

Abstract
Infection of C57BL/6 mice with Plasmodium berghei ANKA induces a fatal neurological disease commonly referred to as experimental cerebral malaria. The onset of neurological symptoms and mortality depend on pathogenic CD8+ T cells and elevated parasite burdens in the brain. Here we provide clear evidence of liver damage in this model, which precedes and is independent of the onset of neurological symptoms. Large numbers of parasite-specific CD8+ T cells accumulated in the liver following P. berghei ANKA infection. However, systemic depletion of these cells at various times during infection, while preventing neurological symptoms, failed to protect against liver damage or ameliorate it once established. In contrast, rapid, drug-mediated removal of parasites prevented hepatic injury if administered early and quickly resolved liver damage if administered after the onset of clinical symptoms. These data indicate that CD8+ T cell-mediated immune pathology occurs in the brain but not the liver, while parasite-dependent pathology occurs in both organs during P. berghei ANKA infection. Therefore, we show that P. berghei ANKA infection of C57BL/6 mice is a multiorgan disease driven by the accumulation of parasites, which is also characterized by organ-specific CD8+ T cell-mediated pathology.
INTRODUCTION
Severe malarial syndromes, including severe anemia, hyperparasitemia, cerebral malaria, acute respiratory distress, and clinical jaundice (37), differ significantly in terms of the target organs of disease, with the brain, lungs, kidney, and liver all variously affected. Nevertheless, a common feature of all severe malaria syndromes is that parasite burdens are significantly higher than in patients suffering uncomplicated malaria (9). This implies that parasite burdens play a crucial role in causing cerebral, respiratory, and hepatic disease during malaria.
Numerous experimental mouse models have been employed to study the pathogenesis of severe malaria syndromes. For example, infection with Plasmodium chabaudi and Plasmodium yoelii strains can induce hyperparasitemia and severe anemia (20–22), while infection with Plasmodium berghei NK65 induces immune-mediated liver damage (1, 2, 13, 40). Infection of C57BL/6 mice with P. berghei ANKA induces a fatal cerebral disease characterized by breakdown of the blood-brain barrier and onset of neurological symptoms mediated by CD8+ T cells (8, 15, 39). This model is commonly referred to as experimental cerebral malaria (ECM) (10). However, acute lung injury has also been reported in ECM (11, 12, 23, 29, 32). In addition, one study reported liver damage in this model, although the mechanisms of pathogenesis and their relationship to cerebral disease were not investigated (18).
Recent work from our laboratory and others has shown that parasite burden is crucial in triggering the onset of neurological symptoms in ECM (3, 5, 25). We have also shown that parasites accumulate in many tissues, including the liver, which is one of the sites of greatest parasite tissue sequestration in ECM (3). In this study, we examined the relative roles of parasites and CD8+ T cells in causing and sustaining liver damage following P. berghei ANKA infection. We show that CD8+ T cell-mediated immune pathology is organ specific, occurring in the brain but not the liver, while parasite-driven pathology is evident in both organs.
MATERIALS AND METHODS
Mice and ethics statement.
Female C57BL/6 mice and congenic CD45.1+ C57BL/6 mice aged 6 to 8 weeks were purchased from the Australian Resource Centre (Canning Vale, Perth, Western Australia) and maintained under conventional conditions. OTI mice (16) and perforin-deficient mice were bred and maintained in-house. This study was carried out in strict accordance with guidelines from the National Health and Medical Research Council of Australia, as detailed in the document Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 7th ed. (2004). All animal procedures and protocols were approved (A02-633 M) and monitored by the Queensland Institute of Medical Research Animal Ethics Committee.
Parasites and infections.
P. berghei ANKA lines were used in all experiments after one in vivo passage in mice. A transgenic P. berghei ANKA (231c1l) line expressing luciferase and green fluorescent protein under the control of the EF1-α promoter (P. berghei ANKA-luc) was used for experiments involving in vivo imaging (3, 4). Transgenic P. berghei ANKA strains expressing model T cell epitopes (P. berghei ANKA-Ova) and control strains were maintained and used as previously reported (24). All mice were infected with 105 parasitized red blood cells (pRBCs) intravenously (i.v.) via the lateral tail vein. Blood parasitemia was monitored by examination of Diff-Quick (Lab Aids, Narrabeen, NSW, Australia)-stained thin blood smears obtained from tail bleeds. Sodium artesunate (Gulin Pharmaceutical Co., Ltd., Gulin, Guangxi, China) was prepared according to the manufacturer's instructions, diluted in saline, and administered to mice twice with an interval of 12 h at 10 mg/kg and subsequently daily via intraperitoneal (i.p.) injection at the same dose.
Disease assessment.
Mice were monitored twice daily after day 5 postinfection (p.i.), and clinical ECM was evaluated (3, 4, 31). Clinical ECM scores were defined by the presentation of the following signs: ruffled fur, hunching, wobbly gait, limb paralysis, convulsions, and coma. Each sign was given a score of 1. Animals with severe ECM (cumulative score of 4) were sacrificed by CO2 asphyxiation according to ethics guidelines, and the following time point was given a score of 5 to denote death.
Liver enzyme analysis.
Heparinized blood was immediately centrifuged for 10 min at 300 × g. Plasma was refrigerated and tested within 12 h of receipt. Plasma samples were processed in a single batch for determination of alanine aminotransferase (ALT) and aspartate transaminase (AST) levels using a Beckman Unicell DxC800 analyzer.
Liver histological examination.
Formalin-fixed, paraffin-embedded liver sections were stained with hematoxylin and eosin or with picrosirius red to detect collagen protein deposition.
Antibodies and other reagents.
Allophycocyanin (APC)- or Pacific Blue (PB)-conjugated anti-T cell receptor β (TCRβ) (H57-597) chain, phycoerythrin (PE)-Cy5- or PE-conjugated anti-CD4 (GK1.5), PE-Cy5-conjugated anti-CD8α (53-6.7), and PE- or fluorescein isothiocyanate-conjugated anti-CD45.1 (A20), were purchased from Biolegend (San Diego, CA) or BD Biosciences (Franklin Lakes, NJ). PE-conjugated anti-human granzyme B (GzmB) monoclonal antibody (MAb) (cross-reactive with mouse GzmB; GB12) was purchased from Caltag Laboratories. Anti-CD8β (53-5.8), anti-CD4 (YTS.191), and isotype control MAbs were purified from culture supernatants by protein G column purification (Amersham, Uppsala, Sweden) followed by endotoxin removal (Mustang membranes; PallLife Sciences, East Hills, NY). Purified control rat IgG was also used in some experiments and was purchased from Sigma-Aldrich (Castle Hill, NSW, Australia). CD8+ T cell depletion (>90% efficiency in the liver) was achieved by administration of 0.5 mg of 53-5.8 MAb intraperitoneally, followed by a 0.2-mg i.p. dose 3 days afterwards if required.
Preparation of liver and brain mononuclear cells.
Liver and brain mononuclear cells were isolated as described previously (14, 35, 36). Briefly, tissue was passed through a 200-μm metal sieve in RPMI 1640 tissue culture medium supplemented with 2% (vol/vol) fetal calf serum (wash buffer) and washed twice with wash buffer. The cell pellet was resuspended in 33% (vol/vol) Percoll in phosphate-buffered saline (PBS) and centrifuged at 693 × g for 12 min at room temperature. Supernatant containing unwanted cells and debris was removed, the leukocyte pellet was washed once in wash buffer, and red blood cells were lysed using a hypotonic buffer (Sigma) and washed and resuspended in RPMI 1640 medium supplemented with 5% (vol/vol) fetal calf serum.
Flow cytometric analysis.
For the staining of cell surface antigens, cells were incubated with fluorochrome-conjugated MAbs on ice for 20 min, washed twice, and fixed in 2% (wt/vol) paraformaldehyde in PBS. Alternatively, samples to be stained intracellularly were fixed and permeabilized using BD Cytofix/Cytoperm kits according to the manufacturer's instructions. Data were acquired on a FACSCanto II flow cytometer (BD Biosciences) and analyzed using FlowJo software (Treestar, Ashland, OR).
In vivo bioluminescence imaging.
Luciferase-expressing P. berghei ANKA pRBCs were visualized by imaging whole bodies or dissected organs with an I-CCD photon-counting video camera and in vivo imaging system (IVIS 100; Xenogen, Alameda, CA) (3, 4, 31). Mice were anesthetized with fluorothane and injected intraperitoneally with 0.1 ml of 5 mg/ml d-luciferin firefly potassium salt (Xenogen). Five minutes afterwards, images were captured on the IVIS 100 according to the manufacturer's instructions. Parasites were visualized in the liver and brain after removal from mice that had been perfused via the heart with 10 ml of cold PBS. Bioluminescence generated by luciferase transgenic P. berghei ANKA in mouse liver or brain tissue was measured according to the manufacturer's instructions. The unit of measurement was photons/s/cm2/steradian (sr).
Statistical analysis.
Differences in cell numbers, whole body and organ bioluminescence, and serum ALT and AST levels were analyzed using the Mann-Whitney nonparametric test. For all statistical tests, a P value of <0.05 was considered significant.
RESULTS
Large numbers of activated, antigen-specific CD8+ T cells accumulate in the liver during ECM.
At the onset of severe neurological symptoms in C57BL/6 mice infected with P. berghei ANKA, CD8+ T cells were recruited to the brain (Fig. 1 A) (8, 15, 39), and intriguingly, the number of splenic CD8+ T cells was lower than that in naïve mice, to a degree that could not be explained solely by recruitment to the brain. Strikingly, we discovered a 10-fold increase in the number of CD8+ T cells in the liver at this time (Fig. 1A). Remarkably, of the combined number of CD8+ T cells in the spleen, brain, and liver, 2.4% ± 0.2% (mean ± standard error of the mean [SEM]) resided in the livers of naïve mice, while this proportion was increased to 72.3% ± 3.7% in mice suffering ECM symptoms. These data indicate that the liver becomes a major depot of CD8+ T cells during the symptomatic stages of ECM.
Fig. 1.

Large numbers of antigen-specific CD8+ T cells accumulate in the liver during ECM. (A) C57BL/6 mice (n = 5) were infected with P. berghei ANKA-luc, and 6 days later, when mice displayed ECM symptoms, CD8+ T cells in the brain, spleen, and liver were enumerated in these and naïve mice by flow cytometry. Data are representative of three independent experiments. (B) C57BL/6 mice (n = 5) were adoptively transferred by i.v. injection with 10,000 CD8+ MACS-purified, congenically marked CD45.1+ OTI cells and 2 h later were infected with P. berghei ANKA-Ova or left uninfected. Six days later, when infected mice displayed ECM symptoms, CD8+ CD45.1+ OTI T cells in the brain, spleen, and liver were enumerated by flow cytometry. Data are representative of two independent experiments. *, P < 0.05 (Mann-Whitney test) relative to corresponding naïve group.
To determine whether antigen-specific CD8+ T cells migrate to the liver at ECM onset, we employed a transgenic P. berghei ANKA line (P. berghei ANKA-Ova) engineered to express the chicken egg ovalbumin CD8+ T cell epitope SIINFEKL (24). We transferred 104 SIINFEKL-specific CD8+ T (OTI) cells into mice prior to infection with P. berghei ANKA-Ova. We observed no expansion of OTI cells in uninfected mice (Fig. 1B) or mice infected with non-Ova-expressing P. berghei ANKA (data not shown), while significant numbers of OTI cells were found in the spleens, brains, and, importantly, livers of P. berghei ANKA-Ova infected mice. Thus, antigen-specific CD8+ T cells accumulate in the liver following P. berghei ANKA infection.
To examine the activation status of CD8+ T cells recruited to the liver, we next determined their expression of the canonical cytolytic molecule granzyme B (GzmB). In naïve mice, CD8+ T cells in the spleen (Fig. 2) and liver (data not shown) expressed very low levels of GzmB. In mice displaying early symptoms of ECM (clinical score of 1 or 2), a clear population of GzmB+ CD8+ T cells was detected in the spleen (Fig. 2). However, the proportion of CD8+ T cells expressing this molecule was significantly higher (approaching 60%) in both the brain and the liver. Taken together, these data suggest that CD8+ T cells recruited to the liver during P. berghei ANKA infection not are only antigen specific but also display a activated phenotype.
Fig. 2.

The majority of liver-recruited CD8+ T cells express granzyme B during P. berghei ANKA infection. C57BL/6 mice (n = 5 or 6), were infected with P. berghei ANKA-luc or left naïve. At 6 days p.i., when infected mice displayed clinical scores of 1 or 2, splenic, liver, and brain CD8+ T cells were assessed for GzmB expression by intracellular staining and flow cytometric analysis. Representative fluorescence-activated cell sorter (FACS) histograms are gated on CD8+ TCRβ+ cells, and numbers alongside each gate depict the percentage of GzmB+ cells. **, P < 0.01 (Mann-Whitney test) relative to naïve spleen group. These data are summarized in the adjacent graph and are representative of two independent experiments performed.
Liver damage occurs in ECM prior to the onset of neurological symptoms.
Given that antigen-specific GzmB+ CD8+ T cells were found in the brain and the liver on day 6 p.i., at the onset of neurological symptoms, we next investigated whether liver pathology was evident in these mice. Mice displayed no clinical symptoms and exhibited normal serum levels of the liver enzymes alanine transaminase (ALT) and aspartate transaminase (AST) at 4 days p.i. compared to naïve mice (Fig. 3). Mice infected for 5 days with P. berghei ANKA also displayed no clinical symptoms but had significantly elevated ALT/AST levels, indicative of liver damage. There was a trend for ALT/AST levels to be further increased on day 6 p.i. at the onset of severe neurological symptoms (Fig. 3).
Fig. 3.
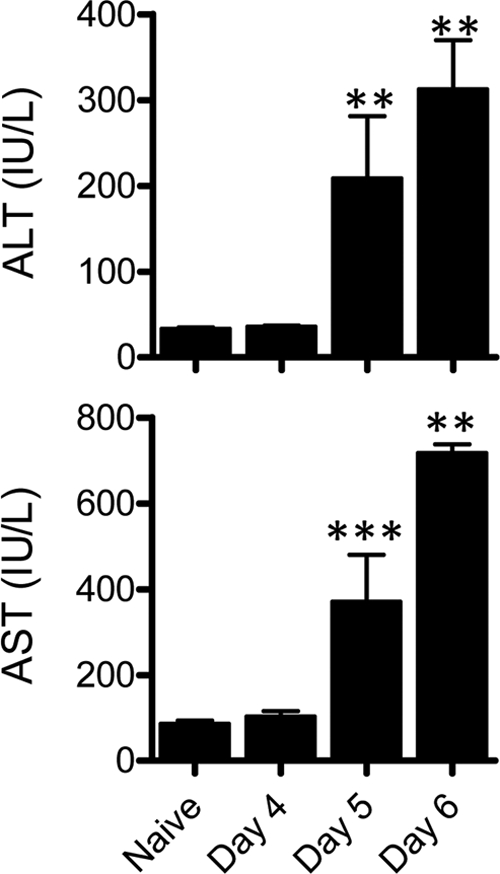
Liver damage occurs in ECM prior to the onset of neurological symptoms. C57BL/6 mice (n = 6 to 11) were infected with P. berghei ANKA-luc, and 4, 5, or 6 days later plasma samples from these and naïve mice were assessed for liver ALT and AST levels. **, P < 0.01; ***, P < 0.001 (Mann-Whitney test, relative to naïve mice). Data are representative of three independent experiments.
Given the elevated ALT/AST levels on day 5 p.i., we next assessed liver CD8+ T cell recruitment at this time point and discovered a small, though statistically significant, increase in the number of these cells, which was substantially increased by day 6 p.i. (Fig. 4 A). We next examined the extent of liver damage histologically on days 5 and 6 p.i. (Fig. 4B). Consistent with our flow cytometric analysis on day 5 p.i. (Fig. 4A), leukocytes were not readily observed by histology in the livers of either naïve mice or those infected for 5 days (Fig. 4B). However, on day 6 p.i. a striking accumulation of leukocytes and parasitized red blood cells was evident in the lumen of essentially all blood vessels examined (Fig. 4B). It is important to note that no large areas of severe necrotic damage or granulomatous lesion formation were observed in infected mice at any time point. These data suggest that the majority of CD8+ T cells recruited to the liver remain confined to the lumen of blood vessels and do not infiltrate significantly into the liver parenchyma. We also examined liver sections for evidence of liver fibrosis (Fig. 4B). In naïve mice, collagen was detected in the normal structural components of major blood vessels. In mice infected for 5 days, however, we observed evidence of additional collagen deposition within the liver parenchyma, a further indicator of stress and damage in this organ. By day 5 p.i. we also observed punctuate black deposits, which were absent from naïve livers, within certain cells, which we inferred to be malarial hemozoin pigment. The degree of collagen and malarial pigment deposition appeared to be more evident 24 h later, on day 6 p.i. Taken together these data provide clear evidence not only that liver damage and CD8+ T cell recruitment occur during ECM but, importantly, that they precede the onset of CD8+ T cell-mediated neurological symptoms.
Fig. 4.

Histopathological assessment of liver damage during P. berghei ANKA (PbA) infection. C57BL/6 mice (n = 5 or 6) were infected with P. berghei ANKA-luc or left naïve. (A) At 5 or 6 days after infection, liver-resident CD8+ T cells were enumerated by flow cytometry. *, P < 0.05; **, P < 0.01 (Mann-Whitney test, relative to naïve group). (B) At 5 or 6 days after infection, paraffin-embedded tissues were sectioned and stained with hematoxylin and eosin (upper panels) or with picrosirius red (lower panels). Collagen is depicted by red staining and malaria pigment by punctuate black staining in the lower panels. Bars, 10 μm. These data are representative of two independent experiments performed.
Parasite burden mediates liver damage independently of CD8+ T cells.
Given that disease severity in ECM correlates with whole-body parasite burdens (3–5, 9, 25, 27), we next determined the relative role of parasites and CD8+ T cells in causing liver damage following P. berghei ANKA infection. We treated mice from day 3 postinfection (after dendritic cells have primed the pathogenic CD8+ T cell response [24, 38]), either with artesunate to remove parasites or with anti-CD8β or anti-CD4 depleting monoclonal antibodies to remove their respective T cell populations. Three days later, control infected mice and anti-CD4-treated mice exhibited severe disease symptoms as expected, while those treated with artesunate or depleted of CD8+ T cells had no clinical signs of illness (data not shown). Mice were then assessed for whole-body and liver parasite burdens, as well as for evidence of liver damage (Fig. 5). Infected mice treated with artesunate or depleted of CD8+ T cells displayed substantially reduced parasite burdens, both in the whole body (Fig. 5A) and in the liver (Fig. 5B), compared with infected control mice or anti-CD4-treated mice. Artesunate-treated mice exhibited no liver damage, with ALT and AST levels comparable to those in naïve mice. In stark contrast, CD4+ T cell-depleted or CD8+ T cell-depleted mice displayed liver damage similar to that in control infected mice (Fig. 5C). There was a trend for CD8+ T cell depletion to further increase ALT levels compared to those in control treated or anti-CD4-treated mice. These data clearly show that whereas high parasite burdens are associated with liver damage, CD8+ T cells are not responsible for disease in this organ.
Fig. 5.

Parasites, not CD8+ or CD4+ T cells, mediate liver damage following P. berghei ANKA infection. C57BL/6 mice (n = 5) were infected with P. berghei ANKA-luc, and 3 days later infected mice, as well as naïve controls, were treated with control rat IgG, anti-CD4 MAb, anti-CD8β MAb, or artesunate. (A and B) A further 3 days later, when control rat IgG-treated mice were displaying ECM symptoms, mice were assessed for whole-body parasite burden (A) and liver parasite burden (B) by bioluminescence imaging. (C) Plasma samples were also tested for liver enzyme ALT and AST levels. **, P < 0.01 (Mann-Whitney test) for comparison of each treatment group to the control rat IgG treated group. These data are representative of two independent experiments.
We also examined the relative roles of parasites and CD8+ T cells in liver damage in mice treated with artesunate or depleted of CD8+ T cells at 6 days following P. berghei ANKA infection, after the onset of severe neurological symptoms. Both treatments ameliorated ECM symptoms within 24 h, as expected (data not shown), and reduced parasite burdens in the whole body and liver (Fig. 6). However, while artesunate treatment reduced ALT and AST levels to those observed in naïve mice by 4 days posttreatment, CD8+ T cell depletion did not reduce liver damage, with a trend toward increased liver damage over the treatment period (Fig. 6). These data demonstrate that reducing the parasite load by artesunate treatment can reverse liver damage in P. berghei ANKA-infected mice suffering severe neurological symptoms. In contrast, although CD8+ T cell depletion reversed neurological symptoms and reduced parasite burdens, it did not resolve liver damage.
Fig. 6.

Therapeutic artesunate treatment, but not CD8+ T cell depletion, ameliorates established liver damage following P. berghei ANKA infection. C57BL/6 mice (n = 5) were infected with P. berghei ANKA-luc, and 6 days later infected mice displaying ECM symptoms, as well as naïve controls, were treated with control rat IgG, anti-CD8β MAb, or artesunate. (A and B) Mice were then assessed at 1 day and 4 days after treatment for whole-body parasite burden (A) and liver parasite burden (B) by bioluminescence imaging. (C) Plasma samples were also tested for liver enzyme ALT and AST levels: **, P < 0.01 (Mann-Whitney test) for comparison of artesunate group to anti-CD8β MAb group at 4 days posttreatment. These data are representative of two independent experiments.
Finally, to determine whether liver damage could occur in the complete absence of neurological symptoms, we infected ECM-resistant, perforin-deficient C57BL/6 mice and assessed liver damage (Fig. 7) at a time when control, wild-type infected mice were displaying clinical symptoms (data not shown). ALT and AST levels were significantly elevated in P. berghei ANKA-infected perforin-deficient mice compared to uninfected control mice, demonstrating that P. berghei ANKA-mediated liver damage occurs regardless of whether neurological symptoms are induced or not.
Fig. 7.
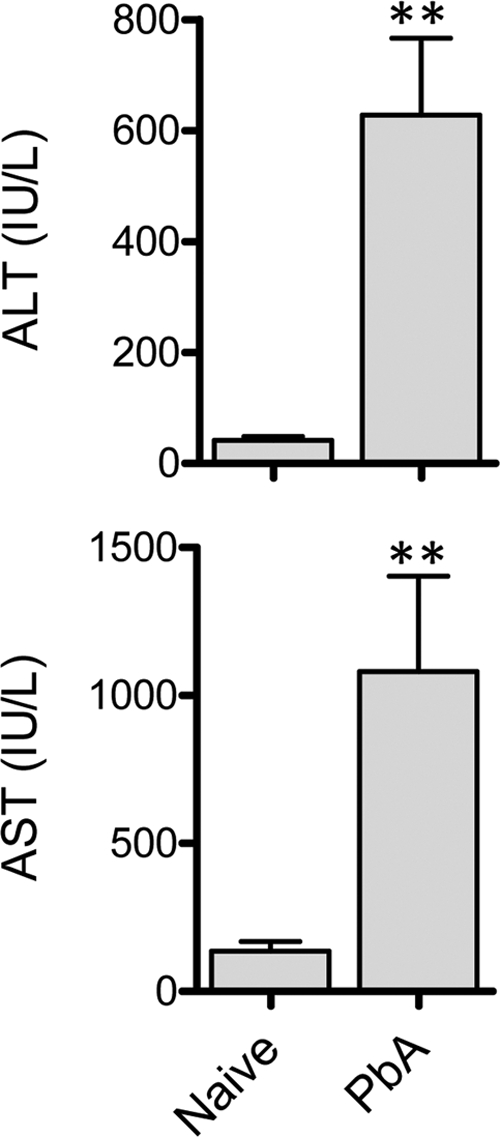
Perforin-deficient mice exhibit liver damage during P. berghei ANKA infection. C57BL/6 perforin-deficient mice (n = 5 or 6) were infected with P. berghei ANKA-luc or left naïve. Six days later, when these mice displayed no clinical symptoms of disease, plasma samples were tested for liver enzyme ALT and AST levels. **, P < 0.01 (Mann-Whitney test) relative to naïve perforin-deficient mice.
DISCUSSION
Using an ECM model, we show that large numbers of parasite-specific CD8+ T cells accumulate in the liver, but unlike in the brain, they play no role in causing liver tissue damage associated with this infection. Instead, our data indicate that parasites are the major promoters of hepatic injury in this malaria model. Critically, our data support the idea that P. berghei ANKA infection causes a parasite-driven multiorgan disease but that CD8+ T cell-mediated immune pathology occurs in an organ-specific manner.
The mechanisms by which CD8+ T cells accumulate in such large numbers in the liver during P. berghei ANKA infection remain unknown. One possibility is that the substantial increase in ICAM-1 and PECAM expression that occurs following P. berghei ANKA infection may mediate adherence of CD8+ T cells to liver endothelium (6). One further explanation for the large numbers of CD8+ T cells in the liver is that they are undergoing activation-induced cell death and are therefore being removed by the liver (17, 26). Whether CD8+ T cells in the liver are preferentially undergoing apoptosis compared to brain-recruited cells remains to be studied.
It is not yet clear if the parasite-specific CD8+ T cell response in the liver differs from that in the brain, where it clearly mediates neurological symptoms and death. Our data indicate that liver CD8+ T cells express similar amounts of granzyme B as brain-recruited CD8+ T cells (data not shown). This suggests that liver- and brain-recruited CD8+ T cells are similarly activated and therefore have the same capacity to cause immune pathology. In that case, liver and brain tissue may display differential susceptibility to cytotoxic T cell-mediated killing, perhaps involving an as-yet-unidentified cross-presenting cell in the brain that triggers CD8+ T cell-mediated killing and that is absent from the liver.
Liver damage during blood-stage malaria infection has been modeled in mice using the P. berghei NK65 strain (1, 2, 13). interleukin-12 (IL-12), MyD88, and NKT cells play a role in mediating liver damage in this model (1, 2, 40), while IL-27 prevents liver immune pathology in another rodent malaria model (13). A previous report demonstrated that CTLA-4 blockade exacerbated liver damage during ECM in an IL-12- and gamma interferon (IFN-γ)-dependent manner but did not examine the immune cells involved (18). However, while we have found no evidence that CD8+ T cells or CD4+ T cells (data not shown) mediate liver damage in ECM, it remains to be established whether other immune cells contribute to liver damage during P. berghei ANKA infection.
The observation that efficient removal of parasites by artesunate ameliorates liver damage highlights its reversible nature during P. berghei ANKA infection. In general, efficient drug treatment during severe malaria in humans also restores serum liver enzymes to levels in healthy controls (28). The mechanisms by which liver damage is induced in human malaria remain unclear. However, a correlation between parasite sequestration in the liver and AST levels has been established (30), suggesting a role for parasites in mediating liver damage in humans. Hepatomegaly is also a common feature of those suffering liver damage during malaria, but at present it is unclear how this is linked to pathology (19, 28, 30, 33, 34). It will be of interest to determine whether leukocytes accumulate in the liver during severe human malaria and, if so, whether activated CD8+ T cells are present within the liver in this population.
It was intriguing to observe a trend for CD8 depletion to further elevate serum liver enzyme levels in P. berghei ANKA-infected mice. A similar trend was noted in infected perforin-deficient mice compared to wild-type mice (data not shown). Further experiments are required to better understand these data. Nevertheless, these data suggest that while CD8+ T cells mediate pathology in the brain in this model, they might protect the liver from excessive pathology. The mechanisms by which CD8+ T cells might do so are unclear, but given their expression of GzmB and a possible requirement for perforin, it is tempting to hypothesize that cytotoxic CD8+ T cells in the liver kill cells undergoing parasite-induced stress and, by doing so, remove cellular sources of liver pathology.
It has recently been demonstrated that late CD8 depletion rapidly reduces parasite burdens during the symptomatic stages of P. berghei ANKA infection (5). These data suggest that CD8+ T cells might impair pathogen clearance, but further experiments are required to determine possible mechanisms. One hypothesis is that cytotoxic T cell-mediated killing of splenic macrophages during murine blood-stage Plasmodium infection prevents these cells from efficiently clearing parasites (7). In conclusion, we have demonstrated here that parasite-specific CD8+ T cells are recruited to the liver and brain following P. berghei ANKA infection of C57BL/6 mice but are responsible for disease only in the brain and not in the liver. Instead, parasites accumulated in tissue play a critical role in driving multiorgan pathology during P. berghei ANKA infection.
ACKNOWLEDGMENTS
We thank Brooke Berry, Andrew Ong, George Marshall, and Jim Case at the Queensland Health Clinical and Statewide Services, Pathology Queensland, for conducting liver function tests. We also thank Histology Laboratory, Animal Facility, and Flow Cytometry Facility of the Queensland Institute of Medical Research for their services and assistance. We thank William R. Heath (University of Melbourne) for providing P. berghei ANKA-Ova transgenic parasites.
This work was funded by grants awarded by the National Health and Medical Research Council of Australia.
Footnotes
Published ahead of print on 22 February 2011.
REFERENCES
- 1. Adachi K., et al. 2001. Plasmodium berghei infection in mice induces liver injury by an IL-12- and Toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J. Immunol. 167:5928–5934 [DOI] [PubMed] [Google Scholar]
- 2. Adachi K., et al. 2004. Contribution of CD1d-unrestricted hepatic DX5+ NKT cells to liver injury in Plasmodium berghei-parasitized erythrocyte-injected mice. Int. Immunol. 16:787–798 [DOI] [PubMed] [Google Scholar]
- 3. Amante F. H., et al. 2010. Immune-mediated mechanisms of parasite tissue sequestration during experimental cerebral malaria. J. Immunol. 185:3632–3642 [DOI] [PubMed] [Google Scholar]
- 4. Amante F. H., et al. 2007. A role for natural regulatory T cells in the pathogenesis of experimental cerebral malaria. Am. J. Pathol. 171:548–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baptista F. G., et al. 2010. Accumulation of Plasmodium berghei-infected red blood cells in the brain is crucial for the development of cerebral malaria in mice. Infect. Immun. 78:4033–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bauer P. R., Van Der Heyde H. C., Sun G., Specian R. D., Granger D. N. 2002. Regulation of endothelial cell adhesion molecule expression in an experimental model of cerebral malaria. Microcirculation 9:463–470 [DOI] [PubMed] [Google Scholar]
- 7. Beattie L., Engwerda C. R., Wykes M., Good M. F. 2006. CD8+ T lymphocyte-mediated loss of marginal metallophilic macrophages following infection with Plasmodium chabaudi chabaudi AS. J. Immunol. 177:2518–2526 [DOI] [PubMed] [Google Scholar]
- 8. Belnoue E., et al. 2002. On the pathogenic role of brain-sequestered alphabeta CD8+ T cells in experimental cerebral malaria. J. Immunol. 169:6369–6375 [DOI] [PubMed] [Google Scholar]
- 9. Dondorp A. M., et al. 2005. Estimation of the total parasite biomass in acute falciparum malaria from plasma PfHRP2. PLoS Med. 2:e204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Engwerda C., Belnoue E., Gruner A. C., Renia L. 2005. Experimental models of cerebral malaria. Curr. Top. Microbiol. Immunol. 297:103–143 [PubMed] [Google Scholar]
- 11. Epiphanio S., et al. 2010. VEGF promotes malaria-associated acute lung injury in mice. PLoS Pathog. 6:e1000916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Favre N., et al. 1999. Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microbes Infect. 1:961–968 [DOI] [PubMed] [Google Scholar]
- 13. Findlay E. G., et al. 2010. Essential role for IL-27 receptor signaling in prevention of Th1-mediated immunopathology during malaria infection. J. Immunol. 185:2482–2492 [DOI] [PubMed] [Google Scholar]
- 14. Haque A., et al. 2010. Therapeutic glucocorticoid-induced TNF receptor-mediated amplification of CD4+ T cell responses enhances antiparasitic immunity. J. Immunol. 184:2583–2592 [DOI] [PubMed] [Google Scholar]
- 15. Hermsen C., van de Wiel T., Mommers E., Sauerwein R., Eling W. 1997. Depletion of CD4+ or CD8+ T-cells prevents Plasmodium berghei induced cerebral malaria in end-stage disease. Parasitology 114:7–12 [DOI] [PubMed] [Google Scholar]
- 16. Hogquist K. A., et al. 1994. T cell receptor antagonist peptides induce positive selection. Cell 76:17–27 [DOI] [PubMed] [Google Scholar]
- 17. Huang L., Soldevila G., Leeker M., Flavell R., Crispe I. N. 1994. The liver eliminates T cells undergoing antigen-triggered apoptosis in vivo. Immunity 1:741–749 [DOI] [PubMed] [Google Scholar]
- 18. Jacobs T., Plate T., Gaworski I., Fleischer B. 2004. CTLA-4-dependent mechanisms prevent T cell induced-liver pathology during the erythrocyte stage of Plasmodium berghei malaria. Eur. J. Immunol. 34:972–980 [DOI] [PubMed] [Google Scholar]
- 19. Kochar D. K., et al. 2003. Malarial hepatitis. J. Assoc. Physicians India 51:1069–1072 [PubMed] [Google Scholar]
- 20. Lamb T. J., Brown D. E., Potocnik A. J., Langhorne J. 2006. Insights into the immunopathogenesis of malaria using mouse models. Expert Rev. Mol. Med. 8:1–22 [DOI] [PubMed] [Google Scholar]
- 21. Langhorne J., Quin S. J., Sanni L. A. 2002. Mouse models of blood-stage malaria infections: immune responses and cytokines involved in protection and pathology. Chem. Immunol. 80:204–228 [DOI] [PubMed] [Google Scholar]
- 22. Li C., Seixas E., Langhorne J. 2001. Rodent malarias: the mouse as a model for understanding immune responses and pathology induced by the erythrocytic stages of the parasite. Med. Microbiol. Immunol. 189:115–126 [DOI] [PubMed] [Google Scholar]
- 23. Lovegrove F. E., et al. 2008. Parasite burden and CD36-mediated sequestration are determinants of acute lung injury in an experimental malaria model. PLoS Pathog. 4:e1000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lundie R. J., et al. 2008. Blood-stage Plasmodium infection induces CD8+ T lymphocytes to parasite-expressed antigens, largely regulated by CD8alpha+ dendritic cells. Proc. Natl. Acad. Sci. U. S. A. 105:14509–14514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McQuillan J. A., et al. 2011. Coincident parasite and CD8 T cell sequestration is required for development of experimental cerebral malaria. Int. J. Parasitol. 41:155–163 [DOI] [PubMed] [Google Scholar]
- 26. Mehal W. Z., Juedes A. E., Crispe I. N. 1999. Selective retention of activated CD8+ T cells by the normal liver. J. Immunol. 163:3202–3210 [PubMed] [Google Scholar]
- 27. Nie C. Q., et al. 2009. IP-10-mediated T cell homing promotes cerebral inflammation over splenic immunity to malaria infection. PLoS Pathog. 5:e1000369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patwari A., Aneja S., Berry A. M., Ghosh S. 1979. Hepatic dysfunction in childhood malaria. Arch. Dis. Child. 54:139–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Piguet P. F., et al. 2001. Role of CD40-CVD40L in mouse severe malaria. Am. J. Pathol. 159:733–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Prommano O., et al. 2005. A quantitative ultrastructural study of the liver and the spleen in fatal falciparum malaria. Southeast Asian J. Trop. Med. Public Health 36:1359–1370 [PubMed] [Google Scholar]
- 31. Randall L. M., et al. 2008. Common strategies to prevent and modulate experimental cerebral malaria in mouse strains with different susceptibilities. Infect. Immun. 76:3312–3320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Senaldi G., Vesin C., Chang R., Grau G. E., Piguet P. F. 1994. Role of polymorphonuclear neutrophil leukocytes and their integrin CD11a (LFA-1) in the pathogenesis of severe murine malaria. Infect. Immun. 62:1144–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shah S., et al. 2009. Malarial hepatopathy in falciparum malaria. J. Coll. Physicians Surg. Pak. 19:367–370 [PubMed] [Google Scholar]
- 34. Sowunmi A. 1996. Hepatomegaly in acute falciparum malaria in children. Trans. R. Soc. Trop. Med. Hyg. 90:540–542 [DOI] [PubMed] [Google Scholar]
- 35. Stanley A. C., et al. 2008. VCAM-1 and VLA-4 modulate dendritic cell IL-12p40 production in experimental visceral leishmaniasis. PLoS Pathog. 4:e1000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stanley A. C., et al. 2008. Activation of invariant NKT cells exacerbates experimental visceral leishmaniasis. PLoS Pathog. 4:e1000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. WHO 2010. Guideline for the treatment of malaria, 2nd ed. WHO, Geneva, Switzerland [Google Scholar]
- 38. Wilson N. S., et al. 2006. Systemic activation of dendritic cells by Toll-like receptor ligands or malaria infection impairs cross-presentation and antiviral immunity. Nat. Immunol. 7:165–172 [DOI] [PubMed] [Google Scholar]
- 39. Yanez D. M., Manning D. D., Cooley A. J., Weidanz W. P., van der Heyde H. C. 1996. Participation of lymphocyte subpopulations in the pathogenesis of experimental murine cerebral malaria. J. Immunol. 157:1620–1624 [PubMed] [Google Scholar]
- 40. Yoshimoto T., et al. 1998. A pathogenic role of IL-12 in blood-stage murine malaria lethal strain Plasmodium berghei NK65 infection. J. Immunol. 160:5500–5505 [PubMed] [Google Scholar]