

Abstract
Legionella pneumophila is the etiological agent of Legionnaires' disease. Crucial to the pathogenesis of this intracellular pathogen is its ability to subvert host cell defenses, permitting intracellular replication in specialized vacuoles within host cells. The Dot/Icm type IV secretion system (T4SS), which translocates a large number of bacterial effectors into host cell, is absolutely required for rerouting the Legionella phagosome. Many Legionella effectors display distinctive eukaryotic domains, among which are protein kinase domains. In silico analysis and in vitro phosphorylation assays identified five functional protein kinases, LegK1 to LegK5, encoded by the epidemic L. pneumophila Lens strain. Except for LegK5, the Legionella protein kinases are all T4SS effectors. LegK2 plays a key role in bacterial virulence, as demonstrated by gene inactivation. The legK2 mutant containing vacuoles displays less-efficient recruitment of endoplasmic reticulum markers, which results in delayed intracellular replication. Considering that a kinase-dead substitution mutant of legK2 exhibits the same virulence defects, we highlight here a new molecular mechanism, namely, protein phosphorylation, developed by L. pneumophila to establish a replicative niche and evade host cell defenses.
INTRODUCTION
Legionella pneumophila is the most common causative agent of the atypical and severe pneumonia legionellosis. Pathogenic Legionella strains emerge from the environment after intracellular multiplication in phagocytic protozoans. It has been proposed that its ability to exploit the basic cellular mechanisms of numerous protozoal hosts also enables Legionella to infect human macrophages of alveolar lungs and consequently to cause disease (29, 63).
Mechanisms of infection of protozoans and macrophages by L. pneumophila are quite similar (24). Upon internalization of L. pneumophila, the bacteria containing phagosome avoids fusion with lysosomes and recruits mitochondria and endoplasmic reticulum (ER) to establish a replication niche. The Dot/Icm type IV secretion system (T4SS), which translocates bacterial proteins called “effectors” into host cell, is absolutely required to reprogram the endosomal-lysosomal degradation pathway of the phagocytic cell and to lead to bacterial intracellular survival and successful replication (3, 5, 64, 67). Multiple approaches have successfully identified more than 100 effector proteins (reviewed by reference 22). Many Legionella effectors display distinctive eukaryotic domains, such as those involved in protein-protein interactions (e.g., ankyrin repeats, leucine-rich repeats, and coiled coils), and domains with more defined activities (protein kinases, ubiquitin ligases, sphingosine-1-phosphate lyases, phospholipases, and guanine nucleotide exchange factors). In spite of the fact that L. pneumophila seems well equipped with proteins adapted to interfere with a wide variety of host cell processes, only a minority of these effectors have been functionally characterized (7, 18, 21, 30, 40, 44, 48, 49, 54, 56, 61, 72, 73).
The genomes of the five sequenced L. pneumophila strains—Philadelphia, Lens, Paris, Corby, and Alcoy—have been reported to encode four eukaryote-like serine/threonine kinases (8, 15, 16, 26). Phosphorylation-dephosphorylation of proteins at serine/threonine/tyrosine residues represents a powerful regulatory mechanism of cellular activity. Indeed, intensive research has revealed that eukaryotes contain numerous interconnected signal transduction networks in which protein phosphorylation plays a dominant role. For many years, however, phosphorylation at serine/threonine/tyrosine has been considered a new addition to the cell's regulatory arsenal and believed to be exclusive to eukaryotes. Due in large part to genomic sequencing programs, some “classical” serine/threonine/tyrosine protein kinases have been identified in several bacterial genomes, but the function of most of them remains unknown (13, 19). Interestingly, some of these enzymes have been recently described to play a critical role in virulence of several pathogenic bacteria: PknG of Mycobacterium tuberculosis (71), YpkA of Yersinia pseudotuberculosis (4), and SteC of Salmonella enterica serovar Typhimurium (59) are translocated into the host cell cytoplasm to modulate eukaryotic signal transduction to the bacteria advantage and thus contribute to virulence. Moreover, StkP of Streptococcus pneumoniae is a global regulator of gene expression that positively controls virulence for lung infection and bloodstream invasion (20, 66), SP-STK of Streptococcus pyogenes has been reported to have pleiotropic effect, including the expression of major virulence factors (32), and PrkC of Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence (37).
We investigated the role of bacterial protein kinases in the virulence of L. pneumophila. We report that the five putative protein kinases identified by in silico analysis of the L. pneumophila genomes are functional in terms of phosphorylation; actually, they can autophosphorylate and undergo in vitro phosphorylation of classical eukaryotic protein kinase substrates. Interestingly, we demonstrate that the protein kinase LegK2 plays a key role in the virulence of Legionella toward amoebae. More precisely, we observed that the legK2 mutant is impaired in ER recruitment and intracellular replication. Since LegK2 is translocated into the host cell cytoplasm during infection, we assume that this protein kinase would interfere with host cell signal transduction pathways to subvert host cell defenses to the bacteria benefit, thus resulting in an effective infectious cycle and virulence.
MATERIALS AND METHODS
Growth of bacteria and phagocytes.
The bacterial strains and plasmids used in the present study are summarized in Table 1.
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant properties | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| XL1-Blue | endA1 gyrA96(Nalr) thi-1 recA1 relA1 lac glnV44 F′[::Tn10 proAB+lacIqΔ(lacZ)M15] hsdR17(rK+ mK−) | Stratagene |
| BL21(DE3)(pREP4-groESL) | F−ompT gal dcm lon hsdSB(rB− mB−) λ(DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | 2 |
| L. pneumophila | ||
| CIP 108286 | Virulent L. pneumophila serogroup 1, strain Lens | 8 |
| ΔdotA mutant | Lens lpl2613::Km | 23 |
| ΔlegK1 mutant | Lens lpl1545::Km | This study |
| ΔlegK2 mutant | Lens lpl2066::Km | This study |
| ΔlegK3 mutant | Lens lpl2481::Km | This study |
| ΔlegK4 mutant | Lens lpl0262::Km | This study |
| ΔlegK5 mutant | Lens lpl2476::Km | This study |
| A. castellanii | Environmental isolate | P. Pernin, Faculty of Pharmacy, Université Lyon 1 |
| D. discoideum | ||
| DBS0302388 | Wild-type DH1 strain | 12 |
| DBS0236184 | [act15]:cnxA:GFP | 55 |
| Dd03 | [act15]:legK2(K112M):c-myc | This study |
| J774A.1 | ATCC TIB-67 | |
| Plasmids | ||
| pGEX-6P-3 | E. coli expression vector | 69 |
| pGEX-legK1 | lpl1545 inserted in BamHI-SalI of pGEX-6P-3 for GST-LegK1 overproduction | This study |
| pGEX-legK2 | lpl2066 inserted in BamHI-SalI of pGEX-6P-3 for GST-LegK2 overproduction | This study |
| pGEX-legK3 | lpl2481 inserted in BamHI-SalI of pGEX-6P-3 for GST-LegK3 overproduction | This study |
| pQE30 | E. coli expression vector | Qiagen |
| pQE30-legK4 | lpl0262 inserted in BamHI-SalI of pQE30 for 6His-LegK4 overproduction | This study |
| pQE30-legK5 | lpl2476 inserted in PstI-SacI of pQE30 for 6His-LegK5 overproduction | This study |
| pKD13 | Used for amplification of Km | 14 |
| pAV695 | sacB, OriT OriV, cml (pCDP05 with a deletion of a 4.3-kb fragment) | Modified from reference 60 |
| p695-legK1::Km | pAV695 derivative carrying lpl1545 interrupted by Km | This study |
| p695-legK2::Km | pAV695 derivative carrying lpl2066 interrupted by Km | This study |
| p695-legK3::Km | pAV695 derivative carrying lpl2481 interrupted by Km | This study |
| p695-legK4::Km | pAV695 derivative carrying lpl0262 interrupted by Km | This study |
| p695-legK5::Km | pAV695 derivative carrying lpl2476 interrupted by Km | This study |
| pXDC50 | Legionella expression vector carrying mCherry | X. Charpentier |
| plegK1 | pXDC50 derivative Legionella expression vector carrying mCherry; expression of legK1 under the control of legK2 promoter (400 bp) | This study |
| plegK2 | pXDC50 derivative Legionella expression vector carrying mCherry; expression of legK2 under its promoter (400 bp) | This study |
| plegK2(K112M) | pXDC50 derivative Legionella expression vector carrying mCherry; expression of legK2(K112M) under its promoter (400 bp) | This study |
| plegK3 | pXDC50 derivative Legionella expression vector carrying mCherry; expression of legK3 under the control of legK2 promoter (400 bp) | This study |
| plegK4 | pXDC50 derivative Legionella expression vector carrying mCherry; expression of legK4 under the control of legK2 promoter (400 bp) | This study |
| plegK5 | pXDC50 derivative Legionella expression vector carrying mCherry; expression of legK5 under the control of legK2 promoter (400 bp) | This study |
| p3041 | pXDC50 derivative Legionella expression vector carrying mCherry and gentamicin resistance gene | This study |
| pEP46 | D. discoideum expression vector carrying legK2(K112M)-c-myc under the control of the actin promoter | This study |
| pEP73 | Legionella expression vector carrying gfp-legK2; production of the GFP-LegK2 hybrid protein | This study |
L. pneumophila strains were grown at 30°C either on buffered charcoal yeast extract (BCYE) agar or in BYE liquid medium; each medium was supplemented with kanamycin at 10 μg ml−1 or chloramphenicol at 5 μg ml−1 when appropriate. Escherichia coli strains were grown at 37°C in LB medium supplemented with ampicillin at 100 μg ml−1, kanamycin at 20 μg ml−1, or chloramphenicol at 5 μg ml−1. E. coli XL1-Blue was used to maintain plasmids and strain E. coli BL21(DE3)(pREP4-groESL) was used for recombinant protein overproduction.
Axenic Acanthamoeba castellanii cells were grown on PYG medium (proteose-yeast extract-glucose medium) at 30°C and split once a week. Dictyostelium discoideum expressing calnexin-green fluorescent protein (GFP) (DBS0236184) was obtained from the Dicty Stock Center (depositor A. Müller-Taubenberger [55]). D. discoideum cells were axenically grown in HL5 medium at 22°C, supplemented with G418 at 20 μg ml−1 when necessary.
J774A.1 macrophages were maintained at 37°C in 5% CO2 in RPMI 1640 medium supplemented with 10% heat-inactivated fetal calf serum.
General DNA techniques.
The oligonucleotides used in the present study are shown in Table 2.
Table 2.
Primers used in this study
| No. | Name | Sequence (5′–3′)a | Description |
|---|---|---|---|
| 1 | N-lpl1545-BamHI | ATAGGATCCCCTCGTACAATGTTTTTTTCC | GST-LegK1 overproduction |
| 2 | C-lpl1545-SalI | ATAGTCGACTTACTCAGCCACTAACCATAAGG | GST-LegK1 overproduction |
| 3 | N-lpl2066-BamHI | ATAGGATCCGTTTATTACATAAATTTGAAGGAAC | GST-LegK2 overproduction |
| 4 | C-lpl2066-SalI | ATAGTCGACTTAGCTTGGGCCTCGCATC | GST-LegK2 overproduction |
| 5 | N-lpl2481-BamHI | ATAGGATCCTTTGATAGAAATATAAAAGAAATAATC | GST-LegK3 overproduction |
| 6 | C-lpl2481-SalI | ATAGTCGACTTATAATTCAAAGCCTGAAT | GST-LegK3 overproduction |
| 7 | N-lpl0262-BamHI | ATAGGATCCAAATTGCTTCGGTTTCATGAATT | 6His-LegK4 overproduction |
| 8 | C-lpl0262-SalI | ATAGTCGACTTAATATGGCAAAATGATGACGT | 6His-LegK4 overproduction |
| 9 | N-lpl2476-SacI | ATAGAGCTCGGGATTATCATGGCTACAGT | 6His-LegK5 overproduction |
| 10 | C-lpl2476-PstI | ATACTGCAGTTATTTTATGAAATCGGCCTTTA | 6His-LegK5 overproduction |
| 11 | Kan-FRTS | CACGTCGACAGCGATTGTGTAGGCTGGAGC | Km amplification |
| 12 | Kan-FRTR | GGGGATCCGTCGACCTGC | Km amplification |
| 13 | P1-dotA-NotI | AAAAGCGGCCGCGCTCTCGCTGAAAGTGGCTC | lpl2613 deletion |
| 14 | P2-dotA-SalI | CGCGTCGACTGCTTGCAAGCTCTTGGTTG | lpl2613 deletion |
| 15 | P3-dotA-SalI | CGCGTCGACGCCATTTCCTACATCCAATCG | lpl2613 deletion |
| 16 | P4-dotA-NotI | AAAAGCGGCCGCCCGGTTTAGAGCTTGGTCCA | lpl2613 deletion |
| 17 | P1-legK1-NotI | AAAAGCGGCCGCCGTTGATGCCGCTAATCTCC | lpl1545 deletion |
| 18 | P2-legK1-SalI | CGCGTCGACCGGTTTAACCGCTATATGCCC | lpl1545 deletion |
| 19 | P3-legK1-SalI | CGCGTCGACAAGGCTATCAAGCAGTTTTCCC | lpl1545 deletion |
| 20 | P4-legK1-NotI | AAAAGCGGCCGCTTTGAGAAAATAATCCCAGGCG | lpl1545 deletion |
| 21 | P1-legK2-NotI | AAAAGCGGCCGCAATTTGAAGGAACAACCCTTACCTC | lpl2066 deletion |
| 22 | P2-legK2-SalI | CGCGTCGACAAGTTTTTCCAGGACACATCCCT | lpl2066 deletion |
| 23 | P3-legK2-SalI | CGCGTCGACTTCGAATTTACAGGCTTACAAGGATC | lpl2066 deletion |
| 24 | P4-legK2-NotI | AAAAGCGGCCGCGCCTCGCATCAATGAAGGTG | lpl2066 deletion |
| 25 | P1-legK3-NotI | AAAAGCGGCCGATTTACTTCCGGGCACTGG | lpl2481 deletion |
| 26 | P2-legK3-SalI | CGCGTCGACGGCATTCGTTTCATCGTCAG | lpl2481 deletion |
| 27 | P3-legK3-SalI | CGCGTCGACAGCAACTTGCGTCCATTACG | lpl2481 deletion |
| 28 | P4-legK3-NotI | AAAAGCGGCCAGCTTCGCTTGCATGCAAA | lpl2481 deletion |
| 29 | P1-legK4-NotI | AAAAGCGGCCGCAAGCCAATCATCGTTCCCAC | lpl0262 deletion |
| 30 | P2-legK4-SalI | CGCGTCGACGTTCTGGTGCTAAATAGCTTGCG | lpl0262 deletion |
| 31 | P3-legK4-SalI | CGCGTCGACCTGCCACATCAAGTCCCCTC | lpl0262 deletion |
| 32 | P4-legK4-NotI | AAAAGCGGCCGCTGGCAAAATGATGACGTTGC | lpl0262 deletion |
| 33 | P1-legK5-NotI | AAAAGCGGCCGCCATGGCTACAGTAGATTCCG | lpl2476 deletion |
| 34 | P2-legK5-SalI | CGCGTCGACGAACGTTAGCTTCACGCTCT | lpl2476 deletion |
| 35 | P3-legK5-SalI | CGCGTCGACCACTGAAAATCGCGGATATCG | lpl2476 deletion |
| 36 | P4-legK5-NotI | AAAAGCGGCCGCGTTCCATGTCAATTTTAGGGC | lpl2476 deletion |
| 37 | 5-promolpl2066-SacI | ATAGAGCTCCAGGGTAACTGAATAAGCCC | lpl2066 complementation |
| 38 | 3-promolpl2066-KpnI | CGGGGTACCTCTCCTACAAATCAATTGCC | lpl2066 complementation |
| 39 | 5′SphI-promolpl2066 | ATAGCATGCCAGGGTAACTGAATAAGCCC | lpl2066 complementation |
| 40 | lpl2066(K112M)-sens | CCCAAAGAGAATATACTGATGGTTTTATATCAAAATTTTAGTAATGTCG | lpl2066 mutagenesis |
| 41 | lpl2066(K112M)-rev | CGACATTACTAAAATTTTGATATAAAACCATCAGTATATTCTCTTTGGG | lpl2066 mutagenesis |
Restriction enzyme sites are indicated in boldface.
Standard techniques were used for nucleic acid cloning and restriction analysis. Restriction enzymes and T4 DNA ligase were purchased from Fermentas (Saint Rémy-les-Chevreuses, France). Plasmid DNA from E. coli was prepared by rapid alkaline lysis (31). PCR amplifications were carried out with Phusion polymerase as recommended by the manufacturer (Finnzymes, Espoo, Finland). Purification of DNA fragments from agarose gels for subcloning was carried out with a QIAquick gel purification kit (Qiagen, Courtaboeuf, France).
Expression and purification of recombinant proteins.
DNA fragments corresponding to the coding sequences of legK1 (lpl1545), legK2 (lpl2066), legK3 (lpl2481), legK4 (lpl0262), and legK5 (lpl2476) were PCR amplified using genomic DNA of L. pneumophila Lens as the template and the following oligonucleotide pairs: for legK1, 1/2; for legK2, 3/4; for legK3, 5/6; for legK4, 7/8; and for legK5, 9/10. The amplified DNA fragments were digested with BamHI and SalI or with SacI and PstI for legK5 and then inserted into PGEX-6P-3 for legK1, legK2, and legK3 or pQE30 for legK4 and legK5. The resulting plasmids—pGEX-legK1, pGEX-legK2, pGEX-legK3, pQE30-legK4, and pQE30-legK5—were introduced into E. coli strain BL21(DE3)(pREP4-groESL). Transformants were grown at 37°C until cultures reached an optical density at 600 nm (OD600) of 0.7. Gene expressions were induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 4 h at 20°C, and cell lysates were prepared by using a French pressure cell (SLM, Urbana, IL). GST and 6His recombinant proteins were purified under native conditions by affinity chromatography on glutathione-Sepharose beads (GE Healthcare, Orsay, France) and a Ni-NTA column (Qiagen), respectively, according to the manufacturers' recommendations. The purity of the eluted protein was analyzed by SDS-PAGE, and protein concentrations were determined by using a protein assay dye reagent concentrate (Bio-Rad, Marnes-la-Coquette, France).
In vitro phosphorylation assay.
In vitro phosphorylation of about 2 μg of purified recombinant protein was performed for 30 min at 37°C in 20 μl of a buffer containing 25 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 1 mM dithiothreitol, 10 μM ATP, and 5 μCi of [γ-32P]ATP (Perkin-Elmer, Courtaboeuf, France). In some assays, MgCl2 was replaced with MnCl2. In eukaryotic substrate phosphorylation assays, 1 μg of myelin basic protein (MBP) was added. In each case, the reaction was stopped by the addition of an equal volume of 2× Laemmli loading buffer (41).
TEM translocation assays.
J774A.1 cells grown in RPMI 1640 containing 10% fetal calf serum were seeded in a black, clear-bottom 96-well plate at 105 cells per well 24 h prior to infection. L. pneumophila strains carrying the various TEM β-lactamase fusions were grown on BCYE plates containing chloramphenicol. Single colonies were then streaked onto BCYE plates containing chloramphenicol and 0.5 mM IPTG and grown for 24 h to induce production of the hybrid proteins. Then, 10 μl of bacteria resuspended in RPMI at 5 × 108 cells ml−1 was used to infect J774A.1 cells (multiplicity of infection [MOI] = 50). After centrifugation (600 × g, 10 min) to initiate bacterium-cell contact, the plate was shifted to 37°C, followed by incubation for 1 h with CO2 exchange. Cell monolayers were loaded with the fluorescent substrate by adding 20 μl of 6× CCF4/AM solution (LiveBLAzer-FRET B/G loading kit; Invitrogen, Cergy Pontoise, France) containing 15 mM Probenecid (Sigma, St. Quentin Fallavier, France). The cells were incubated for an additional 2 h at room temperature. Fluorescence was quantified on a Victor microplate reader (Perkin-Elmer) with excitation at 405 nm (10-nm band-pass), and emission was detected via 460 nm (40-nm band-pass, blue fluorescence) and 530 nm (30-nm band-pass, green fluorescence) filters. The cells were visualized by fluorescence microscopy using an inverted microscope equipped with the β-lactamase ratiometric filter set (Chroma, Bellows Falls, VT) (17).
Gene inactivation in L. pneumophila.
To obtain L. pneumophila Lens mutants defective for legK genes, a homologous recombination strategy was chosen as previously described (23). The 400-bp upstream and downstream regions of the gene of interest were amplified by PCR, digested by NotI and SalI, and then cloned into pAV695. A kanamycin resistance cassette was amplified from pKD13 (with the primers 11 and 12), digested by SalI, and cloned inside the encoding sequence of each legK gene in pAV695. The resulting construct was introduced into L. pneumophila for chromosomal recombination.
Construction of a kinase-dead LegK2-producing strain.
In order to obtain the catalytic mutant of LegK2, namely, LegK2(K112M) defective in phosphate donor ATP binding, substitution in the legK2 gene was performed with a QuikChange II site-directed mutagenesis kit (Stratagene) using primers 40 and 41 listed in Table 2, and the pXDC50 derivative vector in which legK2 is cloned under its own promoter, namely, plegK2 plasmid, as a template (Table 1). The final construct was used to transform the L. pneumophila ΔlegK2 mutant strain.
Pigment production.
For quantifying pigment accumulation, 1-ml portions of the samples, obtained from 5-day-old BYE cultures grown at 30°C, were centrifuged at 16,000 × g for 10 min, and the OD550 of the supernatants was measured (65).
Cytotoxicity to A. castellanii.
For measurement of the number of viable A. castellanii cells remaining after infection, the monolayers were treated with 10% Alamar blue (Invitrogen, Cergy Pontoise, France). At the time point indicated, the monolayers were washed four times with protease-yeast extract (PY), and then 100 μl of PY containing 10% (vol/vol) of Alamar blue was added to each well. After an overnight incubation, the OD570 values were determined. The relative degree of amoeba mortality was expressed as the ratio of the OD value of infected monolayer to that of uninfected one, calculated as [1 − (mean ODinfected/mean ODuninfected)] × 100.
Measurement of released extracellular bacteria.
L. pneumophila was grown on BCYE agar for 5 days at 30°C prior to infection. A. castellanii cells were seeded in multiwell plates to a final concentration of 5 × 105 cells ml−1. After adhesion, the cells were infected at an MOI of 10 with L. pneumophila. The plates were centrifuged at 880 × g for 10 min, followed by incubation for 1 h at 30°C. Monolayers were then washed four times with PY to remove extracellular bacteria. The time at the end of the final wash was the initial time point. After 24 h, extracellular bacteria were numbered by plating them on BCYE.
Intracellular growth in A. castellanii.
Intracellular growth was monitored as previously described (36). L. pneumophila cells harboring a fluorescent mCherry protein-producing plasmid were grown on BCYE agar containing 0.5 mM IPTG and chloramphenicol for 5 days at 30°C. A. castellanii cells were grown as adherent cells in 96-well microplates (105 cells/well) and then infected with 106 fluorescent legionellae (MOI = 10). Infection was synchronized by spinning the bacteria at 880 × g for 10 min. Intracellular growth was automatically monitored by measuring the fluorescence of mCherry at an excitation of 587 nm and an emission of 610 nm in a Xenius Safas plate reader every hour for 66 h (Safas, Monaco). Fluorescence data were subjected to background subtractions (uninfected cells).
Bacteria uptake assay by trypan blue quenching.
L. pneumophila phagocytosis was measured by trypan blue quenching as previously described (10). Briefly, bacteria from overnight cultures in BYE were labeled with 5,6-carboxyfluorescein succinimidyl ester. A. castellanii cells were grown as adherent cells in 96-well microplates (5 × 105 cells/well) and then infected with 107 fluorescent legionellae (MOI = 20). After centrifugation (880 × g for 10 min) to initiate cell-bacterium contact, the plate was incubated at 30°C for 30 min. The medium was then replaced by 50 μl per well of trypan blue solution to quench the fluorescence of noninternalized bacteria. After 1 min of incubation, the fluorescence of internalized bacteria was measured on a Xenius Safas plate reader (Safas, Monaco) with an excitation of 485 nm and an emission of 530 nm.
Recruitment of the ER to LCV in D. discoideum.
D. discoideum cells producing calnexin-GFP were used to analyze by fluorescence microscopy the recruitment of ER to Legionella-containing vacuole (LCV) that harbor mCherry-labeled L. pneumophila. D. discoideum cells were seeded onto sterile glass coverslips in 6-well plates at 5 × 106 per well in HL5 and allowed to adhere overnight. Monolayers were infected at an MOI of 100 with bacteria grown for 5 days at 30°C. The plates were spun at 880 × g for 10 min, followed by incubation at 25°C. The monolayers were then washed two times with 2 ml of SorC buffer (2 mM Na2HPO4, 15 mM KH2PO4, 50 mM CaCl2; pH 6.0) and fixed with 3.7% paraformaldehyde (30 min, 4°C). Coverslips were examined with an inverted confocal microscope (Axiovert 200M; Zeiss, Thornwood, NJ) equipped with a ×63 phase-contrast objective lens (Plan Neofluar [Zeiss]; aperture, 1.4, oil).
Localization of LegK2 within the host cell.
The ΔlegK2 mutant strain was cotransformed with p3041 and pEP73 plasmids (Table 1), which encode mCherry and the GFP-LegK2 hybrid protein, respectively. The obtained strain was used to infect D. discoideum DH1 at an MOI of 100 for 1 h. Fluorescent GFP-LegK2 protein and mCherry-labeled bacteria were observed with an inverted confocal microscope (Axiovert 200M; Zeiss) equipped with a ×63 phase-contrast objective (Plan Neofluar). Alternatively, D. discoideum DH1 harboring pEP46 vector (Table 1), which ectopically express legK2(K112M)-c-myc, were seeded onto coverslips and infected with mCherry-labeled L. pneumophila legK2 and dotA mutant strains or E. coli XL1-Blue as described above. After 1 h of infection, the cells were fixed, permeabilized, and blocked with phosphate-buffered saline supplemented with 0.2% bovine serum albumin. The coverslips were then stained with α-c-Myc monoclonal antibody (1/5,000, clone 9E10; Sigma) for 1 h and visualized with Alexa Fluor 488-conjugated secondary antibody (Molecular Probes, Eugene, OR) at a 1/300 dilution for 30 min. Microscopic observations of mCherry-labeled bacteria and LegK2 protein were performed with an inverted confocal microscope (Axiovert 200M).
Statistical analysis.
The results were statistically analyzed by using a Student t test. The t test results obtained correspond to the comparison between legK mutants with the parental strain values in the same conditions.
RESULTS
L. pneumophila Lens genome encodes five putative protein kinases.
In silico analysis of the epidemic L. pneumophila Lens strain genome revealed five genes encoding putative protein kinases. These genes, which we named legK1 to legK5 for Legionella eukaryotic gene kinases, as proposed earlier (16), encode 529, 545, 462, 961, and 453 amino acids proteins with calculated molecular masses of 61, 62, 53, 109, and 51 kDa, respectively (Table 3). Alignment with several prokaryotic and eukaryotic protein kinases revealed residues that are highly conserved in the Hanks subdomains characterizing the eukaryote-like protein kinase family (28). These include the glycine-rich loop and the invariant lysine in subdomains I and II, which are essential for binding and correct orientation of the phosphate donor ATP (Fig. 1 A). Consensus sequences in subdomains VI, VII, and VIII, involved in phosphotransfer and substrate recognition are also conserved in LegK1, LegK3, and LegK4; LegK2 and LegK5 lack subdomain VIII (Fig. 1A). Further primary sequence analysis predicted an EF-hand domain in LegK5, which is known to be involved in calcium binding (53). Moreover, hydropathy profiles suggest a transmembrane helix between amino acids 478 and 498 of LegK1, which is consistent with a membrane location of LegK1, whereas LegK2 to LegK5 are likely cytoplasmic (Table 3).
Table 3.
L. pneumophila Lens putative protein kinases

Domains inferred from electronic annotations: TM, transmembrane helix; STPK, serine/threonine protein kinase domain; YPK, tyrosine protein kinase domain; Ca-depPK, calmodulin-dependent protein kinase domain; EF-HAND-1, specific protein domain; AA, amino acid position of the first PK domain.
Fig. 1.

Putative protein kinases encoded by the L. pneumophila genome. (A) Multiple sequence alignment of the protein kinase domains of LegK1 to LegK5. The highly conserved amino acid residues of the Hanks subdomains I, II, VIB, VII, and VIII are indicated in boldface. (B) Schematic representations of the legK5 genomic region. The GC content of each gene of the region is indicated in the upper scheme. The genes conserved in the five sg1 L. pneumophila strains Lens, Paris, Philadelphia, Corby, and Alcoy are represented in gray, while the genes specific to L. pneumophila Lens are represented in black. Encoded proteins are named below.
The protein kinases encoding genes legK1 to legK5 are distributed in various regions of the L. pneumophila Lens chromosome. Organization of the legK4 region is remarkable: legK4 is located immediately upstream a gene (lpl0263) that encodes a protein with a high degree of similarity (39% identity) with the C-terminal domain (from amino acid 323 to amino acid 910) of LegK4. This genetic organization is conserved in the five sequenced L. pneumophila strains and suggests a partial duplication of legK4 homologous genes. On the other hand, whereas genes legK1 to legK4 are conserved in all L. pneumophila sequenced strains, legK5 is specific of strain Lens. It is located in a chromosomal region containing four genes that are absent in Philadelphia, Paris, Corby, and Alcoy genomes (Fig. 1B). Compared to the average genomic GC content of 38.4% for Lens strain (8), legK5 and its neighboring genes have a significantly lower GC content of 35% (legK5), 34% (lpl2477), and 36% (lpl2478). Moreover, lpl2478 gene encodes a putative transposase, and the region is flanked by two homologous genes, namely, lpl2475 and lpl2479 (Fig. 1B). These observations suggest that the legK5 chromosomal region has been acquired in Lens genome by a small insertion event associated with horizontal transfer.
LegK2 to LegK5 display autokinase and protein kinase activities.
To determine whether LegK1 to LegK5 are functional protein kinases, each protein was overproduced and purified as a recombinant protein fused to either six-histidine or glutathione S-transferase tags. The legK4 and legK5 genes were PCR amplified using genomic DNA of L. pneumophila strain Lens and cloned into the expression vector pQE30 to allow the synthesis of N-terminal six-histidine-tagged proteins. The recombinant proteins, with respective molecular masses of 110 and 52 kDa, were overproduced in the E. coli strain BL21(DE3)(pREP4-groESL) and highly enriched by a single-step chromatography on Ni-NTA matrix (Fig. 2 A). The full-length legK2 and legK3 genes were synthesized by PCR amplification and cloned into pGEX-6P-3 plasmid to produce GST N-terminal fused proteins. The truncated legK1 gene (nucleotides 4 to 1431) lacking the DNA fragment encoding the putative transmembrane helix was cloned into the same expression vector. The GST-tagged recombinant LegK1 (87 kDa), LegK2 (88 kDa), and LegK3 (79 kDa) proteins were purified to partial homogeneity from the E. coli strain BL21(DE3) (pREP4-groESL) by using a glutathione-Sepharose 4B matrix (Fig. 2A).
Fig. 2.

Biochemical activities of recombinant LegK proteins. (A) SDS-PAGE analysis of purified GST-LegK1, GST-LegK2, GST-LegK3, 6His-LegK4, and 6His-LegK5 after staining with Coomassie blue. Molecular mass standards are indicated on the left. (B) Effects of cations on autokinase activities of GST-LegK2, GST-LegK3, 6His-LegK4, and 6His-LegK5. Purified LegK proteins were subjected to in vitro autophosphorylation assays in the presence of [γ-32P]ATP and Mg2+ or Mn2+. Phosphoproteins were separated by SDS-PAGE and then revealed by autoradiography. (C) Protein kinase activities of LegK proteins. The eukaryotic substrate myelin basic protein (MBP) was incubated with each LegK recombinant protein in the presence of [γ-32P]ATP. Phosphoproteins were visualized by autoradiography after SDS-PAGE separation.
The purified tagged LegK1-LegK5 proteins were then separately assayed for autokinase activity. Each recombinant protein was incubated with the phosphate donor [γ-32P]ATP and then separated by SDS-PAGE and analyzed by autoradiography (Fig. 2B). Except for GST-LegK1, all Legionella recombinant kinases were significantly radiolabeled and thus undergo autophosphorylation. Their autokinase activities are differently dependent of magnesium or manganese ions. Although LegK4 used these ions as cofactor with the same efficiency, LegK2 and LegK5 autophosphorylated more efficiently in the presence of manganese; conversely, the radiolabeling of LegK3 was higher in the presence of magnesium (Fig. 2B).
The recombinant LegK1 to LegK5 proteins were further characterized by studying their ability to phosphorylate proteins. They were assayed for in vitro phosphorylation of the general eukaryotic protein kinase substrate, myelin basic protein (MBP), in the presence of [γ-32P]ATP. Except for GST-LegK1, a radiolabeled signal corresponding to the expected 18-kDa molecular mass of MBP was detected, thus demonstrating that LegK2, LegK3, LegK4, and LegK5 are able to phosphorylate protein substrates such as MBP (Fig. 2C).
LegK1 to LegK4 proteins are effectors of the Dot/Icm T4SS.
Most of the eukaryote-like proteins encoded by L. pneumophila genome are translocated into the host cell cytoplasm, where they may interfere with host cell functions, thus allowing bacterial evasion of the endosomal pathway (17). Although LegK1, LegK2, and LegK3 have been recently identified as Dot/Icm T4SS effectors (17, 25, 68), the translocation of LegK4 and LegK5 has not been documented yet. In order to determine whether these protein kinases are translocated during the infectious cycle, the TEM-1 reporter system (β-lactamase) was used (17). Briefly, the TEM-1 was fused to the N-terminal of LegK4 and LegK5, and L. pneumophila strains efficiently expressing these fusions (Fig. 3 A) were used to infect J774 macrophages. Host cells were then loaded with CCF4, which emits green fluorescence (520 nm) due to fluorescence resonance energy transfer between the two fluorophores of this substrate when excited at 409 nm (Fig. 3B, uninfected). When the hybrid protein was translocated, as expected for the fusion with the known Dot/Icm effector VipA, it cleaved the β-lactam ring of CCF4, thus resulting in fluorescence changing from green to blue (447 nm), when excited at the same wavelength (Fig. 3B, TEM-VipA). Conversely, when TEM-1 was fused to the housekeeping protein enoyl-acyl coenzyme A reductase FabI, cells still developed green fluorescence (Fig. 3B, TEM-FabI). Emission of blue fluorescence was observed when cells were infected with wild-type L. pneumophila strains expressing the TEM-LegK4 fusion protein, while green fluorescence was still emitted when cells were infected with bacteria containing TEM-LegK5 fusion. These analyses establish that LegK4, but not LegK5, is translocated into host macrophages during L. pneumophila infection. When a dotA mutant L. pneumophila strain producing TEM-LegK4 infected macrophages, no change in fluorescence was observed [Fig. 3B, ΔdotA(TEM-LegK4)]. This result demonstrates that translocation of the LegK4 protein kinase is Dot/Icm T4SS dependent.
Fig. 3.
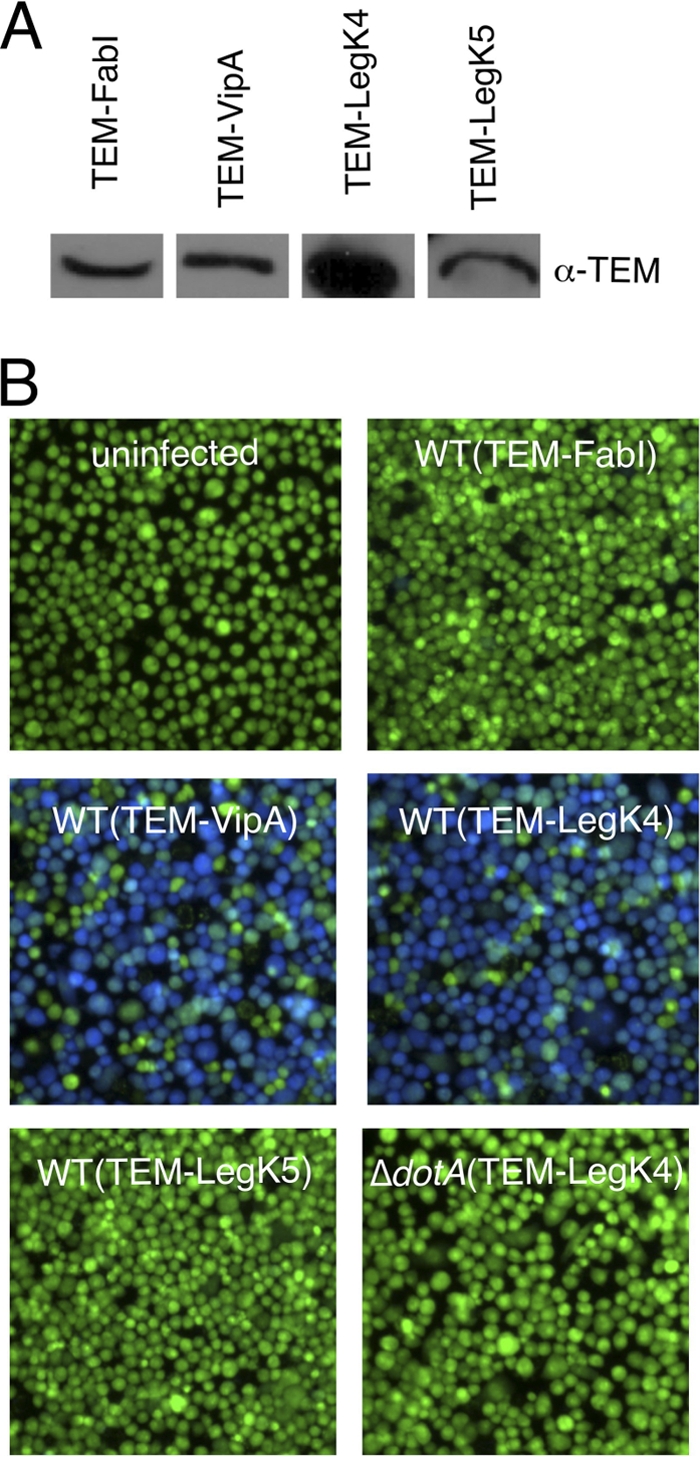
Dot/Icm-dependent translocation of LegK4 into J774 cells. (A) Western blot analysis of TEM fusion expression detected with an α-TEM antibody. (B) J774 cells were infected with L. pneumophila wild-type or dotA mutant strains harboring TEM-FabI, TEM-VipA, TEM-LegK4, and TEM-LegK5 expression plasmids at an MOI of 50. Infected cells were loaded with CCF4/AM, and translocation was determined by a comparison of cleaved to uncleaved CCF4 that gives blue and green fluorescence, respectively. Images were obtained by using epifluorescence microscopy on individual assay wells.
Inactivation of legK1 to legK5 genes has no effect on medium growth neither intracellular life cycle switch of Legionella.
To identify the physiological role of Legionella protein kinases, each legK gene was separately inactivated, as described in Materials and Methods. Genes were partially deleted and replaced by a unique insertion of a kanamycin cassette (legK::kan), verified by PCR and Southern hybridization (data not shown). The five isogenic mutants displayed similar growth characteristics in liquid medium compared to the parental strain L. pneumophila Lens, and analysis of mutant cell morphologies by light microscopy did not reveal any major differences (data not shown).
Successful infection of host cells is strictly linked to a precise timing of the life cycle of Legionella, namely, to the switch from the replicative to transmissive bacteria. Intracellular acquired transmissive traits can be mimicked by stationary medium growth phase (52). To check whether the LegK protein kinases are involved in the regulatory processes that control the intracellular life cycle of Legionella, each legK mutant was assayed for the characteristic trait of the postexponential growth phase, pigment production (65). The five legK mutants accumulated pigment during growth, as did the wild-type strain (data not shown).
LegK2 protein kinase activity plays a key role in the virulence of Legionella toward amoeba.
In order to assess the virulence of each legK mutant strain, Acanthamoeba castellanii amoebae were infected at an MOI of 10 with the wild-type L. pneumophila Lens or the isogenic legK::kan mutants, and the viability of amoeba cells present in infected monolayers at 48 h postinfection was quantified by using the Alamar blue dye. The cytotoxicity of the legK2 mutant strain toward amoeba, estimated at 30%, was significantly lower (P < 0.01) than the value of 90% measured for the parental strain. Conversely, legK1, legK3, legK4, and legK5 mutants displayed similar cytotoxicity toward amoebae compared to the wild-type strain (Fig. 4 A).
Fig. 4.

Role of LegK proteins in virulence toward A. castellanii. (A) Cytotoxicity of L. pneumophila. Cells were infected at an MOI of 10. The viability of amoeba cells present in infected monolayers at 48 h postinfection was quantified by using the Alamar blue dye. These data are representative of two independent experiments done in triplicate. (B) Release of bacteria from amoebae infected with L. pneumophila. After 24 h of infection, the number of extracellular bacteria was evaluated by the standard plate count assay. The results are expressed in CFU ml−1 and are representative of two independent experiments performed in triplicate. Error bars represent the standard deviations. (C) Intracellular growth of L. pneumophila in amoebae. Amoebae were infected at an MOI of 10 by L. pneumophila cells expressing the mCherry gene on a plasmid. Bacteria multiplication was automatically monitored by measuring the fluorescence of mCherry at an excitation of 587 nm and an emission of 610 nm every 2 h for 66 h. Fluorescence data were subjected to background subtractions (uninfected cells).
To further evaluate the virulence of each Legionella strain, extracellular released bacteria were numbered after one infectious cycle, i.e., at 24 h postinfection. Compared to the parental strain, 1.5-log fewer bacteria of the mutant strain legK2 (P = 0.04) were released from A. castellanii at 24 h postinfection. In contrast, extracellular bacteria quantified for the legK1, legK3, legK4, and legK5 mutants were similar to the wild-type strain (Fig. 4B).
To check whether the legK2 phenotype is related to a defect of bacteria egress or intracellular multiplication, the Lens strain or its legK mutant derivatives expressing the fluorescent mCherry protein on a plasmid were used to infect amoebas at an MOI of 10. Bacterial intracellular growth was monitored by fluorescence measurement during a 66 h-time course (Fig. 4C). As expected, wild-type L. pneumophila started efficient intracellular growth (an ∼40-fold increase in fluorescence) from 15 h postinfection, whereas the T4SS dotA mutant failed to replicate. The legK1, legK3, legK4, and legK5 mutants showed intracellular multiplication from the same time, with the same or lightly reduced growth rates compared to Lens strain. On the contrary, legK2 mutant was delayed for intracellular multiplication since it started replicating 48 h after infection. After that, its growth rate was not significantly different (P = 0.02) from that measured for the wild-type strain.
To confirm the link between legK2 deletion and the virulence defect observed, complementation experiments were carried out. Complementation of the legK2 mutant by introducing a ΔmobA RSF1010 derivative vector expressing the intact legK2 gene restored the cytotoxicity and the released extracellular bacteria of the parental strain (Fig. 5 A and B). Complementation assays of the legK2 intracellular multiplication defect were performed by introducing the same vector, producing both the intact LegK2 and the fluorescent mCherry proteins. The intracellular replication kinetics observed for the complemented legK2 strain was similar to that observed for the Lens strain (Fig. 5C).
Fig. 5.

Complementation of the legK2 mutant virulence defect by other legK genes. (A) Cytotoxicity of L. pneumophila measured as described in Fig. 4A. (B) Release of bacteria from amoebae evaluated as described in Fig. 4B. (C) Intracellular growth of L. pneumophila in amoebae monitored as reported in Fig. 4C.
To determine whether the protein kinase activity of LegK2 is critical for Legionella intracellular multiplication, a substitution kinase-dead mutant of LegK2, namely, LegK2(K112M) defective in phosphate donor ATP binding, was generated by site-directed mutagenesis of the legK2 gene cloned on the RSF1010 derivative vector. This construct, which stably produces the same level of kinase-dead LegK2 compared to the wild-type protein (as checked by Western blotting experiments [data not shown]), was used to complement the ΔlegK2 mutant defects in cytotoxicity (Fig. 5A), extracellular bacteria release (Fig. 5B), and intracellular multiplication (Fig. 5C). The substitution legK2 mutant is not able to complement the defects of the deletion legK2 mutant strain, suggesting that the protein kinase activity of LegK2 plays a key role in the virulence of L. pneumophila.
Thus, L. pneumophila encodes five functional protein kinases that play a variable role in bacterial virulence toward amoebae: a nondetectable role for LegK1, LegK3, LegK4, and LegK5, but a key role for LegK2. These results suggest that Legionella protein kinases are differentially expressed during the Legionella infectious cycle or that they are not functionally equal, and they would control various host or bacterial functions. To answer this question, legK gene expression during the exponential and stationary bacterial growth phases, which mimic the replicative and transmissive bacterial forms, respectively, was established by performing real-time quantitative PCR experiments. All the five legK genes were expressed in laboratory growth conditions, and expression patterns displayed a similar slight downregulation of legK1, legK2, legK3, and legK4 genes when bacteria entered the postexponential growth phase (data not shown). Moreover, the legK2 virulence defect was assayed for complementation by vectors expressing legK1, legK3, legK4, or legK5 under the control of the endogenic promoter of the legK2 gene. None of these legK genes expressing plasmids can reverse the virulence phenotype of legK2 mutant, as observed with the cytotoxicity of the corresponding strains (Fig. 5A), the extracellular bacteria numeration (Fig. 5B), and the intracellular replication (Fig. 5C). Surprisingly, expression of these genes even led to an additional loss of virulence. To check whether this observation is restricted to a legK2 genetic background, the same legK genes expressing vectors were transformed into the wild-type strain Lens. Expression of each legK gene on this plasmid resulted in a loss of virulence (data not shown). Further experiments are under way to mechanistically examine this result, but we assume that this effect most likely results from nonspecific factors, such as the elevated expression level of legK genes, which may interfere with the translocation of other T4SS substrates. Taken together, these data demonstrate that LegK2 plays a unique role in the virulence of L. pneumophila and is not functionally redundant with the four tested Legionella protein kinases.
LegK2 protein kinase activity is required for ER recruitment on the LCV.
To further characterize the virulence defect of legK2 mutant strain, the infectious cycle step altered was investigated. Intracellular survival at the onset of the A. castellanii infection was assessed by counting the viable intracellular bacteria at 1 h 20 min, 2 h, and 4 h postinfection. The level of viable intracellular legK2 cells in amoebae corresponds to <10% of the parental strain (P = 0.02), as early as 1 h 20 postinfection (Fig. 6 A). This result points out the major role of the protein kinase LegK2 in the early steps of the infectious cycle of L. pneumophila, namely, in bacterial uptake or in bacterial intracellular survival.
Fig. 6.

LegK2 is required for ER recruitment on LCV. (A) Uptake and survival ability of legK2 mutant strain. A. castellanii cells were infected at an MOI of 10 with wild-type, dotA mutant, and legK2 mutant strains. After different periods of contact with L. pneumophila, monolayers were treated for 1 h with gentamicin to kill adherent bacteria and disrupted with 0.04% Triton X-100. Viable intracellular bacteria were diluted and plated onto BCYE agar plates for colony enumeration. The results are expressed as a relative value (%) compared to a control invasion experiment with the wild-type strain. These data are representative of three independent experiments performed in triplicate; error bars represent the standard deviations. (B) Bacterial uptake assay. A. castellanii cells were infected with fluorescein-labeled Legionella at an MOI of 20, in the presence of cytochalasin D when indicated (+ CytoD). After 30 min of incubation, the medium was replaced by trypan blue solution to quench the fluorescence of noninternalized bacteria. The fluorescence of internalized bacteria was measured using an excitation of 485 nm and an emission of 530 nm. Fluorescence data were corrected for differences in labeling efficiency between the tested strains. Labeling efficiencies between strains varied by ca. 10%. (C) Recruitment of calnexin-GFP. Fifty Legionella containing vacuoles were scored from each sample by confocal laser scanning micrographs of calnexin-GFP-labeled D. discoideum AX3 infected at an MOI of 100 with mCherry-labeled L. pneumophila. Calnexin-positive vacuoles were numbered for amoeba cells infected by the wild-type L. pneumophila Lens strain, its derivative dotA and legK2 mutant strains, and the complemented legK2(plegK2) and legK2(plegK2K112M) mutant strains. The data are representative of three independent experiments; each error bar represents the standard deviation. (D) Localization of ectopically produced LegK2(K112M)-c-Myc in D. discoideum cells during infection. Confocal laser scanning micrographs of DH1 cells expressing legK2(K112M)-c-myc, either uninfected or infected with mCherry-labeled L. pneumophila legK2 mutant and dotA mutant strains or XL1-Blue E. coli. LegK2(K112M)-c-Myc was detected by an α-c-Myc antibody. The experiments were reproduced twice with similar results.
Bacteria uptake in amoebae was measured by trypan blue quenching of fluorescein-labeled L. pneumophila. Wild-type strain or legK2 derivative were fluorescein-labeled and used to infect A. castellanii. After contact, trypan blue was added to quench the fluorescence of noninternalized bacteria. Measured fluorescence is representative of bacterial uptake, as demonstrated by the effect of the actin polymerization inhibitor, cytochalasin D (Fig. 6B). The fluorescence of internalized legK2 mutant was similar to that measured for the wild-type strain (Fig. 6B), thus suggesting that LegK2 is not required for bacterial uptake but is more likely required for subsequent infectious cycle steps, namely, host cell vesicular trafficking control.
The unique intracellular fate of L. pneumophila involves ER recruitment to the LCV to evade the endocytic pathway and reside in a specialized vacuole, allowing intracellular replication. Recruitment of the ER by L. pneumophila was observed in the model amoeba Dictyostelium discoideum expressing the specific marker of ER, namely, calnexin fused to the GFP. Amoebae were infected at an MOI of 100 with wild-type or legK2 mutant strains that express the red fluorescent protein (mCherry), and calnexin-positive LCVs were visually scored by confocal microscopy at 1 and 4 h postinfection. In D. discoideum infected with the parental Legionella strain Lens, ca. 70% of the LCVs stained positive for calnexin-GFP as early as 1 h after infection. On the contrary, only 12 and 13% (P < 0.01) of the legK2 mutant containing vacuoles were positive for calnexin at 1 and 4 h postinfection, respectively. The legK2 ER recruitment defect is partially complemented by legK2-expressing RSF1010 derivative vector, since 49 and 51% (P= 0.01) of the legK2(plegK2) strain containing vacuoles were labeled by calnexin at 1 and 4 h postinfection, respectively. The incomplete complementation of the mutant could result from an elevated level of legK2 gene expression on the plasmid, which may interfere with the translocation of other T4SS substrates required for ER recruitment. Together, these results indicate that in the absence of LegK2, ER is recruited to LCVs less efficiently. To identify whether the protein kinase activity of LegK2 is involved in ER recruitment, complementation assays of the deleted legK2 mutant defect were performed with the substitution mutant legK2(K112M)-expressing vector. About 11% of the legK2(plegK2K112M) mutant-containing vacuoles were stained with the ER marker calnexin at 1 and 4 h postinfection, which is very similar to that observed for the legK2 mutant (Fig. 6C). The lack of complementation by the legK2 catalytic mutant points out the major role of LegK2 protein kinase activity in ER recruitment on the LCV.
To determine whether LegK2 anchors to the phagosomal membrane to recruit ER on the LCV, cellular localization of LegK2 during infection was investigated. Legionella cells producing the hybrid protein GFP-LegK2 were used to infect amoeba cells, and cellular localization of the hybrid protein was observed by confocal microscopy. Despite a good level of gfp-legK2 expression in bacteria, we could not detect GFP-LegK2 protein either in the host cell cytoplasm or on the LCV, probably due to a low level of translocation into host cells (data not shown). To circumvent this difficulty, a strain of the amoeba D. discoideum that ectopically produces the C-terminal c-Myc-tagged LegK2(K112M) protein was constructed. Ectopic expression of the catalytic legK2 mutant, rather than the wild-type legK2 strain, was preferred to avoid possible aspecific effects of an active protein kinase overproduction on the amoeba physiology and to facilitate stable binding of LegK2 to its putative host cell interactants (35, 58), which may be crucial for LegK2 localization. Immunofluorescence observations suggested a diffused host cell cytoplasm localization of LegK2(K112M)-c-Myc (Fig. 6D). To determine whether the localization of LegK2(K112M) could be dependent on other T4SS effectors, localization of LegK2(K112M)-c-Myc was investigated during infection of amoebae by a ΔlegK2 strain of L. pneumophila. Interestingly, infection triggered the localization of LegK2(K112M)-c-Myc into the plasma membrane (Fig. 6D). Surprisingly, the same observation was made by incubating amoebae with Escherichia coli, suggesting that localization of LegK2(K112M) to the plasma membrane may be dependent on bacterial phagocytosis rather than on T4SS effector translocation and efficient Legionella infection (Fig. 6D). Thus, when LegK2 is ectopically overproduced in amoebae, it is associated with the plasma membrane but not with the phagosomal membrane.
DISCUSSION
The genomes of the five sequenced L. pneumophila strains are characterized by an unusual abundance of eucaryote-like proteins encoding genes; among them four protein kinases encoding genes have been identified (8). Our extensive in silico analysis of the epidemic strain Lens genome identified one more protein kinase encoding gene, namely, legK5 gene. It is noteworthy that legK5 is specific to this strain. Interestingly, the LegK5 protein is homologous (50.99% identity) to a protein (UniprotKB entry number C6N1E1) from the strictly intracellular amoebal pathogen Legionella drancourtii (42). Moreover, lpl2477, located immediately downstream legK5, encodes a protein that displays 27% identity with an uncharacterized protein (UniProtKB entry number Q1RJU6) from Rickettsia belii, another intracellular bacteria that infects amoebae. Finally, the GC content of the legK5 genomic region and the occurrence of a transposase-encoding gene strongly suggest that legK5 has been acquired by a small insertion event associated with horizontal transfer during co-evolution with other bacteria in amoebae; it might represent a new instance of genomic recombination in amoebae, which have been considered a gene melting pot for evolution in a recent review (50). Multiple sequence comparisons of the kinase domains of LegK1 to LegK5 with eukaryotic and procaryotic protein kinase domains revealed that LegK1 and LegK3 cluster in the group of eukaryotic protein kinases, close to protein kinases from protozoa (data not shown), as previously reported (8, 46). This observation raises the possibility of their initial acquisition by L. pneumophila from the distant eukaryotic organisms that are natural hosts for Legionella. In contrast, LegK2, LegK4, and LegK5 are not closely similar to eukaryotic protein kinases or prokaryotic enzymes. It is noteworthy that protein kinases have been identified in intracellular bacteria such as Mycobacterium tuberculosis (1, 9) and nonpathogenic species such as Myxococcus xanthus and Anabaena cyanobacteria (47, 57, 74). The occurrence of protein kinase-encoding genes in free nonpathogenic bacterial species and in-depth analysis of GC content of various protein kinase encoding genes led researchers previously to conclude that bacterial protein kinases (“eukaryote-like”) existed before the divergence between eukaryotes and prokaryotes during evolution (27). Thus, even though the presence of horizontally acquired eukaryotic genes in amoeba pathogens such as L. pneumophila appears increasingly evident, the precise origin of protein kinase-encoding genes in L. pneumophila genomes has not been completely deciphered.
The eukaryotic kinase family is characterized by 11 conserved sequences, described as the Hanks subdomains, which are involved in ATP binding, and in phosphorylation reaction. LegK2 and LegK5 lack the Hanks subdomain VIII required for substrate recognition and phosphotransfer. However, both LegK2 and LegK5 are able to autophosphorylate and phosphorylate the eukaryotic protein kinase substrate myelin basic protein (MBP). More than 600 bacterial protein kinases have been identified through genome sequencing projects and homology-based comparisons with eukaryotic kinases and, less frequently, through direct experimental evidence (38, 39). Generally, protein kinases from bacteria conserve the nucleotide binding region (subdomains I to III) and the core catalytic domain (subdomain VII known as Brenner's motif H-X-D-X4-N [6]), but some of them display less homology as conserved enzymatic activity; for instance, SteC, which is required for host cell actin remodeling by Salmonella, is a functional protein kinase that exhibits only subdomains I to III involved in ATP binding (59). Moreover, although YihE from E. coli clearly possesses protein kinase activity, sequence analysis reveals little similarity to eukaryotic protein kinases, whereas tridimensional structure analysis reveals a kinase-like fold similar to that of choline kinase and aminoglycoside phosphotransferase (75). Likewise but to a lesser extent, LegK2 and LegK5 are functional in terms of phosphorylation even though they do not display all of the consensus domains of the eukaryotic protein kinase family. In contrast, LegK1 was not in vitro labeled by autophosphorylation and could not phosphorylate the MBP in standard in vitro assay conditions. It can be hypothesized that LegK1 must be activated to be functional. Indeed, it was previously shown that YpkA from Y. pestis is produced as an inactive protein kinase. This essential virulence factor is then translocated by a T3SS into the host cell, where it is activated by the host cell actin (33). On the other hand, we can assume that LegK1 cannot use the MBP as substrate but could phosphorylate other specific protein substrates. Indeed, it has been recently reported that LegK1 can specifically phosphorylate in vitro the eukaryotic IκBα protein but not MBP in cells, thus activating the NF-κB pathway (25).
The bacterial protein kinases LegK1 to LegK4 are translocated into the host cell cytoplasm by the Dot/Icm T4SS during infection of L. pneumophila. Conversely, LegK5 appeared not to be translocated. It is noteworthy that another intracellular pathogen, M. tuberculosis, encodes 11 different protein kinases, among them one that is translocated into the host cell to subvert the host cell defenses (70, 71), while others are tailored for bacterial signal transduction pathways (1, 51). Likewise, LegK5 may be involved in bacterial transduction signaling pathways that control Legionella metabolism rather than in the control of host cell functions.
Interestingly, inactivation and/or kinase-dead substitution of the legK2 gene resulted in a significant decrease in the virulence of L. pneumophila toward amoebae. It is noteworthy that T4SS effectors mutants are usually not altered in Legionella virulence, probably due to the high redundancy of these effectors genes (22). The legK2 mutant virulence defect is not complemented by other legK gene expression vectors, thus showing a unique and key role for the LegK2 protein kinase in the control of the infectious cycle of L. pneumophila. Although the four other LegK proteins did not interfere individually with the overall ability of Legionella to replicate in the host, they might modulate subtle pathways of Legionella infectivity and/or have complementary or redundant cellular and functional targets. Indeed, although LegK1 has been reported to activate the NF-κB pathway and likely plays an important role in modulating macrophages defenses during L. pneumophila infection, deletion of the legK1 gene has no notable effects on its intracellular replication (25, 45).
The legK2 mutant strain is defective for recruiting the ER to the LCV, which results in delayed bacterial intracellular replication. Remodeling its phagosome by the recruitment of ER is a characteristic trait of the unique intracellular fate of L. pneumophila to evade the endocytic pathway (11, 34). Interestingly, two other Dot/Icm substrates, namely, SidC and SidJ, which do not display any homology with protein kinases, have been recently shown to be involved in ER recruitment (43, 62). Mutation in sidJ results in a phenotype very similar to that of the legK2 mutation, i.e., a sidJ mutant is considerably delayed in both ER recruitment on the LCV and intracellular growth. Moreover, SidJ was not associated with the phagosomal membrane. These results led researchers to conclude that SidJ modulates as-yet-unknown host cellular pathways to control the trafficking or the retention of ER-derived vesicles to the LCV (43). It would be interesting to determine whether SidJ and LegK2 function synergistically and target the same or parallel pathways. On the other hand, SidC is an L. pneumophila effector protein that anchors to the host cell lipid phosphatidylinositol-4 phosphate and could act as a “tethering domain” for ER vesicle recruitment to the LCV (62). Indeed, as with sidJ and legK2 mutants, an LCV harboring the sidC mutant recruits ER less efficiently than does the wild type. However, sidC mutant replicates at the same rate as the wild type, while localizing in calnexin-negative LCVs, thus indicating that the interaction with the ER might not be a prerequisite to form a L. pneumophila replication-permissive compartment. In a similar manner, ralF and drrA mutants replicate normally despite an inability to recruit the host GTPase ER-Golgi regulators Rab1 and Arf1 to the LCV, respectively (49, 54). This indicates that L. pneumophila simultaneously targets multiple and yet unknown functionally redundant pathways to contribute to the formation of replication-permissive LCVs, leading to the absence of a phenotype when any single pathway is disrupted. If we consider that a kinase-dead legK2 mutant is altered both in ER recruitment and in intracellular Legionella replication, it can be proposed that the LegK2 protein kinase plays a key role for modulating one or several of the host cell signal transduction pathways that contribute to the formation of the LCV. Interestingly, this result highlights a new molecular mechanism, namely, protein phosphorylation, developed by L. pneumophila to subvert host cell defenses. The relationship between protein phosphorylation and virulence of L. pneumophila will be investigated further by characterizing the host cell pathways controlled by LegK2. Indeed, deciphering the individual contribution of each Legionella effector to the intracellular lifestyle of the bacteria remains a major and perhaps the principal challenge to understanding the molecular basis of Legionella virulence.
ACKNOWLEDGMENTS
This study was supported by the PEPS program from the Centre National de la Recherche Scientifique and the Université Lyon 1. E.H. was supported by a fellowship from the Ministère de l'Enseignement Supérieur et de la Recherche. X.C. is supported by NIH grant AI023549 awarded to Howard Shuman.
We are grateful to N. Bailo for technical assistance and F. Letourneur for helpful discussions about D. discoideum manipulations. We thank the Dicty Stock Center of Columbia University for D. discoideum strains and Howard Shuman for the opportunity to perform translocation assays in his laboratory.
Footnotes
Published ahead of print on 14 February 2011.
REFERENCES
- 1. Alber T. 2009. Signaling mechanisms of the Mycobacterium tuberculosis receptor Ser/Thr protein kinases. Curr. Opin. Struct. Biol. 19:650–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amrein K., et al. 1995. Purification and characterization of recombinant human p50csk protein-tyrosine kinase from an Escherichia coli expression system overproducing the bacterial chaperones GroES and GroEL. Proc. Natl. Acad. Sci. U. S. A. 92:1048–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Andrews H. L., Vogel J. P., Isberg R. R. 1998. Identification of linked Legionella pneumophila genes essential for intracellular growth and evasion of the endocytic pathway. Infect. Immun. 66:950–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barz C., Abahji T. N., Trulzsch K., Heesemann J. 2000. The Yersinia Ser/Thr protein kinase YpkA/YopO directly interacts with the small GTPases RhoA and Rac-1. FEBS Lett. 482:139–143 [DOI] [PubMed] [Google Scholar]
- 5. Berger K., Isberg R. 1993. Two distinct defects in intracellular growth complemented by a single genetic locus in Legionella pneumophila. Mol. Microbiol. 7:7–19 [DOI] [PubMed] [Google Scholar]
- 6. Brenner S. 1987. Phosphotransferase sequence homology. Nature 329:21. [DOI] [PubMed] [Google Scholar]
- 7. Brombacher E., et al. 2009. Rab1 guanine nucleotide exchange factor SidM is a major phosphatidylinositol 4-phosphate-binding effector protein of Legionella pneumophila. J. Biol. Chem. 284:4846–4856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cazalet C., et al. 2004. Evidence in the Legionella pneumophila genome for exploitation of host cell functions and high genome plasticity. Nat. Genet. 36:1165–1173 [DOI] [PubMed] [Google Scholar]
- 9. Chao J., et al. 2010. Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochim. Biophys. Acta 1804:620–627 [DOI] [PubMed] [Google Scholar]
- 10. Charpentier X., et al. 2009. Chemical genetics reveals bacterial and host cell functions critical for type IV effector translocation by Legionella pneumophila. PLoS Pathog. 5:e1000501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coers J., Monahan C., Roy C. 1999. Modulation of phagosome biogenesis by Legionella pneumophila creates an organelle permissive for intracellular growth. Nat. Cell Biol. 1:451–453 [DOI] [PubMed] [Google Scholar]
- 12. Cornillon S., et al. 2000. Phg1p is a nine-transmembrane protein superfamily member involved in Dictyostelium adhesion and phagocytosis. J. Biol. Chem. 275:34287–34292 [DOI] [PubMed] [Google Scholar]
- 13. Cozzone A. J. 2005. Role of protein phosphorylation on serine/threonine and tyrosine in the virulence of bacterial pathogens. J. Mol. Microbiol. Biotechnol. 9:198–213 [DOI] [PubMed] [Google Scholar]
- 14. Datsenko K., Wanner B. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. D'Auria G., Jiménez-Hernández N., Peris-Bondia F., Moya A., Latorre A. 2010. Legionella pneumophila pangenome reveals strain-specific virulence factors. BMC Genomics 11:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Felipe K., et al. 2005. Evidence for acquisition of Legionella type IV secretion substrates via interdomain horizontal gene transfer. J. Bacteriol. 187:7716–7726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Felipe K. S., et al. 2008. Legionella eukaryotic-like type IV substrates interfere with organelle trafficking. PLoS Pathog. 4:e1000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Degtyar E., Zusman T., Ehrlich M., Segal G. 2009. A Legionella effector acquired from protozoa is involved in sphingolipids metabolism and is targeted to the host cell mitochondria. Cell Microbiol. 11:1219–1235 [DOI] [PubMed] [Google Scholar]
- 19. Deutscher J., Saier M. H. 2005. Ser/Thr/Tyr protein phosphorylation in bacteria: for long time neglected, now well established. J. Mol. Microbiol. Biotechnol. 9:125–131 [DOI] [PubMed] [Google Scholar]
- 20. Echenique J., Kadioglu A., Romao S., Andrew P. W., Trombe M. C. 2004. Protein serine/threonine kinase StkP positively controls virulence and competence in Streptococcus pneumoniae. Infect. Immun. 72:2434–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ensminger A., Isberg R. 2010. E3 ubiquitin ligase activity and targeting of BAT3 by multiple Legionella pneumophila translocated substrates. Infect. Immun. 78:3905–3919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ensminger A. W., Isberg R. R. 2009. Legionella pneumophila Dot/Icm translocated substrates: a sum of parts. Curr. Opin. Microbiol. 12:67–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferhat M., et al. 2009. The TolC protein of Legionella pneumophila plays a major role in multidrug resistance and the early steps of host invasion. PLoS One 4:e7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gao L. Y., Harb O. S., AbuKwaik Y. 1997. Utilization of similar mechanisms by Legionella pneumophila to parasitize two evolutionarily distant host cells, mammalian macrophages and protozoa. Infect. Immun. 65:4738–4746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ge J., et al. 2009. A Legionella type IV effector activates the NF-κB pathway by phosphorylating the IκB family of inhibitors. Proc. Natl. Acad. Sci. U. S. A. 106:13725–13730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Glöckner G., et al. 2008. Identification and characterization of a new conjugation/type IVA secretion system (trb/tra) of Legionella pneumophila Corby localized on two mobile genomic islands. Int. J. Med. Microbiol. 298:411–428 [DOI] [PubMed] [Google Scholar]
- 27. Han G., Zhang C. 2001. On the origin of Ser/Thr kinases in a prokaryote. FEMS Microbiol. Lett. 200:79–84 [DOI] [PubMed] [Google Scholar]
- 28. Hanks S. 2003. Genomic analysis of the eukaryotic protein kinase superfamily: a perspective. Genome Biol. 4:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harb O., Gao L., Abu Kwaik Y. 2000. From protozoa to mammalian cells: a new paradigm in the life cycle of intracellular bacterial pathogens. Environ. Microbiol. 2:251–265 [DOI] [PubMed] [Google Scholar]
- 30. Ingmundson A., Delprato A., Lambright D., Roy C. 2007. Legionella pneumophila proteins that regulate Rab1 membrane cycling. Nature 450:365–369 [DOI] [PubMed] [Google Scholar]
- 31. Ish-Horowicz D., Burke J. 1981. Rapid and efficient cosmid cloning. Nucleic Acids Res. 9:2989–2998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jin H., Pancholi V. 2006. Identification and biochemical characterization of a eukaryotic-type serine/threonine kinase and its cognate phosphatase in Streptococcus pyogenes: their biological functions and substrate identification. J. Mol. Biol. 357:1351–1372 [DOI] [PubMed] [Google Scholar]
- 33. Juris S., Rudolph A., Huddler D., Orth K., Dixon J. 2000. A distinctive role for the Yersinia protein kinase: actin binding, kinase activation, and cytoskeleton disruption. Proc. Natl. Acad. Sci. U. S. A. 97:9431–9436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kagan J., Roy C. 2002. Legionella phagosomes intercept vesicular traffic from endoplasmic reticulum exit sites. Nat. Cell Biol. 4:945–954 [DOI] [PubMed] [Google Scholar]
- 35. Kawachi H., Fujikawa A., Maeda N., Noda M. 2001. Identification of GIT1/Cat-1 as a substrate molecule of protein tyrosine phosphatase zeta /beta by the yeast substrate-trapping system. Proc. Natl. Acad. Sci. U. S. A. 98:6593–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kim E., Charpentier X., Torres-Urquidy O., McEvoy M., Rensing C. 2009. The metal efflux island of Legionella pneumophila is not required for survival in macrophages and amoebas. FEMS Microbiol. Lett. 301:164–170 [DOI] [PubMed] [Google Scholar]
- 37. Kristich C. J., Wells C. L., Dunny G. M. 2007. A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. Proc. Natl. Acad. Sci. U. S. A. 104:3508–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Krupa A., Abhinandan K., Srinivasan N. 2004. KinG: a database of protein kinases in genomes. Nucleic Acids Res. 32:D153–D155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krupa A., Srinivasan N. 2005. Diversity in domain architectures of Ser/Thr kinases and their homologues in prokaryotes. BMC Genomics 6:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kubori T., Hyakutake A., Nagai H. 2008. Legionella translocates an E3 ubiquitin ligase that has multiple U-boxes with distinct functions. Mol. Microbiol. 67:1307–1319 [DOI] [PubMed] [Google Scholar]
- 41. Laemmli U. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 42. La Scola B., et al. 2004. Legionella drancourtii sp. nov., a strictly intracellular amoebal pathogen. Int. J. Syst. Evol. Microbiol. 54:699–703 [DOI] [PubMed] [Google Scholar]
- 43. Liu Y., Luo Z. 2007. The Legionella pneumophila effector SidJ is required for efficient recruitment of endoplasmic reticulum proteins to the bacterial phagosome. Infect. Immun. 75:592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lomma M., et al. 2010. The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell Microbiol. 12:1272–1291 [DOI] [PubMed] [Google Scholar]
- 45. Losick V., Haenssler E., Moy M., Isberg R. 2010. LnaB: a Legionella pneumophila activator of NF-κB. Cell Microbiol. 12:1083–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lurie-Weinberger M., et al. 2010. The origins of eukaryotic-like proteins in Legionella pneumophila. Int. J. Med. Microbiol. 300:470–481 [DOI] [PubMed] [Google Scholar]
- 47. Lux R., Shi W. 2005. A novel bacterial signaling system with a combination of a Ser/Thr kinase cascade and a His/Asp two-component system. Mol. Microbiol. 58:345–348 [DOI] [PubMed] [Google Scholar]
- 48. Machner M., Isberg R. 2007. A bifunctional bacterial protein links GDI displacement to Rab1 activation. Science 318:974–977 [DOI] [PubMed] [Google Scholar]
- 49. Machner M., Isberg R. 2006. Targeting of host Rab GTPase function by the intravacuolar pathogen Legionella pneumophila. Dev. Cell 11:47–56 [DOI] [PubMed] [Google Scholar]
- 50. Moliner C., Fournier P., Raoult D. 2010. Genome analysis of microorganisms living in amoebae reveals a melting pot of evolution. FEMS Microbiol. Rev. 34:281–294 [DOI] [PubMed] [Google Scholar]
- 51. Molle V., Kremer L. 2010. Division and cell envelope regulation by Ser/Thr phosphorylation: Mycobacterium shows the way. Mol. Microbiol. 75:1064–1077 [DOI] [PubMed] [Google Scholar]
- 52. Molofsky A., Swanson M. 2004. Differentiate to thrive: lessons from the Legionella pneumophila life cycle. Mol. Microbiol. 53:29–40 [DOI] [PubMed] [Google Scholar]
- 53. Moncrief N., Kretsinger R., Goodman M. 1990. Evolution of EF-hand calcium-modulated proteins. I. Relationships based on amino acid sequences. J. Mol. Evol. 30:522–562 [DOI] [PubMed] [Google Scholar]
- 54. Murata T., et al. 2006. The Legionella pneumophila effector protein DrrA is a Rab1 guanine nucleotide-exchange factor. Nat. Cell Biol. 8:971–977 [DOI] [PubMed] [Google Scholar]
- 55. Müller-Taubenberger A., et al. 2001. Calreticulin and calnexin in the endoplasmic reticulum are important for phagocytosis. EMBO J. 20:6772–6782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nagai H., Kagan J., Zhu X., Kahn R., Roy C. 2002. A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679–682 [DOI] [PubMed] [Google Scholar]
- 57. Nariya H., Inouye S. 2006. A protein Ser/Thr kinase cascade negatively regulates the DNA-binding activity of MrpC, a smaller form of which may be necessary for the Myxococcus xanthus development. Mol. Microbiol. 60:1205–1217 [DOI] [PubMed] [Google Scholar]
- 58. Patel R., et al. 1999. DRBP76, a double-stranded RNA-binding nuclear protein, is phosphorylated by the interferon-induced protein kinase, PKR. J. Biol. Chem. 274:20432–20437 [DOI] [PubMed] [Google Scholar]
- 59. Poh J., et al. 2008. SteC is a Salmonella kinase required for SPI-2-dependent F-actin remodeling. Cell. Microbiol. 10:20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pope C., Dhand L., Cianciotto N. 1994. Random mutagenesis of Legionella pneumophila with mini-Tn10. FEMS Microbiol. Lett. 124:107–111 [DOI] [PubMed] [Google Scholar]
- 61. Price C., et al. 2009. Molecular mimicry by an F-box effector of Legionella pneumophila hijacks a conserved polyubiquitination machinery within macrophages and protozoa. PLoS Pathog. 5:e1000704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ragaz C., et al. 2008. The Legionella pneumophila phosphatidylinositol-4 phosphate-binding type IV substrate SidC recruits endoplasmic reticulum vesicles to a replication-permissive vacuole. Cell Microbiol. 10:2416–2433 [DOI] [PubMed] [Google Scholar]
- 63. Rowbotham T. 1980. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33:1179–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Roy C. R., Berger K. H., Isberg R. R. 1998. Legionella pneumophila DotA protein is required for early phagosome trafficking decisions that occur within minutes of bacterial uptake. Mol. Microbiol. 28:663–674 [DOI] [PubMed] [Google Scholar]
- 65. Sahr T., et al. 2009. Two small ncRNAs jointly govern virulence and transmission in Legionella pneumophila. Mol. Microbiol. 72:741–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Saskova L., Novakova L., Basler M., Branny P. 2007. Eukaryotic-type serine/threonine protein kinase StkP is a global regulator of gene expression in Streptococcus pneumoniae. J. Bacteriol. 189:4168–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Segal G., Purcell M., Shuman H. A. 1998. Host cell killing and bacterial conjugation require overlapping sets of genes within a 22-kb region of the Legionella pneumophila genome. Proc. Natl. Acad. Sci. U. S. A. 95:1669–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shin S., Roy C. R. 2008. Host cell processes that influence the intracellular survival of Legionella pneumophila. Cell. Microbiol. 10:1209–1220 [DOI] [PubMed] [Google Scholar]
- 69. Smith D., Johnson K. 1988. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene 67:31–40 [DOI] [PubMed] [Google Scholar]
- 70. Tiwari D., et al. 2009. Key residues in Mycobacterium tuberculosis protein kinase G play a role in regulating kinase activity and survival in the host. J. Biol. Chem. 284:27467–27479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Walburger A., et al. 2004. Protein kinase G from pathogenic mycobacteria promotes survival within macrophages. Science 304:1800–1804 [DOI] [PubMed] [Google Scholar]
- 72. Weber S., Ragaz C., Reus K., Nyfeler Y., Hilbi H. 2006. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2:e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xu L., et al. 2010. Inhibition of host vacuolar H+-ATPase activity by a Legionella pneumophila effector. PLoS Pathog. 6:e1000822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Zhang C., Jang J., Sakr S., Wang L. 2005. Protein phosphorylation on Ser, Thr, and Tyr residues in cyanobacteria. J. Mol. Microbiol. Biotechnol. 9:154–166 [DOI] [PubMed] [Google Scholar]
- 75. Zheng J., He C., Singh V., Martin N., Jia Z. 2007. Crystal structure of a novel prokaryotic Ser/Thr kinase and its implication in the Cpx stress response pathway. Mol. Microbiol. 63:1360–1371 [DOI] [PubMed] [Google Scholar]