

Abstract
Spergualin is a natural product that exhibits immunosuppressive, anti-tumor and anti-bacterial activities. Its derivatives, such as 15-deoxyspergualin (15-DSG), have been clinically approved for acute allograft rejection. However, the reported syntheses are cumbersome (> 10 steps) and they suffer from low overall yields (~ 0.3 to 18%). Moreover, spergualin and its derivatives are chemically unstable and rapidly hydrolyzed in aqueous buffer. Here, we have re-explored these issues and report a modified synthetic route with significantly improved overall yield (~31 to 47%). The key transformation is a microwave-accelerated Ugi multi-component reaction that is used to generate the peptoid core in a single step. Using the products of this route, we found that modifications of the hemiaminal significantly increased chemical stability. Thus, we anticipate that this synthetic route will improve access to biologically active 15-DSG derivatives.
Keywords: polyamine, chemical stability, combinatorial synthesis, immunosuppression, peptoid
Spergualin is a natural product derived from Bacillus laterosporus, which has gathered significant medical interest because of its potent immunosuppressive, anti-tumor and antibacterial activities.1–4 Early structural studies revealed that spergualin is composed of three key regions: a peptoid core, guanidylated alkyl group and spermidine-derived polyamine (Figure 1)5 and each of these modules are thought to be required for biological activity.6–8 However, attempts to further characterize the pharmacology of spergualin were complicated by its rapid hydrolysis in aqueous buffers. Thus, a major goal of early synthetic efforts was to modify the most labile regions, such as the hydroxyl at position 15.8 These efforts yielded the significantly more stable (−)-15-deoxyspergualin (15-DSG) (Figure 1), which was granted clinical approval to treat acute allograft rejection.9 More recent efforts have yielded additional derivatives, such as tresperimus (Figure 1), in which a portion of the unstable peptoid is replaced with a carbamate.10, 11 However, although these derivatives are an improvement on spergualin, they are still relatively unstable, with short half-lives (t1/2) in neutral and basic conditions.10 In addition, they are only weakly orally bioavailable (<5%) and metabolized rapidly, with terminal half-lives of only 1–2 hours.12
Figure 1.

Chemical structures of the natural product, spergualin, and two of its derivatives. A schematic of the architecture is shown and the positions of carbons 11 and 15 are indicated.
One major challenge in the search for improved spergualin derivatives is that the reported synthetic routes are cumbersome and low yielding. For example, the first attempts produced 15-DSG in only 0.3% yield in more than 10 steps, starting from L-lysine and 1-amino-propanol (Figure 2A).6, 7, 13 Later efforts mildly improved the yield of 15-DSG (to ~7% to 18%) by starting with 7-bromoheptanenitrile and a protected spermidine derivative (Figure 2B), but these convergent routes remain protracted (> 10 steps) and challenging.14–16
Figure 2.

Comparison of the retrosynthetic analyses. Known convergent routes to spergualin (A) and 15-DSG (B) proceed through a series of more than 10 convergent steps. (C) The proposed route employs the Ugi multi-component reaction to generate the peptoid and guanidylated regions in a single step. See the text for references.
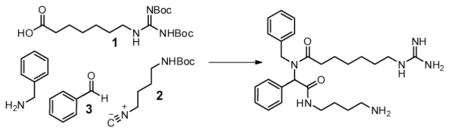
We envisioned an alternative synthetic approach. Specifically, our retrosynthetic analysis employs the Ugi multicomponent reaction to assemble the core of spergualin in a single step (Figure 2C). The Ugi reaction proceeds through the condensation of a carboxylic acid, amine, isocyanide, and aldehyde (or ketone).17, 18 Thus, in addition to the potential advantages gained from the concise creation of the peptoid, this route is also expected to allow access to combinatorial diversity through varying the identity of the four modules, including the guanidinylated amino acid (1), protected isocyanide (2) and aldehyde (3).
Many conditions for the Ugi reaction have been reported, with the most recent studies focusing on the use of microwave irradiation to improve yield and reaction times.19, 20 Guided by these efforts, we first tested the feasibility of the approach using the commercially available isocyanide, 1-isocyanopentane, as a model reactant. Briefly, an equimolar solution of benyzlamine, benzaldehyde, 6-guanidinoheptanoic acid,21 and 1-isocyanopentane was heated in methanol at 120 °C for 20 minutes in a microwave reactor. These conditions yielded the product in good (70%) yield. Encouraged by this observation, we next substituted the simple isocyanide with the desired, tert-butyl (4-isocyanobutyl)carbamate (2).22 Unfortunately, we found that this change dropped the yield to 11%. To solve this issue, we reinvestigated the reaction conditions, focusing on the variables of time, temperature, and solvent (Table 1). This effort revealed that the yield was particularly sensitive to solvent and that DMF tended to be the best choice (average purified yields between 36–46%).
Table 1.
Optimization of the Ugi reaction conditions.
 | ||||
|---|---|---|---|---|
| entry | solvent | temp (°C) | time (min) | yield (%) |
| 1 | MeOH | 100 | 20 | 2–10 |
| 2 | MeOH | 100 | 30 | <2 |
| 3 | MeOH | 90 | 20 | 36 |
| 4 | MeOH | 90 | 30 | 22 |
| 5 | MeOH | 110 | 20 | <2 |
| 6 | MeOH | 120 | 10 | 12 |
| 7 | MeOH | 130 | 10 | n.d. |
| 8 | MeOH | rt | 240 | 35 |
| 9 | EtOH | 90 | 20 | 27 |
| 10 | EtOH | 90 | 40 | 17 |
| 11 | DMF | 100 | 20 | 46 |
| 12 | DMF | 200 | 20 | 36 |
| 13 | DMF | rt | 240 | 43 |
Using the optimized Ugi conditions (DMF, 100 °C, 20 minutes), we then pursued the synthesis of 15-DSG derivatives. Although this reaction is often used in the assembly of large, combinatorial libraries, our first efforts focused on producing a small number of focused derivatives to test specific issues related to chemical stability (see below). Towards that goal, we combined four aldehydes (3a-d) with 7-guanidinoheptanoic acid (1), benzylamine, and tert-butyl (4-isocyanobutyl)carbamate (2) to produce intermediates 4a-d after Boc deprotection. In the next step, we envisioned using a reductive amination to complete the sperimidine module. Towards that goal, we first assembled Fmoc-3-amino-1-propanal (5) from Fmoc-β-alanine, using the method of More and Finney.23 Then, compound 5 (0.05 mmol, 1 equiv) was reacted with 4a-d (0.05 mmol, 1 equiv) in 2 mL THF and NaBH(OAc)3 (0.07 mmol, 1.4 equiv) for 1.5 hours to produce intermediates 6a-d. Importantly, NaBH(OAc)3 was chosen for this step because, unlike NaBH3CN, it doesn’t require low pH.24 This was an important consideration because of the sensitivity of spergualin analogs to degradation. Following the reductive amination, we sought to remove the final protecting groups. However, we found that common ways of removing Fmoc, such as 20% piperidine in DMF, caused significant hydrolysis. Therefore, we employed the alternative, Tris-amine resin (50 equiv) in CHCl3 for 20 minutes.25 Finally, the benzyl group was readily removed under mild conditions using ammonium cerium (IV) nitrate (CAN).26 Following a final purification by alumina chromatography, the products 7a-d were isolated as racemic mixtures in 31 to 47% overall yield (Figure 3).
Figure 3.

Synthesis of spergualin analogs. The Ugi condensation of aldehydes 3a-d, followed by Boc deprotection, produced the intermediates 4a-d in good yield. Reductive amination with the aldehyde 5, followed by deprotection of the final Fmoc and benzyl groups yielded the final products 7a-d in overall, purified yields from 31 to 47 %.
With these compounds in hand, we wanted to examine their stability in aqueous buffers. As mentioned above, one of the biggest challenges in studies of spergualin and its analogs is that they are prone to hydrolysis. Previous work had suggested that removal of the labile 15-hydroxyl group could improve stability.11 However, Lebreton et al. found that even the clinically approved 15-DSG had a t1/2 of only ~2 hrs in pH 10 buffer.10 Under these conditions, degradation is thought to occur via hydrolysis at the hemiaminal (Figure 4C).27 Thus, we reasoned that blocking this degradation route by modifying the C11 position might improve persistence. To test this idea, we first compared the stability of spergualin and 11-methoxy-15-deoxyspergualin (7a) using thin layer chromatography. Consistent with previous findings, the natural product (Sigma Aldritch cat #S5822) is relatively stable under mild acidic conditions (pH 5.0) but it is highly unstable in neutral and basic buffers (Figure 4A). By comparison, compound 7b, with the methoxy substitution at position C11, had significantly improved stability under basic and neutral conditions (Figure 4B). Next, we quantified the t1/2 values of compounds 7a-d and compared them to those of spergualin and 15-DSG. In these studies, we found that the t1/2 values of compound 7a at pH 7.0 and 8.0 were 8- and 120-fold greater than spergualin and at least 4-fold greater than 15-DSG (Figure 4D). Even the relatively simple substitution in 7d, removal of the C11 hydroxyl, greatly improved stability. More striking, bulky substitutions at position 11, as in compound 7b and 7c, greatly improved stability to greater than 2 weeks. Together, these results suggest that multiple types of substitutions at the hemiaminal could provide significant increases in stability under neutral and basic conditions.
Figure 4.

Synthetic derivatives of spergualin are more stable than the natural product. (A) The stability of spergualin was tested by thin layer chromatography. The time points were initiated immediately after dissolution in aqueous buffers at the indicated pH. (B) Stability of 7a under the same conditions. Results are the average of at least independent triplicates and the error bars represent standard error of the mean. (c) Summary of the known hydrolysis products of 15-DSG (D) Table of the stability values for spergualin, 15-DSG and compounds 7a-d. Also included are the relative anti-bacterial activities (+++ MIC < 10 μg/mL; ++ MIC 10–50 μg/mL; + MIC 50–250 μg/mL; − MIC >250 μg/mL).
To understand whether the introduced substitutions might disrupt bioactivity, we tested compounds 7a-d in a series of antibacterial assays against Escherichia coli, Bacillus subtilis and Staphylococcus aureus. Consistent with previous reports, spergualin and 15-DSG had anti-bacterial activity, especially against the gram-positive strains (Figure 4D). We found that compounds 7a-d retained some anti-bacterial activity, although the relative potencies were decreased. Of these compounds, 7d appeared the most promising, with good activity against B. subtilis and S. aureus. Additional studies are clearly needed to understand the relevant structure-activity relationships and to improve compound potency. However, these initial efforts show that spergualin analogs with greatly improved chemical stability retain some bioactivity.
In this work, an improved synthetic method for producing 15-DSG analogs was developed, which increased the overall yields by at least 2-fold and greatly reduced the number of steps. The key transformation is the Ugi reaction to simultaneously generate the peptoid and guanidylated regions. Using this approach, we found that substitutions of the hemiaminal significantly improved chemical stability of these compounds, which is expected to provide a path towards exploration of these compounds as both research probes and therapeutics. The next steps are to improve the in vivo metabolic stability and better understand their pharmacologic activities.
Supplementary Material
Acknowledgments
C.G.E. was supported by a predoctoral fellowship from the Cellular Biotechnology Training Grant (GM008353). M.C.S. was supported by a predoctoral fellowship from the Biogerontology Training Grant (AG000114). This work was additionally supported by grants from the NIH (NS059690) and NSF (MCB-0844512).
Footnotes
Supplementary material is available online.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Takeuchi T, Iinuma H, Kunimoto S, Masuda T, Ishizuka M, Takeuchi M, Hamada M, Naganawa H, Kondo S, Umezawa H. J Antibiot (Tokyo) 1981;34:1619. doi: 10.7164/antibiotics.34.1619. [DOI] [PubMed] [Google Scholar]
- 2.Umezawa H, Ishizuka M, Takeuchi T, Abe F, Nemoto K, Shibuya K, Nakamura T. J Antibiot (Tokyo) 1985;38:283. doi: 10.7164/antibiotics.38.283. [DOI] [PubMed] [Google Scholar]
- 3.Thomas FT, Tepper MA, Thomas JM, Haisch CE. Ann N Y Acad Sci. 1993;685:175. doi: 10.1111/j.1749-6632.1993.tb35863.x. [DOI] [PubMed] [Google Scholar]
- 4.Evans CG, Chang L, Gestwicki JE. J Med Chem. 2010;53:4585. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Umezawa H, Kondo S, Iinuma H, Kunimoto S, Ikeda Y, Iwasawa H, Ikeda D, Takeuchi T. J Antibiot (Tokyo) 1981;34:1622. doi: 10.7164/antibiotics.34.1622. [DOI] [PubMed] [Google Scholar]
- 6.Umeda Y, Moriguchi M, Kuroda H, Nakamura T, Fujii A, Iinuma H, Takeuchi T, Umezawa H. J Antibiot (Tokyo) 1987;40:1303. doi: 10.7164/antibiotics.40.1303. [DOI] [PubMed] [Google Scholar]
- 7.Umeda Y, Moriguchi M, Kuroda H, Nakamura T, Iinuma H, Takeuchi T, Umezawa H. J Antibiot (Tokyo) 1985;38:886. doi: 10.7164/antibiotics.38.886. [DOI] [PubMed] [Google Scholar]
- 8.Umeda Y, Moriguchi M, Ikai K, Kuroda H, Nakamura T, Fujii A, Takeuchi T, Umezawa H. J Antibiot (Tokyo) 1987;40:1316. doi: 10.7164/antibiotics.40.1316. [DOI] [PubMed] [Google Scholar]
- 9.Kaufman DB, Gores PF, Kelley S, Grasela DM, Nadler SG, Ramos E. Transplant Rev. 1996;10:160. [Google Scholar]
- 10.Lebreton L, Annat J, Derrepas P, Dutartre P, Renaut P. J Med Chem. 1999;42:277. doi: 10.1021/jm980431g. [DOI] [PubMed] [Google Scholar]
- 11.Lebreton L, Jost E, Carboni B, Annat J, Vaultier M, Dutartre P, Renaut P. J Med Chem. 1999;42:4749. doi: 10.1021/jm991043x. [DOI] [PubMed] [Google Scholar]
- 12.Ohlman S, Zilg H, Schindel F, Lindholm A. Transpl Int. 1994;7:5. doi: 10.1007/BF00335656. [DOI] [PubMed] [Google Scholar]
- 13.Kondo S, Iwasawa H, Ikeda D, Umeda Y, Ikeda Y, Iinuma H, Umezawa H. J Antibiot (Tokyo) 1981;34:1625. doi: 10.7164/antibiotics.34.1625. [DOI] [PubMed] [Google Scholar]
- 14.Bergeron RJ, McManus JS. J Org Chem. 1987;52:1700. [Google Scholar]
- 15.Durand P, Richard P, Renaut P. J Org Chem. 1998;63:9723. [Google Scholar]
- 16.Durand P, Peralba P, Renaut P. Tetrahedron. 2001;57:2757. [Google Scholar]
- 17.Marcaccini S, Torroba T. Nat Protoc. 2007;2:632. doi: 10.1038/nprot.2007.71. [DOI] [PubMed] [Google Scholar]
- 18.Wang W, Domling A. J Comb Chem. 2009;11:403. doi: 10.1021/cc9000136. [DOI] [PubMed] [Google Scholar]
- 19.Lew A, Krutzik PO, Hart ME, Chamberlin AR. J Comb Chem. 2002;4:95. doi: 10.1021/cc010048o. [DOI] [PubMed] [Google Scholar]
- 20.Ugi I, Heck S. Comb Chem High Throughput Screen. 2001;4:1. doi: 10.2174/1386207013331291. [DOI] [PubMed] [Google Scholar]
- 21.Feichtinger K, Zapf C, Sings HL, Goodman M. J Org Chem. 1998;63:3804. [Google Scholar]
- 22.Xu PZT, Wang W, Zou X, Zhang X, Fu Y. Synthesis. 2003:1171. [Google Scholar]
- 23.More JD, Finney NS. Org Lett. 2002;4:3001. doi: 10.1021/ol026427n. [DOI] [PubMed] [Google Scholar]
- 24.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 25.Carpino LA, Sadat-Aalaee D, Beyermann M. J Org Chem. 1990;55:1673. [Google Scholar]
- 26.Bull SD, Davies SG, Fenton G, Mulvaney AW, Prasad RS, Smith AD. J Chem Soc, Perkin Trans I. 2000;2000:3765. [Google Scholar]
- 27.Nishizawa R, Takei Y, Yoshida M, Tomiyoshi T, Saino T, Nishikawa K, Nemoto K, Takahashi K, Fujii A, Nakamura T, et al. J Antibiot (Tokyo) 1988;41:1629. doi: 10.7164/antibiotics.41.1629. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.