 receptor-like modulator of immune responses. Cytokine Growth Factor Rev. 2004;15:87–95. doi: 10.1016/j.cytogfr.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 9.Tago K, Noda T, Hayakawa M, et al. Tissue distribution and subcellular localization of a variant form of the human ST2 gene product, ST2V. Biochem Biophys Res Commun. 2001;285:1377–83. doi: 10.1006/bbrc.2001.5306. [DOI] [PubMed] [Google Scholar]
- 10.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Kuroiwa K, Arai T, Okazaki H, Minota S, Tominaga S. Identification of human ST2 protein in the sera of patients with autoimmune diseases. Biochem Biophys Res Commun. 2001;284:1104–8. doi: 10.1006/bbrc.2001.5090. [DOI] [PubMed] [Google Scholar]
- 12.Brunner M, Krenn C, Roth G, et al. Increased levels of soluble ST2 protein and IgG1 production in patients with sepsis and trauma. Intensive Care Med. 2004;30:1468–73. doi: 10.1007/s00134-004-2184-x. [DOI] [PubMed] [Google Scholar]
- 13.Becerra A, Warke RV, de Bosch N, Rothman AL, Bosch I. Elevated levels of soluble ST2 protein in dengue virus infected patients. Cytokine. 2008;41:114–20. doi: 10.1016/j.cyto.2007.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimpo M, Morrow DA, Weinberg EO, et al. Serum levels of the interleukin-1 receptor family member ST2 predict mortality and clinical outcome in acute myocardial infarction. Circulation. 2004;109:2186–90. doi: 10.1161/01.CIR.0000127958.21003.5A. [DOI] [PubMed] [Google Scholar]
- 15.Oshikawa K, Yanagisawa K, Tominaga S, Sugiyama Y. Expression and function of the ST2 gene in a murine model of allergic airway inflammation. Clin Exp Allergy. 2002;32:1520–6. doi: 10.1046/j.1365-2745.2002.01494.x. [DOI] [PubMed] [Google Scholar]
- 16.Fagundes CT, Amaral FA, Souza AL, et al. ST2, an IL-1R family member, attenuates inflammation and lethality after intestinal ischemia and reperfusion. J Leukoc Biol. 2007;81:492–9. doi: 10.1189/jlb.0606422. [DOI] [PubMed] [Google Scholar]
- 17.Yin H, Huang BJ, Yang H, et al. Pretreatment with soluble ST2 reduces warm hepatic ischemia/reperfusion injury. Biochem Biophys Res Commun. 2006;351:940–6. doi: 10.1016/j.bbrc.2006.10.166. [DOI] [PubMed] [Google Scholar]
- 18.Sweet MJ, Leung BP, Kang D, et al. A novel pathway regulating lipopolysaccharide-induced shock by ST2/T1 via inhibition of Toll-like receptor 4 expression. J Immunol. 2001;166:6633–9. doi: 10.4049/jimmunol.166.11.6633. [DOI] [PubMed] [Google Scholar]
- 19.Huang BJ, Yin H, Huang YF, et al. Gene therapy using adenoviral vector encoding 4-1BBIg gene significantly prolonged murine cardiac allograft survival. Transpl Immunol. 2006;16:88–94. doi: 10.1016/j.trim.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 20.Szarka RJ, Wang N, Gordon L, Nation PN, Smith RH. A murine model of pulmonary damage induced by lipopolysaccharide via intranasal instillation. J Immunol Methods. 1997;202:49–57. doi: 10.1016/s0022-1759(96)00236-0. [DOI] [PubMed] [Google Scholar]
- 21.Peng X, Hassoun PM, Sammani S, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–51. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 22.Lee WL, Downey GP. Neutrophil activation and acute lung injury. Curr Opin Crit Care. 2001;7:1–7. doi: 10.1097/00075198-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Akira S, Takeda K. Toll-like receptor signaling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 24.Lawrence T, Gilroy DW, Colville-Nash PR, Willoughby DA. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med. 2001;7:1291–7. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 25.Everhart MB, Han W, Sherrill TP, et al. Duration and intensity of NF-kappaB activity determine the severity of endotoxin-induced acute lung injury. J Immunol. 2006;176:4995–5005. doi: 10.4049/jimmunol.176.8.4995. [DOI] [PubMed] [Google Scholar]
- 26.Tajima S, Oshikawa K, Tominaga S, Sugiyama Y. The increase in serum soluble ST2 protein upon acute exacerbation of idiopathic pulmonary fibrosis. Chest. 2003;124:1206–14. doi: 10.1378/chest.124.4.1206. [DOI] [PubMed] [Google Scholar]
- 27.Reutershan J, Morris MA, Burcin TL, et al. Critical role of endothelial CXCR2 in LPS induced neutrophil migration into the lung. J Clin Invest. 2006;116:695–702. doi: 10.1172/JCI27009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sone Y, Serikov VB, Staub NC. Intravascular macrophage depletion attenuates endotoxin lung injury in anesthetized sheep. J Appl Physiol. 1999;87:1354–9. doi: 10.1152/jappl.1999.87.4.1354. [DOI] [PubMed] [Google Scholar]
- 29.Staub NC, Longwortb KE, Serikov V, Jerome EH, Elsasser T. Detergent inhibits 70–90% of responses to intravenous endotoxin in awake sheep. J Appl Physiol. 2001;90:1788–97. doi: 10.1152/jappl.2001.90.5.1788. [DOI] [PubMed] [Google Scholar]
- 30.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lunginjury. Am J Physiol. 2000;279:L1137–45. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 31.Giebelen IA, van Westerloo DJ, Larosa GJ, de Vos AF, van der Poll T. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock. 2007;28:700–3. doi: 10.1097/shk.0b013e318054dd89. [DOI] [PubMed] [Google Scholar]
- 32.Goodman RB, Strieter RM, Martin DP, et al. Inflammatory cytokines in patients with persistence of the acute respiratory distress syndrome. Am J Respir Crit Care Med. 1996;154:602–11. doi: 10.1164/ajrccm.154.3.8810593. [DOI] [PubMed] [Google Scholar]
- 33.Brint EK, Xu D, Liu H, et al. ST2 is an inhibitor of interleukin 1 receptor and Toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol. 2004;5:373–9. doi: 10.1038/ni1050. [DOI] [PubMed] [Google Scholar]
- 34.Espinassous Q, Garcia-de-Paco E, Garcia-Verdugo I, et al. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. J Immunol. 2009;183:1446–55. doi: 10.4049/jimmunol.0803067. [DOI] [PubMed] [Google Scholar]
- 35.Karin M, Lin A. NF-kappaB at the crossroads of life and death. Nat Immunol. 2002;3:221–7. doi: 10.1038/ni0302-221. [DOI] [PubMed] [Google Scholar]
- 36.Garrean S, Gao XP, Brovkovych V, et al. Caveolin-1 regulates NF-κB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol. 2006;177:4853–60. doi: 10.4049/jimmunol.177.7.4853. [DOI] [PubMed] [Google Scholar]