

Abstract
Alphaviruses infect their host by binding cellular receptors and fusing with cell membranes. New structures define the receptor-binding protein of these viruses and its regulation of the membrane-fusion reaction.
Many viruses are enclosed within an envelope – a membrane that is derived from the infected host cell during virus exit. To infect a new host cell, specialized membrane-fusion proteins on the virus envelope fuse it with a membrane of the host cell, delivering the viral genome into the cell. This fusion activity must be deployed only at the correct time during virus entry, and must also be silenced during viral assembly and exit. In this issue, Li et al.7 and Voss et al.8 provide structural insights into the regulation of the membrane-fusion proteins of enveloped alphaviruses during the entry and exit of these viruses in the host cell.
Many alphaviruses are medically relevant; chikungunya virus, for example, is an emerging human pathogen responsible for major recent epidemics9. There are currently no treatments for alphavirus infections, and detailed information on the structure and lifecycle of these viruses is crucial for developing antiviral strategies and vaccines.
But first, a quick glance at what is already known. E1 is the membrane-fusion protein of alphaviruses, and its fusion activity is triggered by the mildly acidic pH of intracellular vesicles1. Structural studies have defined the architecture of the E1 molecule2,3, its arrangement on the virus particle3–5, and the E1 conformational changes that drive membrane fusion6. E1 is tightly associated with another membrane protein, E2, which has been an elusive missing piece of the virus’s structural puzzle. The alphavirus envelope is covered by an organized lattice composed of E2/E1 pairs arranged into 80 trimers or ‘spikes’.
The alphavirus infection cycle begins when E2 binds receptors on the surface of a host cell. This allows internalization of the virus and its transport into the acidic intracellular vesicles. The low pH induces a rearrangement of the E2/E1 pair, allowing E1’s fusion activity to be unleashed12. E1 inserts its hydrophobic fusion loop into the membrane of the host cell vesicle, forms E1 trimers, and refolds to pull the host cell and viral membranes together, thus causing membrane fusion and virus infection6.
In addition to binding the host cell receptor, E2 is also an essential component throughout the virus’s lifecycle. During viral replication, this protein is synthesized as a precursor called p62 (or PE2) and acts to chaperone the folding of its E1 partner. Similar to the vesicular entry pathway, the exit pathway also involves transport through cellular compartments that have an acidic pH. The p62/E1 pair is more acid-resistant than the E2/E1 dimer, and this appears to protect E1 from premature fusion during transport through the exit pathway10. Late in transport, the cellular enzyme furin cleaves p62 to produce the mature E2 protein plus a small peripheral protein, E311. The virus then exits by budding from the cell surface, with some alphavirus species retaining E3 and others releasing it.
Li et al.7 (page XXX) and Voss et al.8 (page YYY) present the molecular structure of the E2/E1 pair and define the mechanisms by which E2 both silences E1 during virus exit and regulates E1’s triggering at low pH during virus entry. Focusing on sindbis virus7 and chikungunya virus8, the authors generated modified versions of p62/E1 proteins that were joined together by flexible linkers and lacked their membrane-anchoring domains. This was critical to stabilizing E2 for structural studies. Each team determined the crystal structures of these protein pairs for their virus and fit it into the molecular outline of the alphavirus particle, previously established by electron microscopy3–5. Voss and colleagues’ chikungunya virus structures define the immature p62/E1 pair and the mature E2/E1 complex with the retained E3, whereas the sindbis virus structures of Li et al. reveal the mature E2/E1 pair (without E3), associated in trimeric spikes as on the surface of the virus.
The new structures show that the mature E2 protein is an elongated molecule containing three domains with immunoglobulin-like folds: the amino-terminal domain A, located at the centre; domain B at the tip; and the carboxy-terminal domain C, located close to the viral membrane.
The chikungunya E2 covers much of its E1 partner on the virus surface, with the hydrophobic fusion loop of E1 clamped in the groove between the domains A and B of E2. The sindbis E2/E1 pair was crystallized at acidic pH, and although it closely resembles the chikungunya E2/E1 structure, the E2 domain B that “caps” the E1 fusion loop is disordered and not visualized. This structure suggests that an early intermediate in the low pH-triggered fusion process is formed by the release of the E2 domain B, exposing the E1 fusion loop. This E2/E1 rearrangement seems to occur through changes in a flexible ribbon-like connector that links domain B to domains A and C, and packs tightly against the underlying E1 protein.
The immature p62/E1 and mature E3/E2/E1 complexes are very similar apart from the tether region that links E3 to E2 and is the site of furin cleavage. This suggests that the major difference producing the increased acid-resistance of the immature spike is that the connector ribbon maintaining the domain-B cap in place is stabilized by interactions of the tethered E3.
The new structures7,8 illuminate key aspects of the alphavirus lifecycle. In addition, the exposed regions of E2 domains A and B contain several sites to which neutralizing antibodies bind, as well as sites implicated in virus–receptor interaction. The structures of these domains therefore can now be used to clarify the mechanisms of virus receptor-binding and neutralization, and to exploit these processes for antiviral and vaccine strategies.
The structures of the p62/E1 and E2/E1 pairs suggest specific residues that may control their dissociation at low pH and explain how p62 and E2 regulate virus fusion. Knowing the details of the p62/E1 interaction will also help to determine how much of the requirement for p62 during E1 synthesis is due to protection of E1 from low pH vs. to direct assistance of p62 with E1 folding. The intriguing ‘uncapped’ structure of the alphavirus spike points out how little is known about downstream fusion intermediates, which must involve considerable movements of E2 and E1 on a highly organized virus particle and will be an exciting area for future work.
Figure 1.
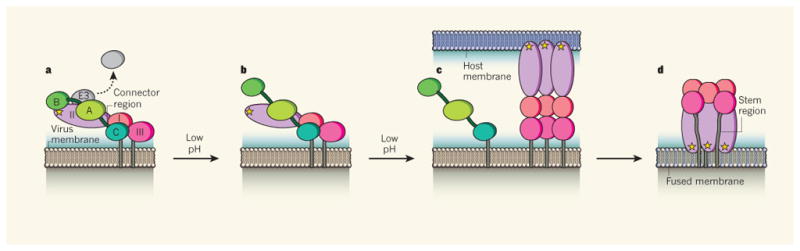
A. A schematic view of the alphavirus p62/E1 proteins. E1 is shown with domains I, II, and III colored in red, yellow, and blue, respectively, and the fusion loop indicated by the green star. The p62 protein is shown with domain A in cyan, domain B in green, domain C in pink, the ribbon-like connector in purple, the E3 region in grey. The virus membrane is shown in brown. Only 1 p62/E1 pair in the trimeric spike is shown.
B. p62 is cleaved by furin, untethering the E3 protein which is released in some alphaviruses.
C. Low pH causes the movement of E2 domain B and the connector, exposing the E1 fusion loop.
D. E1 inserts into the host cell membrane. E2 releases E1 but the timing of release and the location and conformation of E2 at this stage are undefined.
E. E1 forms an extended trimer.
F. E1 refolds to a hairpin-like structure via the movement of domain III and the juxtamembrane stem region (grey). This refolding drives membrane fusion. The fused membrane is shown in purple.
References
- 1.Helenius A, Kartenbeck J, Simons K, Fries E. J Cell Biol. 1980;84:404–420. doi: 10.1083/jcb.84.2.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lescar J, et al. Cell. 2001;105:137–148. doi: 10.1016/s0092-8674(01)00303-8. [DOI] [PubMed] [Google Scholar]
- 3.Roussel A, et al. Structure. 2006;14:75–86. doi: 10.1016/j.str.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 4.Pletnev SV, et al. Cell. 2001;105:127–136. doi: 10.1016/s0092-8674(01)00302-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W, et al. J Virol. 2002;76:11645–11658. doi: 10.1128/JVI.76.22.11645-11658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gibbons DL, et al. Nature. 2004;427:320–325. doi: 10.1038/nature02239. [DOI] [PubMed] [Google Scholar]
- 7.Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. Nature. 2010;XXX:XXX–XXX. doi: 10.1038/nature09546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voss JE, et al. Nature. 2010;XXX:YYY–YYY. [Google Scholar]
- 9.Schwartz O, Albert ML. Nature Rev Micro. 2010;8:491–500. doi: 10.1038/nrmicro2368. [DOI] [PubMed] [Google Scholar]
- 10.Wahlberg JM, Boere WAM, Garoff H. J Virol. 1989;63:4991–4997. doi: 10.1128/jvi.63.12.4991-4997.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Fugere M, Day R, Kielian M. J Virol. 2003;77:2981–2989. doi: 10.1128/JVI.77.5.2981-2989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wahlberg JM, Garoff H. J Cell Biol. 1992;116:339–348. doi: 10.1083/jcb.116.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]