

Abstract
Several functional abnormalities in phagocytes from patients with liver cirrhosis contribute to an increased risk of infection. An increased resting respiratory burst has been observed in neutrophils from cirrhotic patients. We investigated whether an infection in cirrhosis affects the respiratory burst capacity of neutrophils and monocytes in response to Escherichia coli. This study included 45 hospitalized patients with liver cirrhosis and clinical signs of infection, 39 patients with liver cirrhosis in the absence of infection and 29 healthy subjects. Respiratory burst, lipopolysaccharide-binding protein (LBP), and immunoglobulin (Ig)G-autoantibodies against oxidized low-density lipoproteins (ab-oxLDL) were measured. The fraction of neutrophils spontaneously producing reactive oxygen species (ROS) was elevated in liver cirrhosis (P < 0·01). The neutrophil resting burst increased with Child–Pugh stage (P = 0·02) and correlated with augmented ROS release in response to opsonized E. coli (P < 0·05). Although LBP was increased in patients with cirrhosis (P < 0·01), higher LBP levels correlated with a lower resting burst in neutrophils (rs = –0·395; P < 0·01). In the presence of infection, the resting burst was unaltered. However, neutrophil ROS release in response to E. coli was reduced markedly (P = 0·01), and it decreased as serum C-reactive protein (CRP) concentration rose (rs = −0·437; P < 0·01), indicating the development of a sepsis-like immune paralysis. A positive correlation between ab-oxLDL and ROS release was observed (P < 0·01). In conclusion, the respiratory burst increases with severity of liver cirrhosis but is restrained by increasing LBP levels. Augmented ROS release in response to E. coli is accompanied by elevated markers of oxidative damage and becomes exhausted in the presence of infection.
Keywords: cirrhosis, endotoxaemia, infection, neutrophil function, respiratory burst/oxidative state
Introduction
Bacterial infections occur in one-third of all patients hospitalized for decompensated liver cirrhosis [1], leading to an increased risk of sepsis [2], and these infections are associated with a 12-month mortality of 66% [3]. The most common types of infection include spontaneous bacterial peritonitis (SBP), urinary tract infection, pneumonia, septicaemia and skin infection [4]. As part of the innate immune system, neutrophils and monocytes produce reactive oxygen species (ROS) for the purpose of killing invading microorganisms. These ROS may also inflict damage on nearby tissues and are therefore of pathogenic significance [5]. After exposure to an inflammatory stimulus, such as lipopolysaccharide (LPS) or tumour necrosis factor (TNF)-α[6], circulating quiescent neutrophils undergo priming, which enables endothelial adherence and induces hyper-responsiveness to otherwise weak chemotactic stimuli [7,8]. The extensive immunological interplay between neutrophils and monocytes is exemplified by the fact that LPS and proteolytic enzymes from activated neutrophils prime monocytes to produce ROS [9–11] and to release proinflammatory cytokines, including TNF-α[12], which further prime neutrophils [13].
Patients with advanced liver disease are exposed to recurrent episodes of bacterial translocation and endotoxaemia [14], which are accompanied by the presence of bacterial DNA in blood and ascites [15,16], elevated serum LPS levels [17] and an increased concentration of LPS-binding protein (LBP) [18,19]. Several functional abnormalities that may contribute to an increased risk of bacterial infection have been observed in phagocytes from patients with advanced liver disease, including deficiencies in endothelial adhesion, transmigration [20,21] and phagocytic capacity [22,23], despite increased markers of neutrophil activation [24–26]. Mookerjee et al. have demonstrated that a higher neutrophil resting burst, more precisely an increase in the basal fraction of neutrophils producing ROS, in patients with liver cirrhosis and alcoholic hepatitis is a predictor of infection and mortality [27]. However, with respect to anti-bacterial capacity, Panasiuk and co-workers have reported reduced neutrophil ROS production after stimulation with opsonized E. coli and phorbol-12-myristate-13-acetate (PMA), indicating a decrease in the respiratory burst capacity of neutrophils from patients with liver cirrhosis [28].
In this study, we investigated whether an increased resting burst in patients with liver cirrhosis is associated with an impaired respiratory burst capacity in response to bacteria. We hypothesized that, in the presence of subclinical endotoxaemia and clinically manifested infection, phagocytes are less responsive to Escherichia coli and their respiratory burst capacity becomes exhausted, as has been demonstrated previously for critically ill non-cirrhotic patients with systemic inflammatory response syndrome (SIRS) [29] and Gram-negative septicaemia [30]. To confirm our hypothesis and to determine the significance of elevated ROS production, the endotoxaemia marker LBP [31], the acute-phase reactant C-reactive protein (CRP) and immunoglobulin (Ig)G autoantibodies against oxidized low-density lipoproteins as a marker of lipid peroxidation in vivo[32,33] were correlated with respiratory burst activity in neutrophils and monocytes from patients with liver cirrhosis.
Materials and methods
Study subjects
The study included 84 patients with liver cirrhosis who were treated in our department between April 2009 and March 2010, and 29 self-declared healthy individuals. All subjects gave informed consent, and the study was approved by the local ethics committee (1786-05/06). Diagnosis of liver cirrhosis was made by histological criteria when a liver biopsy was performed or by a combination of clinical, biochemical and imaging data. Patients with cirrhosis were not eligible if they received immunosuppressive therapy, underwent surgical intervention within the last month, suffered from gastrointestinal bleeding or underwent an endoscopic intervention within the last 3 days or actively consumed alcohol. Infection at the time of inclusion was defined by the presence of at least one of the following criteria: positive blood cultures (Biomérieux, Durham, NC, USA), positive urinary cultures (UrinAx, Axon Lab AG, Baden, Switzerland), pulmonary infiltrates on a chest X-ray, neutrocytic ascites (neutrophil count ≥ 250/mm3), skin infections, leucocyturia (≥20 cells per high-power field) or the presence of other clinically manifested infections according to clinical, radiological and microbial data. Furthermore, patients who presented with fever (body temperature > 38·5°C), leucocytosis (>12 Gpt/l) or who received antibiotic therapy for >24 h without evidence of organ involvement were considered to have undetermined infection.
The following patient data were collected at baseline: sex, age, aetiology of liver disease, concentrations of CRP, alanine aminotransferase (ALT), international normalized ratio (INR), creatinine and bilirubin, model for end-stage liver disease (MELD) score and Child–Pugh stage.
Blood sampling
In the morning, 9 ml of heparinized whole blood (Sarstedt AG & CO, Nümbrecht, Germany) was collected, stored at room temperature and measured for respiratory burst analysis within 24 h. An additional 9 ml of blood was collected (Sarstedt), centrifuged (1000 rcf, 4°C, 10 min), aliquoted and stored at –80°C in cryovials (Sarstedt) for subsequent protein analysis.
Phagocyte respiratory burst
The respiratory burst of monocytes and neutrophils was examined with the Phagoburst kit (Glycotope, Heidelberg, Germany) after in vitro stimulation according to the manufacturer's protocol. Briefly, 100 µl of heparinized whole blood was incubated for 10 min with 20 µl unlabelled opsonized E. coli (1–2 × 109 bacteria per ml), 20 µl of N-formyl-methionyl-leucyl-phenylalanine (fMLP) (5 µm), 20 µl of PMA (8·1 µm) or 20 µl of phosphate-buffered saline (PBS) as a negative control. The formation of ROS was monitored through the oxidation of dihydrorhodamine 123 to rhodamine. Cytometric analysis was performed with the automated, multi-colour flow cytometry system, fluorescence activated cell sorter (FACS)Calibur (BD Biosciences, San Jose, CA, USA). Neutrophil and monocyte populations were identified by the use of forward- and right-angle light-scatter after gating on human DNA. The fluorescence emission of 10 000 cells per sample was recorded on a logarithmic scale, and a cut-off between cell populations was applied as demonstrated in Fig. 1. Respiratory burst was expressed as a percentage of activated phagocytes and the mean fluorescence intensity (MFI). The integrated mean fluorescence intensity (iMFI) resulted from the product of the frequency of ROS-producing cells and the median fluorescence intensity of the cells [34].
Fig. 1.
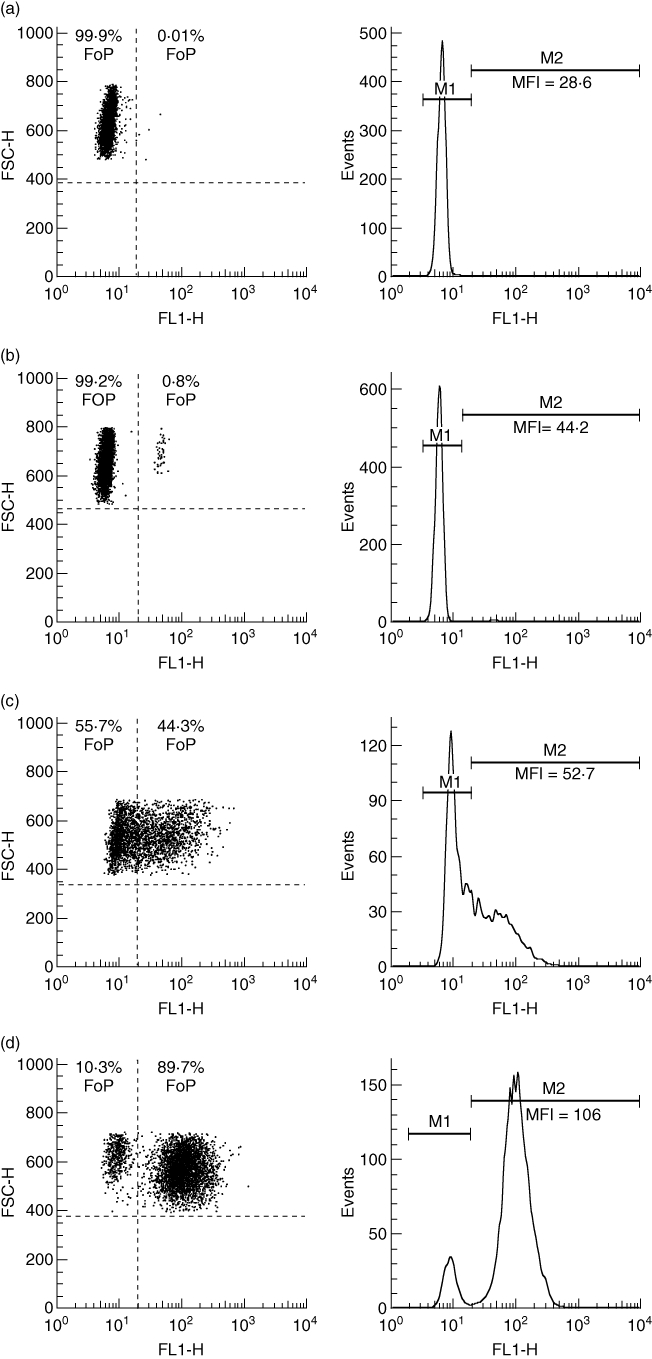
Analysis of resting burst and Escherichia coli-induced respiratory burst in neutrophils. Representative fluorescence activated cell sorter (FACS) plots and histograms of neutrophil respiratory burst in (a) a healthy control with virtually no resting burst [0·1% reactive oxygen species (ROS)-producing cells]; (b) a non-infected patient with liver cirrhosis with an increased basal fraction of ROS-producing neutrophils (0·8%) and increased basal ROS release (MFI 44·2); (c) a healthy control after in vitro incubation with opsonized E. coli; and (d) a non-infected patient with liver cirrhosis after in vitro incubation with opsonized E. coli showing an increased fraction (89·7%) and ROS release (MFI 106). FSC-H: forward-scatter; SSC-H: side-scatter; FoP: frequency of parents; FL1-H: fluorescence intensity; M1: non-activated cell population; M2: activated cell population.
Serum protein analysis
LBP serum concentration was determined in a subset of 59 subjects (11 controls, 21 patients with liver cirrhosis without infection, 27 patients with liver cirrhosis and infection). LBP concentrations were measured in diluted Serum samples duplicate using the HK315 human LBP sandwich enzyme-linked immunosorbent assay (ELISA) (Hycult Biotech, Uden, the Netherlands), according to the manufacturer's instructions. The standard curve was created by sixfold serial dilution of a 50 ng/ml standard solution in duplicate.
The ab-oxLDL serum concentration was analysed in a subset of 60 subjects (11 controls, 21 patients with liver cirrhosis without infection and 28 patients with liver cirrhosis and infection). Serum concentrations of IgG-autoantibodies against oxidized low-density lipoproteins (ab-oxLDL) were measured in duplicate using a commercially available ELISA (Immundiagnostik AG, Bensheim, Germany). The standard curve was created by fivefold serial dilution of a 50 U/l standard solution in duplicates. Measurements were performed at 460 nm in a photometric plate reader (Victor, Wallac, Waltham, MA, USA).
Statistical analysis
Baseline patient characteristics are indicated either by the median and range for continuous variables with non-normal distribution or by frequency for discrete variables. Statistical differences between groups of subjects were analysed by the non-parametric Mann–Whitney U-test, the Kruskal–Wallis test for continuous data or Pearson's χ2 test for discrete data using two-sided tests. Non-parametric measurement of statistical dependence between two variables was performed by calculating Spearman's rank correlation coefficient rho (rs). All statistical calculations were performed using spss version 18 software (SPSS Inc., Chicago, IL, USA).
Results
Patient population
A total of 84 hospitalized patients with cirrhosis were included. The majority of the patients were male Caucasians (77%) who suffered from alcoholic liver cirrhosis (80%) (Table 1). Whereas 39 patients presented without signs of infection at inclusion (group A), 45 patients fulfilled the criteria for infection (group B). Urinary tract infection (n = 12), pneumonia (n = 12), skin infection (n = 10) and spontaneous bacterial peritonitis (n = 5) were the most frequent infections. As expected, patients with signs of infection had higher median leucocyte levels (7·8 Gpt/l versus 5·6 Gpt/l; P < 0·001) and higher median CRP (37·7 mg/l versus 7·5 mg/l; P < 0·001) than non-infected patients. There was no difference in the severity of liver cirrhosis between groups A and B, although a trend towards a higher Child–Pugh score (P = 0·092) and a higher MELD score was observed in group B (P = 0·052). Patients with clinically manifested infection had lower median albumin levels than patients without signs of infection (24 mg/l versus 30 mg/l; P < 0·001). Healthy subjects (14 male and 15 female) without liver cirrhosis were included to serve as the control group. Because controls were not age-matched, the median age in the control group was younger than the median age in the cirrhotic patient group (45 versus 59 years; P < 0·001).
Table 1.
Patient characteristics
| Cirrhosis, no infection (n = 39) | Cirrhosis, infection (n = 45) | P-value | |
|---|---|---|---|
| Years of age, median (range) | 58 (37–82) | 58 (40–80) | 0·437 |
| Male sex, no. (%) | 32 (82) | 33 (73) | 0·341 |
| Aetiology, no. (%) | 0·953 | ||
| Alcohol | 31 (79) | 36 (80) | |
| Virus | 2 (5) | 2 (4) | |
| Cryptogen | 3 (8) | 3 (7) | |
| Other | 3 (8) | 4 (9) | |
| Child–Pugh stage, no. (%) | 0·147 | ||
| A | 5 (13) | 2 (4) | |
| B | 22 (56) | 21 (47) | |
| C | 12 (31) | 22 (49) | 0·092 |
| MELD score, median (range) | 13·5 (8–28) | 15 (7–40) | 0·057 |
| Presence of ascites, no. (%) | 28 (72) | 39 (87) | 0·091 |
| Laboratory values, median (range) | |||
| Bilirubin, µmol/l | 43 (9–504) | 38 (5–531) | 0·920 |
| INR | 1.4 (0·9–2·4) | 1·4 (1·0–2·2) | 0·594 |
| Creatinine, µmol/l | 81 (43–171) | 97 (40–351) | 0·083 |
| ALT, µmol/l × s | 0·65 (0·31–3·63) | 0·60 (0·14–1·46) | 0·238 |
| Leucocyte count, Gpt/l | 5·6 (2·4–10·7) | 7·8 (2·7–32·3) | <0·001 |
| CRP, mg/l | 7·5 (2·0–78·8) | 37·7 (2·0–397·5) | <0·001 |
| Albumin, mg/l | 30 (17–44) | 24 (16–40) | <0·001 |
MELD: model of end-stage liver disease; INR: international normalized ratio; ALT: alanine aminotransferase; CRP: C-reactive protein.
Neutrophil resting burst
The fraction of neutrophils with basal ROS production (median 1·1% versus 0·5%; P < 0·001) was increased in patients with cirrhosis compared to controls, as was ROS release by these cells (median MFI 27·9 versus 23·5; P = 0·013). Based on these results the iMFI, a marker of cumulative neutrophil ROS production, was threefold higher in patients with liver cirrhosis than in controls (median iMFI 0·36 versus 0·12; P < 0·001). The neutrophil resting burst did not differ between infected and non-infected cirrhotic patients (Table 2a).
Table 2.
Respiratory burst in (a) neutrophils and (b) monocytes
| Median (range) | P-value | ||||
|---|---|---|---|---|---|
| Control, no cirrhosis (n = 29) | Group A cirrhosis, no infection (n = 39) | Group B cirrhosis, infection (n = 45) | Control versus group A | Group A versus group B | |
| (a) Neutrophil stimulation | |||||
| Basal | |||||
| ROS-producing cells (%) | 0·5 (0·0–13·2) | 1·0 (0·0–10·9) | 1·3 (0·0–19·5) | 0·004 | 0·425 |
| MFI | 23·5 (14·6–77·0) | 30·3 (16·0–52·2) | 27·0 (12·3–43·1) | 0·003 | 0·117 |
| iMFI | 0·1 (0·0–3·4) | 0·3 (0·0–4·9) | 0·4 (0·0–6·4) | 0·001 | 0·816 |
| fMLP | |||||
| ROS-producing cells (%) | 1·7 (0·3–35·1) | 2·7 (0·6–18·5) | 2·5 (0·0–22·5) | 0·068 | 0·477 |
| MFI | 22·6 (15·8–27·8) | 25·2 (17·8–42·3) | 25·0 (16·7–37·8) | 0·006 | 0·914 |
| iMFI | 0·4 (0·1–7·4) | 0·8 (0·1–7·8) | 0·6 (0·0–6·6) | 0·018 | 0·315 |
| Opsonized E. coli | |||||
| ROS-producing cells (%) | 98·1 (34·6–99·9) | 98·9 (58·1–100·0) | 97·7 (19·9–100·0) | 0·294 | 0·224 |
| MFI | 50·9 (26·8–99·6) | 67·7 (21·8–257·0) | 43·7 (17·2–161·0) | 0·020 | 0·010 |
| iMFI | 50·3 (10·0–98·6) | 65·2 (16·9–257·0) | 43·5 (3·7–144·9) | 0·026 | 0·014 |
| PMA | |||||
| ROS-producing cells (%) | 100 (74·2–100·0) | 100 (23·7–100·0) | 99·9 (56·5–100·0) | 0·881 | 0·187 |
| MFI | 192·0 (22·5–347·0) | 228·0 (18·3–431·0) | 194·0 (16·6–433·0) | 0·385 | 0·062 |
| iMFI | 192·0 (16·7–347·0) | 227·5 (4·3–429·3) | 193·8 (10·7–433·0) | 0·382 | 0·058 |
| (b) Monocyte stimulation | |||||
| Basal | |||||
| ROS-producing cells (%) | 1·1 (0·0–18·5) | 1·3 (0·0–31·5) | 2·3 (0·0–19·0) | 0·423 | 0·326 |
| MFI | 16·3 (12·7–26·9) | 16·7 (13·3–38·2) | 15·4 (14·1–23·0) | 0·963 | 0·072 |
| iMFI | 0·2 (0·0–2·8) | 0·3 (0·0–6·1) | 0·4 (0·0–6·0) | 0·395 | 0·492 |
| fMLP | |||||
| ROS-producing cells (%) | 3·0 (0·0–18·0) | 2·2 (0·0–42·0) | 4·0 (0·0–18·5) | 0·708 | 0·350 |
| MFI | 18·2 (15·4–24·2) | 16·8 (13·9–22·3) | 16·1 (13·0–22·5) | 0·014 | 0·579 |
| iMFI | 0·6 (0·0–3·5) | 0·4 (0·0–6·9) | 0·7 (0·0–3·2) | 0·559 | 0·474 |
| Opsonized E. coli | |||||
| ROS-producing cells (%) | 91·2 (19·0–99·6) | 94·3 (62·5–100·0) | 92·1 (11·8–100·0) | 0·014 | 0·103 |
| MFI | 20·3 (15·8–34·6) | 25·3 (16·0–50·9) | 20·7 (13·8–38·9) | 0·007 | 0·091 |
| iMFI | 18·4 (3·3–32·6) | 22·4 (10·0–50·9) | 20·1 (1·8–38·9) | 0·004 | 0·080 |
| PMA | |||||
| ROS-producing cells (%) | 99·9 (40·9–100·0) | 100·0 (6·4–100·0) | 99·3 (2·6–100·0) | 0·889 | 0·071 |
| MFI | 28·3 (16·3–49·5) | 33·3 (14·8–62·8) | 29·7 (13·9–96·5) | 0·031 | 0·231 |
| iMFI | 28·3 (6·7–49·5) | 33·3 (1·0–62·8) | 28·7 (0·4–96·5) | 0·039 | 0·192 |
MFI: mean fluorescence intensity; iMFI: integrated mean fluorescence intensity; fMLPL N-formyl-methionyl-leucyl-phenylalanine; PMA: phorbol-12-myristate-13-acetate; ROS: reactive oxygen species.
The fraction of ROS-producing neutrophils increased with the severity of cirrhosis from 0·5% in Child–Pugh stage A cirrhosis to 1·1% in stage B cirrhosis and 1·5% in stage C cirrhosis (P = 0·02 by Kruskal–Wallis test) (Fig. 2). Patients with liver cirrhosis had elevated serum LBP levels (median 25·6 µg/ml), compared with those in controls (median 13·8 µg/ml; P = 0·003), which were increased further in cirrhotic patients with infections (median 33·8 µg/ml; P = 0·050) (Fig. 3). Higher serum LBP levels correlated significantly with lower neutrophil resting burst in patients with cirrhosis (rs = −0·395; P = 0·006) (Fig. 4a). This negative correlation between LBP and basal ROS production (MFI) was stronger in cirrhotic patients without an infection (rs = –0·420; P = 0·058) than in cirrhotic patients with an infection (rs = –0·286; P = 0·156).
Fig. 2.
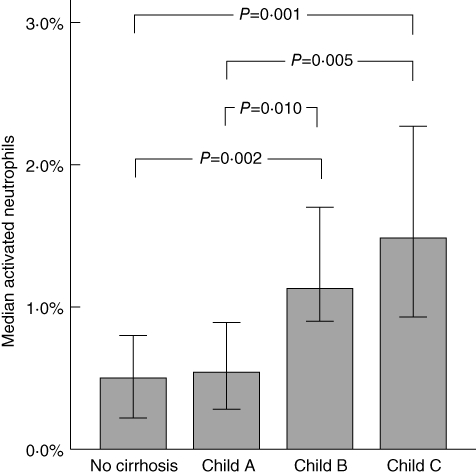
Neutrophil resting burst and severity of liver disease. The bars represent the median basal fraction of reactive oxygen species (ROS)-producing neutrophils in controls (n = 11), patients with Child–Pugh A cirrhosis (n = 7), Child–Pugh B cirrhosis (n = 43) and Child–Pugh C cirrhosis (n = 34). Error bars represent the 95% confidence intervals. Significance levels of the Mann–Whitney U-test are indicated.
Fig. 3.
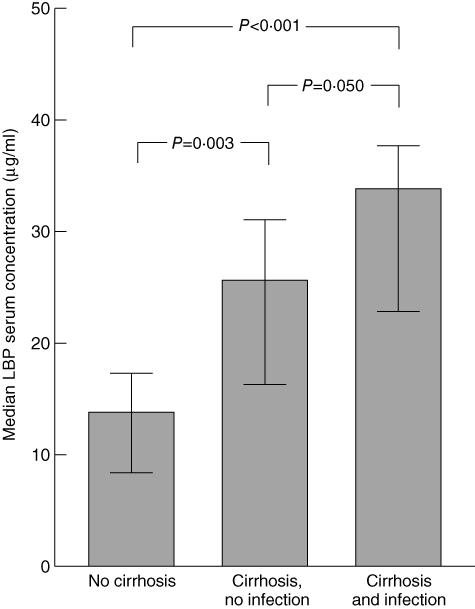
Serum concentrations of lipopolysaccharide-binding protein (LBP). The bars represent median serum concentrations of LBP in controls (n = 11), patients with cirrhosis without infection (n = 21) and patients with cirrhosis and infection (n = 27). Error bars represent the 95% confidence intervals. Significance levels of the Mann–Whitney U-test are indicated.
Fig. 4.
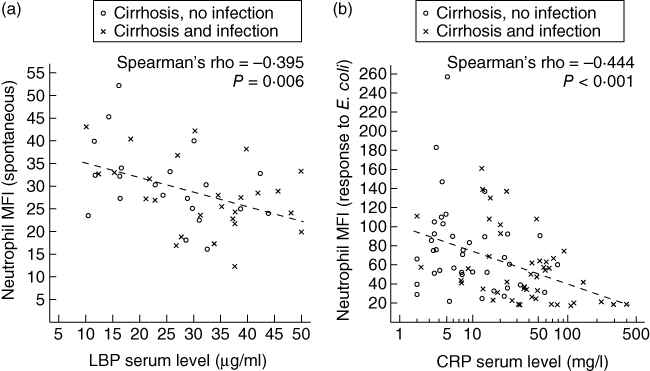
Neutrophil respiratory burst, C-reactive protein (CRP) level and lipopolysaccharide-binding protein (LBP) concentration. The dot plots show the correlations between (a) serum LBP concentration and mean basal reactive oxygen species (ROS) production in neutrophils (n = 47) and (b) serum CRP level and ROS release in neutrophils after in vitro stimulation with opsonized Escherichia coli in patients with liver cirrhosis (n = 84). Spearman's rank correlation coefficients and significance levels of non-parametric correlations are indicated.
Inducible neutrophil respiratory burst
In neutrophils from non-infected patients with liver cirrhosis, basal ROS production and ROS release following incubation of whole blood with opsonized E. coli were correlated positively (rs = 0·325; P = 0·043). The median fraction of ROS-producing neutrophils after stimulation with E. coli was as high in patients with cirrhosis as in controls (Table 2a). Neutrophils from non-infected patients with liver cirrhosis had higher E. coli-induced ROS release than neutrophils from control subjects (MFI 67·7 versus 50·9; P = 0·020). In neutrophils from cirrhotic patients with a clinically manifested infected infection, however, the E. coli-induced ROS release was lower than that in neutrophils from non-infected patients (median MFI 43·7; P = 0·010). Accordingly, increased levels of CRP correlated with lower E. coli-induced ROS release in neutrophils (rs = –0·444; P < 0·001), as displayed in Fig. 4b. This correlation was seen in cirrhotic patients with a clinically manifested infection (rs = –0·437; P = 0·003), but was not significant in non-infected cirrhotic patients (rs = –0·237; P = 0·146).
To differentiate whether patients with a persistently decreased respiratory burst capacity develop infections or whether a clinically manifested infection leads to a transient decrease in ROS release, we evaluated E. coli-induced respiratory burst in seven patients at different infection states. The representative plots of respiratory burst in Fig. 5 demonstrate that, in six of seven patients, E. coli-induced ROS release decreased with infection or regenerated after infection (P = 0·091 by two-sided Wilcoxon's signed-rank test). The time difference between hospitalizations was 36 to 228 days, and the median CRP difference was 26 mg/l.
Fig. 5.
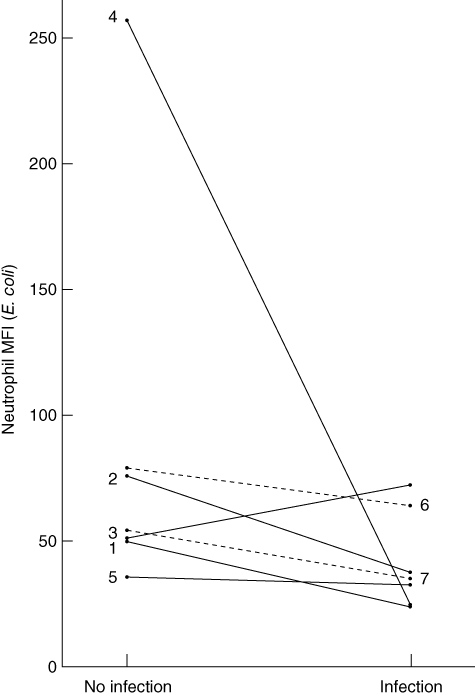
Individual changes in Escherichia coli-induced neutrophil respiratory burst after infection. The figure displays the individual changes in neutrophil reactive oxygen species (ROS) release after E. coli stimulation during infection (n = 7). Patients 1–5 presented without infection at study inclusion but were hospitalized again for infection (two urinary tract infections, pneumonia, spontaneous bacterial peritonitis, skin infection). Patients 6 and 7 presented with infection and were hospitalized again for hepatic decompensation without signs of infection.
To estimate ROS release by primed neutrophils whole blood was incubated with fMLP, the weak chemotactic control stimulus. The fraction of ROS-producing neutrophils after stimulation with fMLP was identical in cirrhotic patients and controls. However, the fMLP-induced ROS release in neutrophils was slightly higher in patients with cirrhosis (median MFI 25·1 versus 22·6; P = 0·005) but did not differ with respect to the presence of an infection (Table 2a). Stimulation with PMA, a non-receptor-dependent activator of protein kinase C, led to identical maximum ROS-release in neutrophils in the three examined groups (Table 2a).
Monocyte respiratory burst
Although basal ROS production by neutrophils and monocytes from patients with cirrhosis correlated positively with each other (rs = 0·414; P < 0·001), the resting burst in monocytes from cirrhotic patients was not elevated compared to controls (Table 2b). We did not observe a difference in basal and fMLP-induced ROS release in monocytes from cirrhotic patients with respect to the state of infection.
As was seen in neutrophils, the fraction of ROS-producing monocytes following incubation with E. coli (median proportion 94·3% versus 91·2%; P = 0·014) and the magnitude of ROS release (median MFI 25·3 versus 20·3; P = 0·007) were increased in non-infected cirrhotic patients compared to controls. Although there was only a statistically insignificant trend towards decreased E. coli-induced ROS release in monocytes from cirrhotic patients with infection compared to monocytes from non-infected cirrhotic patients (Table 2b), higher serum CRP levels were also associated with lower ROS release in monocytes after challenge with E. coli (rs = –0·342; P = 0·001), especially in infected patients (rs = –0·355; P = 0·017).
The monocyte respiratory burst after fMLP stimulation was identical in the three examined groups (Table 2b). After incubation with PMA the ROS release in monocytes from non-infected cirrhotic patients was slightly higher than in monocytes from control subjects (MFI 33·3 versus 28·3; P = 0·031).
Ab-oxLDL and respiratory burst
The median ab-oxLDL concentration was 20·2 U/l in controls, 19·0 U/l in group A and 18·3 U/l in group B. Patients with liver cirrhosis whose phagocytes produced higher amounts of ROS after in vitro stimulation with the strong control stimuli, E. coli and PMA, had higher ab-oxLDL serum levels as analysed using bivariate Spearman's rank correlation (Table 3).
Table 3.
Correlation of immunoglobulin (Ig)G-autoantibody concentration against oxidized low-density lipoproteins (ab-oxLDL) and respiratory burst in patients with liver cirrhosis (n = 49)
| Correlation rs with ab-oxLDL serum levels | P-value | |
|---|---|---|
| Neutrophils (iMFI) | ||
| Basal | 0·079 | 0·588 |
| fMLP | 0·114 | 0·437 |
| Escherichia coli | 0·425 | 0·002 |
| PMA | 0·288 | 0·045 |
| Monocytes (iMFI) | ||
| Basal | 0·301 | 0·036 |
| fMLP | 0·369 | 0·009 |
| E. coli | 0·416 | 0·003 |
| PMA | 0·302 | 0·035 |
rs: Spearman's rank correlation coefficient; iMFI: integrated mean fluorescence intensity; fMLP: N-formyl-methionyl-leucyl-phenylalanine.
Discussion
In this study, we demonstrated that the fraction of ROS-producing circulating neutrophils and basal generation of reactive oxygen species were both increased in patients with liver cirrhosis. This augmented resting burst was accompanied by an increase in ROS release by neutrophils and monocytes in response to E. coli. Whereas the resting burst remained unaltered in the presence of an infection in patients with liver cirrhosis, the respiratory burst capacity of neutrophils in response to opsonized bacteria decreased with infection.
Increased neutrophil resting burst was reported by Mookerjee et al. [27] in patients with cirrhosis and alcoholic hepatitis. Our results verified these findings, although we did see a difference in the absolute frequency of ROS-producing neutrophils, which can be attributed mainly to a stricter gating strategy for FACS analysis in our study (Fig. 1) and to the patient population examined. We confirmed the existence of an elevated neutrophil resting burst in liver cirrhosis by demonstrating an increase in basal neutrophil ROS production. Furthermore, we observed an increase in neutrophil resting burst with increasing severity of liver cirrhosis. Interestingly, we did not see an increase in resting burst in monocytes despite the similarities between activation of monocytes and neutrophils and their immunological interplay [10,11]. However, secretion of TNF-α by activated monocytes might increase resting burst in neutrophils [12,13].
In contrast to the findings of Panasiuk and co-workers [28], who reported a lower respiratory burst in neutrophils from non-infected patients with liver cirrhosis after activation with E. coli and PMA, we observed an increase in ROS release in neutrophils and monocytes after incubation with opsonized E. coli. The elevation in resting burst was correlated positively with the E. coli-induced ROS release in non-infected cirrhotic patients.
In the presence of an infection, ROS production in neutrophils was temporarily decreased. The negative correlation between serum CRP levels and ROS production in neutrophils and monocytes in response to opsonized E. coli indicates the development of a sepsis-like immune paralysis [35,36] as acute-phase reaction increases. However, we can only speculate on the underlying mechanisms. Expression levels of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase subunits, IgG receptors and integrins on neutrophils from patients with cirrhosis during infection need to be measured in subsequent studies to corroborate our findings. Analogous to our data, decreased ROS release in neutrophils after stimulation with E. coli has been reported for non-cirrhotic patients with Gram-negative septicaemia [30]. This infection has similar mechanisms and cytokine composition to the bacterial infection in cirrhotic patients. Stimulation with the strong, receptor-independent stimulus, PMA, which activates NADPH oxidase via protein kinase C, resulted in a non-statistically significant decrease in respiratory burst capacity in infected cirrhotic patients. This result indicates a receptor-mediated mechanism rather than reduced expression of NADPH oxidase in neutrophils from patients with cirrhosis in the presence of an infection. Similarly, the PMA-induced respiratory burst was not altered in a study on non-cirrhotic patients with sepsis [37], whereas a decreased PMA-induced respiratory burst was observed in an animal model of sepsis [38].
It has been demonstrated that neutrophils from septic patients produce more ROS after fMLP stimulation [37,39], which is accompanied by an up-regulation of the G-protein-coupled fMLP-receptor [40]. In our study, ROS production after fMLP stimulation was also increased in the presence of an infection compared to controls, but it was identical to the already increased fMLP response in non-infected cirrhotic patients. Our results suggest that liver cirrhosis and its associated proinflammatory conditions increase the respiratory burst in primed and activated neutrophils, and this is not augmented further in the presence of an infection. However, we cannot exclude any further increase in resting burst with increasing severity of infection and the occurrence of sepsis.
Although several factors, including cytokines, acidosis, hypertonicity and hyperammonaemia, have been reported to prime phagocytes for enhanced release of ROS [12,41], the increased respiratory burst in patients with liver cirrhosis has been attributed mainly to endotoxaemia. Mookerjee and co-workers [27] demonstrated that plasma from cirrhotic patients activates neutrophils in vitro, and the removal of endotoxin from patients' plasma normalized their augmented neutrophil activation. Neutrophil priming by LPS requires the presence of LBP in the plasma and membranous CD14 [42,43], which activates mitogen-activated protein (MAP) kinase p38, leading to production of ROS [43]. LBP is a mainly liver-derived 60 kDa serum glycoprotein that is produced during Gram-negative bacterial infections and endotoxaemia [31]. In cirrhosis, elevated LBP concentrations are associated with higher concentrations of endotoxin, soluble CD14, TNF-α and interleukin-6 [18]. Although we demonstrated that respiratory burst and LBP serum concentrations were increased in patients with liver cirrhosis, LBP levels and respiratory burst were not correlated positively with each other. In contrast, in non-infected patients with cirrhosis, an increase in LBP serum concentrations was associated with a lower resting burst. Although low concentrations of LBP enhance the cellular response to LPS [44], it has been shown that higher concentrations of LBP inhibit phagocyte responses in vitro and in vivo[45,46] and were protective in a mice peritonitis model [45] and a study of patients with severe sepsis [47]. LBP inhibits the endotoxin response by transferring LPS to plasma lipoproteins [48] and also by directly inhibiting the phagocyte response, even after the binding of LPS monomers to CD14 [49]. Thus, we attribute the negative correlation between LBP concentration and resting burst to a reduction in neutrophil response to E. coli in the presence of increasing LBP concentrations. Beyond that, differences in the half-lives of LPS and LBP may lead to an elevation of LBP concentration even after endotoxin clearance, which may aggravate further the LBP-mediated suppression of respiratory burst. In a study by Albillos et al., endotoxin was detected in only one-third of patients with elevated LBP levels [18], suggesting that endotoxaemia is transient, whereas LBP remains elevated for a prolonged period of time.
The production of free reactive oxygen species in neutrophils and monocytes induces LDL oxidation, forming a potent cytotoxic agent that mediates tissue damage [50]. In a cell culture model of hepatic stellate cells, oxidized LDL (oxLDL) stimulated extracellular matrix synthesis via the scavenger receptor CD36 and also mediated fibrosis [51]. Recent data suggest that LPS and oxLDL activate macrophages co-operatively [52] and that oxLDL increases Toll-like receptor expression [53]. The oxidation of LDL induces ab-oxLDL and forms immune complexes that promote the activation of macrophages and the release of proinflammatory cytokines, such as TNF-α, which further primes and activates phagocytes [54]. Ab-oxLDL reflects the immune response to oxLDL over a prolonged period of time and may therefore indicate recurrent episodes of increased respiratory burst. The ab-oxLDL level is an independent predictor of morbidity and mortality in patients with severe chronic heart failure [55]. Although we did not observe any difference in ab-oxLDL concentrations between controls and cirrhotic patients, we observed a significant positive correlation between ROS production in phagocytes and ab-oxLDL concentrations. Although many individual and environmental factors, e.g. nutrition, smoking status, glucose metabolism and chronic inflammatory diseases, are known to influence lipid peroxidation, our results suggest that the increased resting burst and increased respiratory burst capacity in patients with liver cirrhosis are accompanied by the occurrence of oxidative damage in vivo.
Disclosure
All authors declare no potential conflicts of interest.
References
- 1.Borzio M, Salerno F, Piantoni L, et al. Bacterial infection in patients with advanced cirrhosis: a multicentre prospective study. Dig Liver Dis. 2001;33:41–8. doi: 10.1016/s1590-8658(01)80134-1. [DOI] [PubMed] [Google Scholar]
- 2.Foreman MG, Mannino DM, Moss M. Cirrhosis as a risk factor for sepsis and death: analysis of the National Hospital Discharge Survey. Chest. 2003;124:1016–20. doi: 10.1378/chest.124.3.1016. [DOI] [PubMed] [Google Scholar]
- 3.Arvaniti V, D'Amico G, Fede G, et al. Infections in patients with cirrhosis increase mortality 4-fold and should be used in determining prognosis. Gastroenterology. 2010;139:1246–56. doi: 10.1053/j.gastro.2010.06.019. [DOI] [PubMed] [Google Scholar]
- 4.Guerra-Ruiz A, Casafont F, Cobo M, et al. Increased bactericidal/permeability increasing protein in patients with cirrhosis. Liver Int. 2010;30:94–101. doi: 10.1111/j.1478-3231.2009.02121.x. [DOI] [PubMed] [Google Scholar]
- 5.Babior BM. Phagocytes and oxidative stress. Am J Med. 2000;109:33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- 6.Guthrie LA, McPhail LC, Henson PM, Johnston RB. Priming of neutrophils for enhanced release of oxygen metabolites by bacterial lipopolysaccharide. Evidence for increased activity of the superoxide-producing enzyme. J Exp Med. 1984;160:1656–71. doi: 10.1084/jem.160.6.1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheppard FR, Kelher MR, Moore EE, McLaughlin NJD, Banerjee A, Silliman CC. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J Leukoc Biol. 2005;78:1025–42. doi: 10.1189/jlb.0804442. [DOI] [PubMed] [Google Scholar]
- 8.Cowburn AS, Condliffe AM, Farahi N, Summers C, Chilvers ER. Advances in neutrophil biology: clinical implications. Chest. 2008;134:606–12. doi: 10.1378/chest.08-0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Speer CP, Pabst MJ, Hedegaard HB, Rest RF, Johnston RB. Enhanced release of oxygen metabolites by monocyte-derived macrophages exposed to proteolytic enzymes: activity of neutrophil elastase and cathepsin G. J Immunol. 1984;133:2151–6. [PubMed] [Google Scholar]
- 10.Soehnlein O, Kenne E, Rotzius P, Eriksson EE, Lindbom L. Neutrophil secretion products regulate anti-bacterial activity in monocytes and macrophages. Clin Exp Immunol. 2008;151:139–45. doi: 10.1111/j.1365-2249.2007.03532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao X, Carnevale KA, Cathcart MK. Human monocytes use Rac1, not Rac2, in the NADPH oxidase complex. J Biol Chem. 2003;278:40788–92. doi: 10.1074/jbc.M302208200. [DOI] [PubMed] [Google Scholar]
- 12.Pabst M, Pabst K, Handsman D, Beranova-Giorgianni S, Giorgianni F. Proteome of monocyte priming by lipopolysaccharide, including changes in interleukin-1beta and leukocyte elastase inhibitor. Proteome Sci. 2008;6:13. doi: 10.1186/1477-5956-6-13. Available at: http://www.proteomesci.com/content/6/1/13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Leeuwen HJ, van der Tol M, van Strijp JAG, Verhoef J, van Kessel KPM. The role of tumour necrosis factor in the kinetics of lipopolysaccharide-mediated neutrophil priming in whole blood. Clin Exp Immunol. 2005;140:65–72. doi: 10.1111/j.1365-2249.2005.02748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiest R, Garcia-Tsao G. Bacterial translocation (BT) in cirrhosis. Hepatology. 2005;41:422–33. doi: 10.1002/hep.20632. [DOI] [PubMed] [Google Scholar]
- 15.Zapater P, Francés R, González-Navajas JM, et al. Serum and ascitic fluid bacterial DNA: a new independent prognostic factor in noninfected patients with cirrhosis. Hepatology. 2008;48:1924–31. doi: 10.1002/hep.22564. [DOI] [PubMed] [Google Scholar]
- 16.Bruns T, Sachse S, Straube E, et al. Identification of bacterial DNA in neutrocytic and non-neutrocytic cirrhotic ascites by means of a multiplex polymerase chain reaction. Liver Int. 2009;29:1206–14. doi: 10.1111/j.1478-3231.2009.02073.x. [DOI] [PubMed] [Google Scholar]
- 17.Lin C, Tsai I, Ho Y, et al. Endotoxaemia contributes to the immune paralysis in patients with cirrhosis. J Hepatol. 2007;46:816–26. doi: 10.1016/j.jhep.2006.12.018. [DOI] [PubMed] [Google Scholar]
- 18.Albillos A, de la Hera A, González M, et al. Increased lipopolysaccharide binding protein in cirrhotic patients with marked immune and hemodynamic derangement. Hepatology. 2003;37:208–17. doi: 10.1053/jhep.2003.50038. [DOI] [PubMed] [Google Scholar]
- 19.Albillos A, de-la-Hera A, Alvarez-Mon M. Serum lipopolysaccharide-binding protein prediction of severe bacterial infection in cirrhotic patients with ascites. Lancet. 2004;363:1608–10. doi: 10.1016/S0140-6736(04)16206-5. [DOI] [PubMed] [Google Scholar]
- 20.Gomez F, Ruiz P, Schreiber AD. Impaired function of macrophage Fc gamma receptors and bacterial infection in alcoholic cirrhosis. N Engl J Med. 1994;331:1122–8. doi: 10.1056/NEJM199410273311704. [DOI] [PubMed] [Google Scholar]
- 21.Fiuza C, Salcedo M, Clemente G, Tellado JM. Granulocyte colony-stimulating factor improves deficient in vitro neutrophil transendothelial migration in patients with advanced liver disease. Clin Diagn Lab Immunol. 2002;9:433–9. doi: 10.1128/CDLI.9.2.433-439.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajkovic IA, Williams R. Mechanisms of abnormalities in host defences against bacterial infection in liver disease. Clin Sci. 1985;68:247–53. doi: 10.1042/cs0680247. [DOI] [PubMed] [Google Scholar]
- 23.Fiuza C, Salcedo M, Clemente G, Tellado JM. In vivo neutrophil dysfunction in cirrhotic patients with advanced liver disease. J Infect Dis. 2000;182:526–33. doi: 10.1086/315742. [DOI] [PubMed] [Google Scholar]
- 24.Stanley AJ, MacGregor IR, Dillon JF, Bouchier IA, Hayes PC. Neutrophil activation in chronic liver disease. Eur J Gastroenterol Hepatol. 1996;8:135–8. doi: 10.1097/00042737-199602000-00008. [DOI] [PubMed] [Google Scholar]
- 25.Masini E, Mugnai L, Foschi M, Laffi G, Gentilini P, Mannaioni PF. Changes in the production of nitric oxide and superoxide by inflammatory cells in liver cirrhosis. Int Arch Allergy Immunol. 1995;107:197–8. doi: 10.1159/000236975. [DOI] [PubMed] [Google Scholar]
- 26.Taïeb J, Mathurin P, Elbim C, et al. Blood neutrophil functions and cytokine release in severe alcoholic hepatitis: effect of corticosteroids. J Hepatol. 2000;32:579–86. doi: 10.1016/s0168-8278(00)80219-6. [DOI] [PubMed] [Google Scholar]
- 27.Mookerjee RP, Stadlbauer V, Lidder S, et al. Neutrophil dysfunction in alcoholic hepatitis superimposed on cirrhosis is reversible and predicts the outcome. Hepatology. 2007;46:831–40. doi: 10.1002/hep.21737. [DOI] [PubMed] [Google Scholar]
- 28.Panasiuk A, Wysocka J, Maciorkowska E, et al. Phagocytic and oxidative burst activity of neutrophils in the end stage of liver cirrhosis. World J Gastroenterol. 2005;11:7661–5. doi: 10.3748/wjg.v11.i48.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fung YL, Fraser JF, Wood P, Minchinton RM, Silliman CC. The systemic inflammatory response syndrome induces functional changes and relative hyporesponsiveness in neutrophils. J Crit Care. 2008;23:542–9. doi: 10.1016/j.jcrc.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Wenisch C, Parschalk B, Patruta S, Brustbauer R, Graninger W. Effect of polyclonal immunoglobulins on neutrophil phagocytic capacity and reactive oxygen production in patients with Gram-negative septicaemia. Infection. 1999;27:183–6. doi: 10.1007/BF02561525. [DOI] [PubMed] [Google Scholar]
- 31.Schumann RR, Leong SR, Flaggs GW, et al. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–31. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 32.Maggi E, Marchesi E, Ravetta V, Martignoni A, Finardi G, Bellomo G. Presence of autoantibodies against oxidatively modified low-density lipoprotein in essential hypertension: a biochemical signature of an enhanced in vivo low-density lipoprotein oxidation. J Hypertens. 1995;13:129–38. [PubMed] [Google Scholar]
- 33.Shoenfeld Y, Wu R, Dearing LD, Matsuura E. Are anti-oxidized low-density lipoprotein antibodies pathogenic or protective? Circulation. 2004;110:2552–8. doi: 10.1161/01.CIR.0000143225.07377.EA. [DOI] [PubMed] [Google Scholar]
- 34.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–58. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 35.Czermak BJ, Sarma V, Pierson CL, et al. Protective effects of C5a blockade in sepsis. Nat Med. 1999;5:788–92. doi: 10.1038/10512. [DOI] [PubMed] [Google Scholar]
- 36.Reddy RC, Chen GH, Tekchandani PK, Standiford TJ. Sepsis-induced immunosuppression: from bad to worse. Immunol Res. 2001;24:273–87. doi: 10.1385/IR:24:3:273. [DOI] [PubMed] [Google Scholar]
- 37.Kaufmann I, Hoelzl A, Schliephake F, et al. Polymorphonuclear leukocyte dysfunction syndrome in patients with increasing sepsis severity. Shock. 2006;26:254–61. doi: 10.1097/01.shk.0000223131.64512.7a. [DOI] [PubMed] [Google Scholar]
- 38.Yekebas EF, Eisenberger CF, Ohnesorge H, et al. Attenuation of sepsis-related immunoparalysis by continuous veno–venous hemofiltration in experimental porcine pancreatitis. Crit Care Med. 2001;29:1423–30. doi: 10.1097/00003246-200107000-00021. [DOI] [PubMed] [Google Scholar]
- 39.Tennenberg SD, Solomkin JS. Neutrophil activation in sepsis. The relationship between fmet-leu-phe receptor mobilization and oxidative activity. Arch Surg. 1988;123:171–5. doi: 10.1001/archsurg.1988.01400260051005. [DOI] [PubMed] [Google Scholar]
- 40.Tschaikowsky K, Sittl R, Braun GG, Hering W, Rügheimer E. Increased fMet-Leu-Phe receptor expression and altered superoxide production of neutrophil granulocytes in septic and posttraumatic patients. Clin Invest. 1993;72:18–25. doi: 10.1007/BF00231111. [DOI] [PubMed] [Google Scholar]
- 41.Shawcross DL, Wright GAK, Stadlbauer V, et al. Ammonia impairs neutrophil phagocytic function in liver disease. Hepatology. 2008;48:1202–12. doi: 10.1002/hep.22474. [DOI] [PubMed] [Google Scholar]
- 42.Sibelius U, Hattar K, Hoffmann S, et al. Distinct pathways of lipopolysaccharide priming of human neutrophil respiratory burst: role of lipid mediator synthesis and sensitivity to interleukin-10. Crit Care Med. 2002;30:2306–12. doi: 10.1097/00003246-200210000-00020. [DOI] [PubMed] [Google Scholar]
- 43.Yan SR, Al-Hertani W, Byers D, Bortolussi R. Lipopolysaccharide-binding protein- and CD14-dependent activation of mitogen-activated protein kinase p38 by lipopolysaccharide in human neutrophils is associated with priming of respiratory burst. Infect Immun. 2002;70:4068–74. doi: 10.1128/IAI.70.8.4068-4074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hailman E, Vasselon T, Kelley M, et al. Stimulation of macrophages and neutrophils by complexes of lipopolysaccharide and soluble CD14. J Immunol. 1996;156:4384–90. [PubMed] [Google Scholar]
- 45.Lamping N, Dettmer R, Schröder NW, et al. LPS-binding protein protects mice from septic shock caused by LPS or Gram-negative bacteria. J Clin Invest. 1998;101:2065–71. doi: 10.1172/JCI2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zweigner J, Gramm HJ, Singer OC, Wegscheider K, Schumann RR. High concentrations of lipopolysaccharide-binding protein in serum of patients with severe sepsis or septic shock inhibit the lipopolysaccharide response in human monocytes. Blood. 2001;98:3800–8. doi: 10.1182/blood.v98.13.3800. [DOI] [PubMed] [Google Scholar]
- 47.Opal SM, Scannon PJ, Vincent JL, et al. Relationship between plasma levels of lipopolysaccharide (LPS) and LPS-binding protein in patients with severe sepsis and septic shock. J Infect Dis. 1999;180:1584–9. doi: 10.1086/315093. [DOI] [PubMed] [Google Scholar]
- 48.Vesy CJ, Kitchens RL, Wolfbauer G, Albers JJ, Munford RS. Lipopolysaccharide-binding protein and phospholipid transfer protein release lipopolysaccharides from Gram-negative bacterial membranes. Infect Immun. 2000;68:2410–17. doi: 10.1128/iai.68.5.2410-2417.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thompson PA, Tobias PS, Viriyakosol S, Kirkland TN, Kitchens RL. Lipopolysaccharide (LPS)-binding protein inhibits responses to cell-bound LPS. J Biol Chem. 2003;278:28367–71. doi: 10.1074/jbc.M302921200. [DOI] [PubMed] [Google Scholar]
- 50.Cathcart MK, Morel DW, Chisolm GM. Monocytes and neutrophils oxidize low density lipoprotein making it cytotoxic. J Leukoc Biol. 1985;38:341–50. doi: 10.1002/jlb.38.2.341. [DOI] [PubMed] [Google Scholar]
- 51.Schneiderhan W, Schmid-Kotsas A, Zhao J, et al. Oxidized low-density lipoproteins bind to the scavenger receptor, CD36, of hepatic stellate cells and stimulate extracellular matrix synthesis. Hepatology. 2001;34:729–37. doi: 10.1053/jhep.2001.27828. [DOI] [PubMed] [Google Scholar]
- 52.Wiesner P, Choi S, Almazan F, et al. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor kappab and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxaemia. Circ Res. 2010;107:56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geng H, Wang A, Rong G, et al. The effects of ox-LDL in human atherosclerosis may be mediated in part via the toll-like receptor 4 pathway. Mol Cell Biochem. 2010;342:201–6. doi: 10.1007/s11010-010-0484-8. [DOI] [PubMed] [Google Scholar]
- 54.Lopes-Virella MF. Interactions between bacterial lipopolysaccharides and serum lipoproteins and their possible role in coronary heart disease. Eur Heart J. 1993;14:118–24. [PubMed] [Google Scholar]
- 55.Charach G, George J, Afek A, et al. Antibodies to oxidized LDL as predictors of morbidity and mortality in patients with chronic heart failure. J Card Fail. 2009;15:770–4. doi: 10.1016/j.cardfail.2009.05.009. [DOI] [PubMed] [Google Scholar]