

Abstract
We investigated the early innate immune responses induced in human intestinal epithelial cells (IEC) by the three defined Toxoplasma gondii genotype strains. Transcriptome analysis revealed that among differentially expressed genes, β-defensins distinguished the most IEC infected by fast- or slow-replicating T. gondii genotypes. Although β-defensin 1 and 3 genes were not expressed in host cells at early time points postinfection, the slow-replicating type II and III parasites induced high levels of β-defensin 2 gene expression. Notably, no β-defensin 2 gene expression occurred early after infection with the fast-replicating type I parasite. However, activation of this gene in IEC by poly(I:C) treatment prior to infection substantially decreased parasite viability, and pretreatment of parasites with synthetic β-defensin 2 significantly reduced their infectivity of IEC. These findings strongly support the modulation of early β-defensin 2 expression as a mechanism used by type I T. gondii parasites to mediate immune evasion.
INTRODUCTION
Toxoplasma gondii is a highly prevalent apicomplexan parasite that infects the human population, cattle, and poultry (12, 30, 46). Molecular epidemiological studies of a wide collection of human and animal isolates of T. gondii obtained from Europe and North America have revealed the predominance of three major clonal lineages classified as genotypes I, II, and III. Genetic studies of T. gondii suggest that differences in the immune responses and, consequently, the clinical features of the infection can be linked with the parasite genotype (1, 2, 21). Thus, understanding the genetic factors influencing T. gondii virulence could contribute to the development of therapeutics aimed at curing the disease. In mice, virulence is strictly associated with parasite genotype. Type I infections can cause 100% lethality with 1 parasite and are therefore considered highly virulent, whereas infections with type II and type III, which can cause 50% lethality with approximately 104 and 106 parasites, respectively, are considered less virulent (40). The outcome of Toxoplasma infection in mice infected with type I parasites is mainly characterized by widespread parasite dissemination, massive proinflammatory cytokine production, and rapid death, regardless of the genetic background of the mouse, while less virulent strains achieve this effect with a high dose of inoculation and the effect is dependent on host genetic background (14, 19).
T. gondii bradyzoites infect intestinal epithelial cells (IEC), and this dormant form of the parasite rapidly transforms into active tachyzoites responsible for the dissemination of the infection throughout the body. Infected IEC induce innate immune responses via the expression of a wide range of sensors/receptors that recognize molecular patterns on pathogens invading the gut mucosa and transduce NF-κB activating signals (36). These signals induce the transcription of genes coding for antimicrobial peptides, cytokines, and chemokines. Antimicrobial peptides are evolutionarily conserved components of the innate immune system (50). There is evidence that expression of human β-defensin 1 (HBD1) and HBD4 genes in intestinal epithelial cells is constitutive (31, 41), whereas HBD2 and HBD3 gene expression is inducible in response to various signals, such as bacteria, pathogen-associated molecular patterns, or proinflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) (17, 18, 42). Defensins are initially synthesized as prepropeptides and are posttranslationally processed into mature active peptides (15). HBDs possess antimicrobial activity against a wide range of bacteria (8, 18), fungi (13), and viruses (32). In addition, defensins have chemoattractant properties on different cell types, such as monocytes, T lymphocytes, and dendritic cells (DC) (25). The antimicrobial activity of defensins is essentially mediated by the permeabilization of target membranes and may also coincide with inhibition of RNA, DNA, and protein synthesis in pathogens (6).
In the present study, we investigated the early innate mechanisms activated in human IEC against T. gondii of the three defined genotypes. Our study demonstrates that the type I (RH) parasites induce poor early innate immunity in human IEC, which includes a failure to induce β-defensin 2 expression.
(This work was presented in part at the 2nd European Congress of Immunology, Berlin, Germany, 13 to 16 September 2009.)
MATERIALS AND METHODS
Cells.
Human foreskin fibroblast cells (HFF-1) obtained from the ATCC were used to passage T. gondii tachyzoites. The human ileocecal adenocarcinoma cell line (HCT-8) was obtained from the ECACC (Sigma Aldrich). Cells were cultured in Dulbecco's modified Eagle's medium (DMEM)-nutrient mixture Ham's F-12 medium (Invitrogen Life Technologies) supplemented with 10% fetal bovine serum (FBS) (Biochrom AG, Germany) and maintained at 37°C and 5% CO2. Human small intestine primary epithelial cells were obtained from Innoprot (Bizkaia, Spain). These cells were expanded on precoated culture flasks with Matrigel (40 μg/ml) and collagen IV (30 μg/ml) in DMEM-Ham's F-12 medium supplemented with 2% fetal calf serum (FCS), glutamine (2.5 mM), penicillin (100 U/ml), streptomycin (100 μg/ml), epidermal growth factor (20 ng/ml), transferrin (5 μg/ml), insulin (5 μg/ml), hydrocortisone (1 μg/ml), and retinoic acid (300 ng/ml).
Toxoplasma gondii genotypes.
Toxoplasma gondii type I (RH) and type II (76K) strains were obtained from the Toxoplasma Reference Laboratory at our institute, and the type III (NED) strain was obtained from the National Toxoplasma Reference Laboratory (Limoge, France). Bradyzoites were obtained from brain cysts of Swiss mice infected with the 76K strain. These parasites were purified by continuous density gradient centrifugation as described by Cornelissen et al. (10).
Infection of IEC monolayers and parasite quantification.
Intestinal epithelial cell monolayers were infected with each of the three freshly isolated T. gondii genotypes at a multiplicity of infection (MOI) of 1. DNA was extracted from infected cells to study parasite replication. The T. gondii target DNA was AF146527, a 529-bp fragment repeated 200- to 300-fold in the T. gondii genome. Quantitative real-time PCR was performed as previously described (20). Cycle threshold (CT) values were converted to parasite numbers from CT values for T. gondii standards generated with a range of parasite numbers. In order to determine parasite-induced host cell cytotoxicity, lactate dehydrogenase (LDH) in culture supernatants from IEC monolayers was measured (CytoTox; Promega).
[3H]uracil staining and replication assay.
T. gondii expresses uracil phosphoribosyl transferase, an enzyme that salvages uracil to produce uracil monophosphate and hence can be used as a marker for parasite survival and growth (9). In microtiter culture plates, IEC were infected with T. gondii parasites at selective time points, followed by the addition of 2.5 μCi of [3H]uracil/well (Amersham). Cells were analyzed for radioactivity as measured in counts per minute.
Reporter gene assay.
Intestinal epithelial cells were cultured in 24-well plates, followed by transient transfection with 1 μg plasmid encoding the reporter gene luciferase activated by NF-κB (pIL8-κB-luc) and 40 ng of pRL-TK (internal control reporter-Renilla luciferase) (44) using Fugene 6 (Roche) transfection reagent. Cells were then infected with the three genotypes of T. gondii tachyzoites. Twenty-four hours later, the reporter gene activity in cell lysates was detected by a dual-luciferase reporter assay system (Promega) and measured with a Lumat LB 9507 luminometer (Berthold Technologies, Germany).
Cytokine assays.
HCT-8 cell culture supernatants were collected every 8 h for a period of 48 h following infection with the three T. gondii genotype strains. The presence of IL-8 and IL-6 in culture supernatants was determined by a cytokine-specific sandwich enzyme-linked immunosorbent assay (ELISA) (eBioscience). Optical densities were converted to concentrations using a range of cytokine standards of known concentrations.
TaqMan low-density array.
Synthesis of cDNA was performed using a cDNA Quantitect kit (Qiagen). Quantitative real-time PCR (TaqMan) assays were performed using customized 48-gene-format TaqMan low-density arrays on an Applied Biosystems PRISM 7900 sequence detection system. Specific primers and probes were all designed and prepared by Applied Biosystems. The 48-gene-format low-density array contained 44 selected target genes and 4 housekeeping genes (the 18S, Rplp1, hypoxanthine phosphoribosyltransferase [HPRT], and β-actin genes) and was run on samples from noninfected (n = 3), type I (n = 3), type II (n = 3), and type III (n = 3) T. gondii-infected samples. The 2ΔΔCt method was used to calculate relative gene expression, using 18S as the housekeeping gene.
Quantitative reverse transcriptase PCR (RT PCR).
RNA was extracted from infected and noninfected IEC by use of an RNeasy minikit (Qiagen). cDNA synthesis was performed using a RevertAid H Minus cDNA synthesis kit (Fermentas). PCR mixtures (20 μl) containing 1 μl of cDNA, specific primers for β-defensins (43), and SYBR green supermix IQ (Quanta, VWR) were run in triplicate. Standard curves were generated from serial dilutions of PCR products, which were purified using a QIAquick kit (Qiagen). CT values were used to calculate the relative expression levels of genes (2ΔΔCt method) by use of the ribosomal 18S gene as the reference gene.
Statistical analysis.
A one-way analysis of variance (ANOVA) was used to compare the IEC responses elicited by the three different T. gondii genotypes using GraphPAd InStat software.
RESULTS
Intracellular replication of T. gondii and host cell cytotoxicity.
In order to verify that HCT-8 cells are a suitable in vitro model for evaluating T. gondii genotype-specific differences, we first determined the intracellular replication of these parasites in this cell line. Measurement of parasite replication at 8-h intervals for a period of 48 h after infection demonstrated a significant difference in replication rate between the three genotypes from 24 h postinfection (Fig. 1A). The type I (RH) parasite load was significantly higher (P < 0.001) than the loads of type II (76K) and type III (NED) T. gondii. Remarkably, type I parasite replication was exponential from 32 h postinfection onwards, whereas type II and type III parasites showed intermediate and low replication levels, as evidenced by gradual increases in parasite load (Fig. 1A). Second, we determined if the parasite replication had a deleterious effect on host cells. LDH in culture supernatants was measured as an indicator of host cell cytotoxicity. A direct correlation between parasite replication and IEC cytotoxicity was observed with type I and type II T. gondii strains by comparing their respective correlation coefficients (P values of 0.6 and 0.2) (Fig. 1B). Similar to the replication of parasites, the host cell cytotoxicities induced by the three T. gondii genotype strains were in the order RH > 76K > NED. Importantly, significant LDH release was detected only in supernatants from 32 h postinfection, correlating with the high parasite load observed in IEC.
Fig. 1.
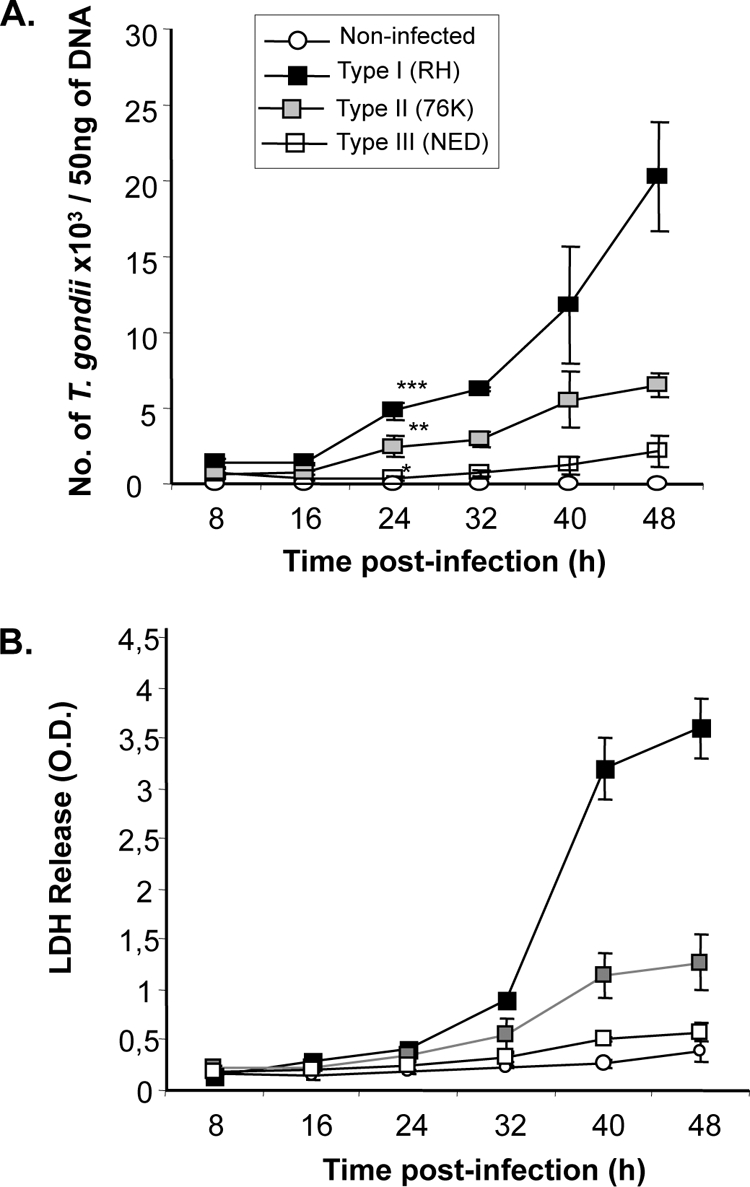
T. gondii replication is correlated with IEC cytotoxicity. (A) Comparison of levels of replication of the three T. gondii genotype strains. Human IEC cultured in 24-well plates at a density of 3 × 105 cells/well were left noninfected or infected with either type I (RH), type II (76K), or type III (NED) parasites at a MOI of 1. DNA was extracted from noninfected and infected cells at 8-h intervals for a period of 48 h, and a quantitative PCR was performed using T. gondii-specific primers and probe (see Materials and Methods). Results are expressed as numbers of T. gondii parasites/50 ng of DNA. ***, P < 0.0001; **, P < 0.001; and *, P < 0.05. Data are representative of 3 experiments, performed in triplicates at each time point. (B) Distinct levels of IEC cytotoxicity are induced by the three T. gondii genotype strains. Cell cultures were set up as described above, and supernatants were collected from noninfected/infected cells at 8-h time intervals. Lactate dehydrogenase in culture supernatants was measured using a CytoTox kit (Promega), and optical densities (O.D.) were measured at 490 nm. Data are representative of two experiments performed in triplicate at each time point.
Type I and type III T. gondii parasites induce significantly lower levels of NF-κB activation and cytokine production in IEC than type II T. gondii.
Previous reports on the capacity of T. gondii to activate host cells have provided conflicting results, largely because of the use of different hosts and/or cell types; moreover, these studies were performed only with the RH strain of T. gondii (7, 24, 29, 38). We aimed to compare the levels of NF-κB activation induced in human IEC by the three different T. gondii genotype strains. Although our experiments indicated that each of the three T. gondii genotype strains elicited activation of NF-κB in comparison to the level for noninfected cells (for type I, P < 0.001; for type II, P < 0.0001; and for type III, P < 0.05), the magnitudes of activity between strains were significantly different (Fig. 2A). We observed that type II parasites, which displayed intermediate replication, induced higher levels of NF-κB activity in IEC than type I and III parasites, confirming the observation of Robben and colleagues in human macrophages (33). We further examined whether the distinct levels of NF-κB activity could be correlated with downstream cytokine production. Type II parasites were seen to activate IEC to secrete significantly higher levels of IL-8 than type I and type III strains from 8 h until 48 h postinfection (Fig. 2B). However, a gradual increase in IL-8 secretion occurred over time following infection with type I and type III parasites. Likewise, the production of IL-6 following type II 76K strain infection was also significantly higher than that induced by type I and type III parasites. Moreover, the levels of IL-6 produced at 8 h postinfection remained unchanged over the 48-h period (Fig. 2C).
Fig. 2.

Type I and type III T. gondii strains induce low levels of NF-κB activation and poor cytokine secretion in human IEC compared to type I parasites. (A) NF-κB activation. IEC were infected by each T. gondii genotype strain at a MOI of 5 or stimulated with the TLR3 ligand [poly(I:C); 50 μg/ml]. NF-κB activation was measured by a reporter gene assay at 24 h postinfection. Results were expressed as fold NF-κB inductions obtained after the number of relative light units for infected cells was normalized to that for noninfected cells. ***, P < 0.0001; **, P < 0.001; *, P < 0.05. Data are representative of three experiments, performed in triplicate at each time point (B and C) Differential induction of IL-8 and IL-6 secretion by the three T. gondii genotypes strains. Cells were cultured in 24-well plates (3 × 105 cells/well) and infected with the three T. gondii genotypes at a MOI of 1. Cytokine secretion in culture supernatants from infected cell cultures collected at 8-h intervals for a period of 48 h was measured by a sandwich ELISA. Optical densities obtained were converted to cytokine concentrations using standard curves of known concentration for each cytokine. Data are representative of three experiments performed in triplicate at each time point.
Type I parasites induce expression of significantly fewer innate genes in IEC than type II and type III strains.
The marked differences in NF-κB activity and cytokine production induced in IEC by the three T. gondii genotype strains led us to investigate the expression of innate immune response genes following infection. RNA isolated from infected or noninfected IEC was analyzed by microfluidic arrays for the expression of innate immune response genes. Of note, in response to type II (76K) parasites, IEC induced higher levels of expression of IL-1β, IL-8, TNF-α, CCL20, thymic stromal lymphopoietin (TSLP), and CXCL10 after 3 h of infection than type I parasites (Fig. 3). At 8 h postinfection, type I parasites specifically downmodulated the expression of IL-1β, IL-6, and ΗΒD2 mRNA (Fig. 3). The expression of IL-8, IL-23A, TNF-α, CCL20, TSLP, and CXCL10 mRNA, on the other hand, was further increased by all three strains (Fig. 3). Overall, the gene expression arrays induced by type II and type III parasites were seen to be similar.
Fig. 3.

Differential expression of early innate immune genes induced in human IEC by high- and low-virulence T. gondii strains. Gene expression levels in noninfected (n = 3) and type I (n = 3), type II (n = 3), and type III (n = 3) strain T. gondii-infected IEC at 3 h (A) and 8 h (B) postinfection were compared by using TaqMan Custom Array technology.
β-Defensins are effector molecules that play an important role in early intestinal innate immune defense by their dual function primarily as antimicrobial factors for defending against pathogens and secondarily as chemotactic factors for recruiting cells for adaptive immune responses. Thus, the differences observed in replication capacity between the three T. gondii types could reflect differences in stimulation of IEC for the secretion of antimicrobial immune effectors such as β-defensins. To investigate this possibility, we examined the early expression of the ΗΒD1, -2, and -3 antimicrobial peptide genes in IEC upon infection by the three T. gondii genotype strains. As depicted in Fig. 4B, clear increases in ΗΒD2 mRNA levels occurred after 3 h of infection with type II and type III T. gondii strains but not with type I strains. However, no significant induction of constitutively expressed HBD1 mRNA levels was observed following infection by each of the three T. gondii genotypes (Fig. 4A). ΗΒD3 gene expression, on the other hand, was downregulated by all the three T. gondii genotype strains (Fig. 4C). We also tested the capacity of T. gondii to stimulate the immune response of IEC when the tachyzoites are converted into bradyzoites, a stage known to be involved in the transmission of the infection in vivo. For this purpose, bradyzoites from type II 76K parasite cysts isolated from the brains of infected mice were tested for their capacity to stimulate HBD2 in IEC. Data in Fig. 4D show that 76K bradyzoites were potent stimulators of HBD2 by IEC. Taken together, these results supported the notion that the lower replication rates of type II and III parasites might reflect their capacity to efficiently stimulate IEC to produce the antimicrobial peptide β-defensin 2. Moreover, β-defensin 2 production was also observed with other strains of type II and III genotypes, demonstrating that our results were not specific to 76K and NED strains (see Fig. S1 in the supplemental material).
Fig. 4.

Type I (RH strain) T. gondii suppress ΗΒD2 gene expression in human intestinal epithelial cells. IEC were cultured in 24-well plates at a density of 3 × 105 cells/well and infected with the three T. gondii genotype strains at a MOI of 5. RNA was extracted from infected and noninfected cells at 3, 8, and 24 h postinfection. First-strand cDNA was synthesized using oligo(dT) primers and a RevertAid H Minus cDNA synthesis kit (Fermentas). Quantitative PCR was performed using primers specific for ΗΒD1 (A), ΗΒD2 (B), or ΗΒD3 (C) using SYBR green supermix (Bio-Rad). (D) Bradyzoites obtained from 76K-infected mice were used for infection in IEC, and HBD2 gene expression was measured at 3- and 8-h time points using the method described above. Results are expressed as relative gene expression levels. ***, P < 0.0001. Data are representative of two experiments, each performed in triplicate at each time point.
Evidence for a major antimicrobial activity of ΗΒD2 against T. gondii.
The absence of early ΗΒD2 gene expression in IEC infected with type I T. gondii led us to postulate that its expression could be actively downmodulated by type I parasites in order to gain a foothold in the host. To address this possibility, we firstly examined HBD2 expression in IEC following poly(I:C) treatment, shown by our previous data to induce high NF-κB activity in HCT-8 cells. Poly(I:C) induced a time-dependent increase in ΗΒD2 mRNA expression, whereas ΗΒD1 and HBD3 mRNA remained baseline (Fig. 5A). In order to examine the antimicrobial effect of HBD2, shown to be induced by poly(I:C) treatment of IEC, type I parasites were incubated with cell culture supernatants obtained at 3, 8, and 24 h after poly(I:C) treatment (Fig. 5B). In another set of experiments, parasites were incubated with IEC pretreated with poly(I:C) for identical periods of time (Fig. 5C). In both cases, the viability of type I parasites was seen to decrease in a time-dependent manner correlating with the upregulation of HBD2 gene expression, providing evidence for the antimicrobial role of HBD2. To assess the direct antimicrobial effect of ΗΒD2, parasites were pretreated for 2 h with synthetic ΗΒD2 peptide obtained from Peptonova, Germany, and parasite invasion into IEC was examined. At a 25 μM concentration of HBD2 peptide, a significant decrease in infectivity was observed, as measured by the lower level of [3H]uracil uptake by parasites than by non-HBD2 treated parasites. This effect was observed to be more severe when the concentration of HBD2 was increased to 50 μM (Fig. 5D).
Fig. 5.

Antimicrobial effect of HBD2 against type I T. gondii. (A) Poly(I:C) induces HBD2 gene expression. IEC were stimulated with poly(I:C) (50 μg/ml) for 3 h, 8 h, and 24 h, followed by RNA extraction. First-strand cDNA was synthesized using oligo(dT) primers and a RevertAid H Minus cDNA synthesis kit (Fermentas). Quantitative PCRs for HBD1, HBD2, and HBD3 were performed as described in the legend to Fig. 4. ***, P < 0.0001. (B) Antimicrobial effect of poly(I:C)-induced HBD2 on type I T. gondii. Three-, eight-, and 24-hour culture supernatants of poly(I:C) (50 μg/ml)-induced IEC were incubated for 2 h with type I T. gondii, along with the addition of 2.5 μCi of [3H]uracil/well (Amersham). Supernatants were then analyzed for uptake of [3H]uracil by these parasites. The data are represented as percent [3H]uracil uptake in comparison to the level for non-poly(I:C)-treated counterparts. ***, P < 0.0001; *, P < 0.05. (C) Decrease in viability of type I parasites in IEC pretreated with poly(I:C). IEC were pretreated with poly(I:C) (50 μg/ml) for 3, 8, and 24 h. At each time point, the cells were infected with type I T. gondii, followed by the addition of 2.5 μCi of [3H]uracil/well. After 24 h of infection, cells were analyzed to measure [3H]uracil by intracellular parasites. The data are represented as percent [3H]uracil uptake in comparison to the level for non-poly(I:C)-treated counterparts. ***, P < 0.0001; *, P < 0.05. (D) Antimicrobial effect of synthetic HBD2 peptide on T. gondii parasites. T. gondii parasites were pretreated at 25 μM and 50 μM concentrations of HBD2 (Peptanova, Germany) for 2 h. Parasites were then washed and infected to IEC monolayers along with the addition of 2.5 μCi of [3H]uracil/well (Amersham). The data are represented as percent [3H]uracil uptake by type I parasites compared to the level for non-HBD2-treated counterparts. ***, P < 0.0001. (E) Lack of ability to suppress poly(I:C)-induced HBD2 by type I (RH) parasites. IEC were infected with RH parasites for a period of 24 h, followed by stimulation with poly(I:C) (50 μg/ml) for 3 and 8 h. Cells treated with only poly(I:C) were used as a positive control, and noninfected and nonstimulated cells were used as a negative control. Quantitative PCR was performed on cDNA obtained from these cells for expression of the HBD2 gene.
We further investigated the possibility that the reduced HBD2 production observed after infection with type I parasites might be the reflection of active suppression exerted by the parasite on IEC. To test this hypothesis, we first infected human IEC with RH and then stimulated them with poly(I:C) to stimulate the production of HBD2. As shown in Fig. 5E, poly(I:C) stimulation of IEC infected by the type I RH strain produced as much HBD2 as uninfected cells, demonstrating that failure to produce HBD2 during infection by RH was due to the inability of type I parasites to stimulate innate immunity in IEC.
Lack of HBD2 expression in HIPEC infected by T. gondii type I parasites.
To see if HBD2 expression by T. gondii infection is not restricted to transformed IEC, we further examined the expression of its gene in human primary intestinal epithelial cells (HIPEC). As depicted in Fig. 6A, similar to what was observed in the HCT-8 cell line, both type II and type III parasites stimulated the expression of HBD2 in primary epithelial cells as early as 3 h after infection. The expression was sustained at 8 h postinfection, and then it decreased to levels expressed in noninfected cells. However, HBD2 mRNA levels observed at 3 different time points in type I (RH)-infected cells were below those recorded for noninfected epithelial cells, decreasing 70-fold lower after 24 h. Thus, similar to what was observed in HCT-8 cells, the failure of type I parasite to stimulate the expression of HBD2 was also observed in primary human intestinal epithelial cells.
Fig. 6.

Type I (RH strain) T. gondii suppresses ΗΒD2 gene expression in human primary IEC. (A) Human primary IEC were cultured in 24-well plates at a density of 3 × 105 cells/well and infected with the three T. gondii genotype strains at a MOI of 5. RNA from infected and noninfected cells was extracted at 3, 8, and 24 h postinfection. First-strand cDNA was synthesized using oligo(dT) primers and a RevertAid H Minus cDNA synthesis kit (Fermentas). Quantitative RT-PCR was performed using primers specific for ΗΒD2 using SYBR green supermix (Bio-Rad), as detailed in Materials and Methods. Results are expressed as relative gene expression levels. (B) Replication of parasites in human primary intestinal epithelial cells (HIPEC) and human fibroblast cells (HFF-1). After 24 h of infection with RH, 76K, and NED strains, cells were analyzed for [3H]uracil uptake. The data shown in the figure are represented in counts per minute.
In our last set of experiments, we investigated whether the differences observed in HBD2 production between the different genotypes could influence the replication of the parasite in human primary IEC. As expected, type I parasites, which do not stimulate HBD2 in primary cells, had the highest level of replication in IEC cells (Fig. 6B). On the other hand, type II parasites, which stimulated HBD2 production as early as 3 h after infection, exhibited lower levels of replication. Interestingly, this difference in replication was also observed in the control human fibroblast cell line HFF-1, known for its incapacity to produce β-defensin 2. Taken together, these results suggested that, though the direct effect of HBD2 on parasite replication was demonstrated in our study, factors other than the capacity to stimulate HBD2 could also control parasite growth. Whether these factors are intrinsic to the parasite or depend on the type of host cells remains to be determined.
DISCUSSION
The data presented in this study strongly support the concept that parasite virulence, to a great extent, can be correlated with its capacity to downmodulate the expression of host innate immune genes in an attempt to establish infection. The manipulation of early IEC response by T. gondii in a strain genotype-dependent manner, a major finding of this study, is of consequence and contributes to the understanding of the parasite genotype-dependent pathogenesis of toxoplasmosis previously described (39).
Since T. gondii parasites replicate in small intestinal epithelial cells before disseminating to distant organs of the body, the early response of IEC to T. gondii is likely to influence the subsequent course of infection, as suggested previously (4, 23). Early gene expression in IEC following T. gondii infection was strikingly strain specific. Response to the type I (RH) T. gondii strain was significantly lower, a difference more pronounced at 3 h than at 8 h postinfection. The expression of HBD2 following the interaction of pathogens with cell surface receptors is known to be mediated by IL-1β (27). The impaired expression of IL-1β mRNA induced by type I parasites in our experiments therefore may also explain the low levels of ΗΒD2 mRNA. Likewise, the slower upregulation of chemokine CCL20 and CXCL10 mRNA, known to rapidly recruit immature DC and effector T cells, suggests early evasion of immunosurveillance by type I parasites (5, 48). Evidence for the capacity of type I parasites to distinctly manipulate IEC response as opposed to type II and type III parasites was also revealed by the lower levels of mRNA expression of TSLP known to regulate T-cell differentiation and DC maturation (28). On the other hand, the simultaneous downmodulation of CCR6 mRNA, the receptor for CCL20, by all three genotypes of T. gondii strains in IEC provides evidence for a scenario where CCL20 is primarily available for the early recruitment of CCR6 expressing adaptive immune cells (37).
Our study is the first to demonstrate the capacity of type I T. gondii, known to be virulent, to downmodulate early HBD2 gene expression in IEC. The induction of ΗΒD2 gene expression in IEC via NF-κB activation is regulated by the Toll-like receptor (TLR) and nucleotide olimerization domain as well as by cytokines (11, 16). Previous reports have shown the capacity of lipopolysaccharide (LPS) and peptidoglycan via TLR4 and TLR2, respectively, to induce HBD2 expression in intestinal epithelial cells (47). In this study, we report the capacity of poly(I:C), a TLR3 ligand, to induce HBD2 gene expression. Of note, we found that both HCT-8 cells and human primary intestinal epithelial cells expressed functional TLR3 (data not shown). Although an antimicrobial effect of poly(I:C)-induced HBD2 was observed on both extracellular and intracellular type I parasites, we cannot exclude the possible role of other factors, such as type I interferons, known to be induced through the TLR3 pathway, in the reduction of parasite viability (26). However, pretreatment of parasites with commercial HBD2 peptide decreased the infectivity of type I parasites into IEC, providing additional evidence for the direct antimicrobial effect of this defensin. On the other hand, type II and type III parasites, known to be less virulent, induced the early expression of HBD2, suggesting that they do not use the downmodulation of this gene for immune evasion. This finding suggests that the enhancement of HBD2 expression by type II and III parasites in the gut may be an efficacious intervention for these T. gondii infections, as it has recently been demonstrated for chronic Pseudomonas aeruginosa lung infection (18).
Pathogenic bacteria that invade the gastrointestinal tract and colonize the intestinal mucosa have been shown to interfere with the transcription of ΗΒD genes in epithelial cells (3, 22), and evidence for a role for virulence proteins was provided in the case of Shigella flexneri (43). The exact parasite-dependent mechanism used by type I T. gondii to manipulate the host early innate immune response is unknown. We hypothesize a possible role for early-secreted rhoptry kinases ROP18 and ROP16, previously demonstrated to play major roles in parasite replication and in alteration of host cell transcription capacity, respectively (11, 34, 35, 45). ROP16 from type I parasites phosphorylates STAT3, downmodulating innate immune responses; however, a single-nucleotide polymorphism (SNP) recently discovered in the active domain of this enzyme in the type II strain prevents this activity (49). This could explain the downmodulation of early innate responses elicited by type I parasites in IEC in contrast to the level for type II parasites. ROP16 proteins are virtually identical in type I and type III strains but highly polymorphic in type II parasites (35), possibly providing an explanation for the low levels of IL-6 and IL-8 secreted by IEC in response to the former parasites in contrast to the level for type II parasites.
In summary, this study demonstrates that T. gondii infections activate NF-κB and its downstream innate immune genes in human intestinal epithelial cells and that the magnitude of this activation is T. gondii strain type dependent. Further, we provide evidence that the suppression of β-defensin 2 gene expression is one of the important mechanisms used by type I parasites to evade intestinal innate immune defenses.
Supplementary Material
ACKNOWLEDGMENTS
We thank the 3R Research Foundation, Switzerland (project no. 107-07), the Friends of the Pasteur Institute of Brussels, and the Belgian Federal Public Service (research grant RT-06/5-Alteval-3) for providing funding. For all authors, there is no conflict of interest to disclose.
We thank M. Goldman for providing the NF-κB reporter gene plasmids and A. Legat for help with the NF-κB reporter gene assay.
Footnotes
Supplemental material for this article may be found at http://iai.asm.org/.
Published ahead of print on 7 March 2011.
REFERENCES
- 1. Ajzenberg D., et al. 2004. Genetic diversity, clonality and sexuality in Toxoplasma gondii. Int. J. Parasitol. 34:1185–1196 [DOI] [PubMed] [Google Scholar]
- 2. Ajzenberg D., et al. 2002. Genotype of 86 Toxoplasma gondii isolates associated with human congenital toxoplasmosis, and correlation with clinical findings. J. Infect. Dis. 186:684–689 [DOI] [PubMed] [Google Scholar]
- 3. Bergman P., et al. 2005. Neisseria gonorrhoeae downregulates expression of the human antimicrobial peptide LL-37. Cell. Microbiol. 7:1009–1017 [DOI] [PubMed] [Google Scholar]
- 4. Bout D., Moretto M., Dimier-Poisson I., Gatel D. B. 1999. Interaction between Toxoplasma gondii and enterocyte. Immunobiology 201:225–228 [DOI] [PubMed] [Google Scholar]
- 5. Brand S., et al. 2006. Cell differentiation dependent expressed CCR6 mediates ERK-1/2, SAPK/JNK, and Akt signaling resulting in proliferation and migration of colorectal cancer cells. J. Cell. Biochem. 97:709–723 [DOI] [PubMed] [Google Scholar]
- 6. Brogden K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250 [DOI] [PubMed] [Google Scholar]
- 7. Butcher B. A., Kim L., Johnson P. F., Denkers E. Y. 2001. Toxoplasma gondii tachyzoites inhibit proinflammatory cytokine induction in infected macrophages by preventing nuclear translocation of the transcription factor NF-kappa B. J. Immunol. 167:2193–2201 [DOI] [PubMed] [Google Scholar]
- 8. Chen X., et al. 2005. Synergistic effect of antibacterial agents human beta-defensins, cathelicidin LL-37 and lysozyme against Staphylococcus aureus and Escherichia coli. J. Dermatol. Sci. 40:123–132 [DOI] [PubMed] [Google Scholar]
- 9. Cleary M. D., Meiering C. D., Jan E., Guymon R., Boothroyd J. C. 2005. Biosynthetic labeling of RNA with uracil phosphoribosyltransferase allows cell-specific microarray analysis of mRNA synthesis and decay. Nat. Biotechnol. 23:232–237 [DOI] [PubMed] [Google Scholar]
- 10. Cornelissen A. W., Overdulve J. P., Hoenderboom J. M. 1981. Separation of Isospora (Toxoplasma) gondii cysts and cystozoites from mouse brain tissue by continuous density-gradient centrifugation. Parasitology 83:103–108 [DOI] [PubMed] [Google Scholar]
- 11. Creagh E. M., O'Neill L. A. 2006. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 27:352–357 [DOI] [PubMed] [Google Scholar]
- 12. Dubey J. P. 2008. The history of Toxoplasma gondii—the first 100 years. J. Eukaryot. Microbiol. 55:467–475 [DOI] [PubMed] [Google Scholar]
- 13. Feng Z., Dubyak G. R., Lederman M. M., Weinberg A. 2006. Cutting edge: human beta defensin 3—a novel antagonist of the HIV-1 coreceptor CXCR4. J. Immunol. 177:782–786 [DOI] [PubMed] [Google Scholar]
- 14. Gavrilescu L. C., Denkers E. Y. 2001. IFN-gamma overproduction and high level apoptosis are associated with high but not low virulence Toxoplasma gondii infection. J. Immunol. 167:902–909 [DOI] [PubMed] [Google Scholar]
- 15. Ghosh D. 2002. Paneth cell trypsin is the processing enzyme for human defensin-5. Nat. Immunol. 3:583–590 [DOI] [PubMed] [Google Scholar]
- 16. Ghosh S., Hayden M. S. 2008. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 8:837–848 [DOI] [PubMed] [Google Scholar]
- 17. Harder J., Bartels J., Christophers E., Schroder J. M. 2001. Isolation and characterization of human [beta]-defensin-3, a novel human inducible peptide antibiotic. J. Biol. Chem. 276:5707–5713 [DOI] [PubMed] [Google Scholar]
- 18. Harder J., et al. 2000. Mucoid Pseudomonas aeruginosa, TNF-alpha, and IL-1beta, but not IL-6, induce human beta-defensin-2 in respiratory epithelia. Am. J. Respir. Cell. Mol. Biol. 22:714–721 [DOI] [PubMed] [Google Scholar]
- 19. Hitziger N., Dellacasa I., Albiger B., Barragan A. 2005. Dissemination of Toxoplasma gondii to immunoprivileged organs and role of Toll/interleukin-1 receptor signalling for host resistance assessed by in vivo bioluminescence imaging. Cell. Microbiol. 7:837–848 [DOI] [PubMed] [Google Scholar]
- 20. Homan W. L., Vercammen M., De Braekeleer J., Verschueren H. 2000. Identification of a 200- to 300-fold repetitive 529 bp DNA fragment in Toxoplasma gondii, and its use for diagnostic and quantitative PCR. Int. J. Parasitol. 30:69–75 [DOI] [PubMed] [Google Scholar]
- 21. Howe D. K., Sibley L. D. 1995. Toxoplasma gondii comprises three clonal lineages: correlation of parasite genotype with human disease. J. Infect. Dis. 172:1561–1566 [DOI] [PubMed] [Google Scholar]
- 22. Islam D., et al. 2001. Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat. Med. 7:180–185 [DOI] [PubMed] [Google Scholar]
- 23. Ju C. H., Chockalingam A., Leifer C. A. 2009. Early response of mucosal epithelial cells during Toxoplasma gondii infection. J. Immunol. 183:7420–7427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim J. M., et al. 2001. Nuclear factor-kappa B plays a major role in the regulation of chemokine expression of HeLa cells in response to Toxoplasma gondii infection. Parasitol. Res. 87:758–763 [DOI] [PubMed] [Google Scholar]
- 25. Lai Y., Gallo R. L. 2009. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 30:131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lien E., Golenbock D. T. 2003. Adjuvants and their signaling pathways: beyond TLRs. Nat. Immunol. 4:1162–1164 [DOI] [PubMed] [Google Scholar]
- 27. Liu L., Roberts A. A., Ganz T. 2003. By IL-1 signaling, monocyte-derived cells dramatically enhance the epidermal antimicrobial response to lipopolysaccharide. J. Immunol. 170:575–580 [DOI] [PubMed] [Google Scholar]
- 28. Liu Y. J. 2007. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cellmaturation. Annu. Rev. Immunol. 25:193–219 [DOI] [PubMed] [Google Scholar]
- 29. Molestina R. E., Payne T. M., Coppens I., Sinai A. P. 2003. Activation of NF-kappaB by Toxoplasma gondii correlates with increased expression of antiapoptotic genes and localization of phosphorylated IkappaB to the parasitophorous vacuole membrane. J. Cell Sci. 116:4359–4371 [DOI] [PubMed] [Google Scholar]
- 30. Montoya J. G., Liesenfeld O. 2004. Toxoplasmosis. Lancet 363:1965–1976 [DOI] [PubMed] [Google Scholar]
- 31. O'Neil D. A., et al. 1999. Expression and regulation of the human beta-defensins hBD-1 and hBD-2 in intestinal epithelium. J. Immunol. 163:6718–6724 [PubMed] [Google Scholar]
- 32. Quinones-Mateu M. E., et al. 2003. Human epithelial beta-defensins 2 and 3 inhibit HIV-1 replication. AIDS 17:F39–F48 [DOI] [PubMed] [Google Scholar]
- 33. Robben P. M., et al. 2004. Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J. Immunol. 172:3686–3694 [DOI] [PubMed] [Google Scholar]
- 34. Saeij J. P., et al. 2006. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science 314:1780–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Saeij J. P., et al. 2007. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445:324–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sanderson I. R., Walker W. A. 2007. TLRs in the Gut I. The role of TLRs/Nods in intestinal development and homeostasis. Am. J. Physiol. Gastrointest. Liver Physiol. 292:G6–G10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schutyser E., Struyf S., Van Damme J. 2003. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 14:409–426 [DOI] [PubMed] [Google Scholar]
- 38. Shapira S., Speirs K., Gerstein A., Caamano J., Hunter C. A. 2002. Suppression of NF-kappaB activation by infection with Toxoplasma gondii. J. Infect. Dis. 185(Suppl. 1):S66–S72 [DOI] [PubMed] [Google Scholar]
- 39. Sibley L. D., Ajioka J. W. 2008. Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu. Rev. Microbiol. 62:329–351 [DOI] [PubMed] [Google Scholar]
- 40. Sibley L. D., Boothroyd J. C. 1992. Virulent strains of Toxoplasma gondii comprise a single clonal lineage. Nature 359:82–85 [DOI] [PubMed] [Google Scholar]
- 41. Singh P. K., et al. 1998. Production of beta-defensins by human airway epithelia. Proc. Natl. Acad. Sci. U. S. A. 95:14961–14966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sorensen O. E., et al. 2005. Differential regulation of beta-defensin expression in human skin by microbial stimuli. J. Immunol. 174:4870–4879 [DOI] [PubMed] [Google Scholar]
- 43. Sperandio B., et al. 2008. Virulent Shigella flexneri subverts the host innate immune response through manipulation of antimicrobial peptide gene expression. J. Exp. Med. 205:1121–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tanaka T., et al. 2008. DiC14-amidine cationic liposomes stimulate myeloid dendritic cells through Toll-like receptor 4. Eur. J. Immunol. 38:1351–1357 [DOI] [PubMed] [Google Scholar]
- 45. Taylor S., et al. 2006. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314:1776–1780 [DOI] [PubMed] [Google Scholar]
- 46. Tenter A. M. 2009. Toxoplasma gondii in animals used for human consumption. Mem. Inst. Oswaldo Cruz 104:364–369 [DOI] [PubMed] [Google Scholar]
- 47. Vora P., et al. 2004. Beta-defensin-2 expression is regulated by TLR signaling in intestinal epithelial cells. J. Immunol. 173:5398–5405 [DOI] [PubMed] [Google Scholar]
- 48. Williams I. R. 2006. CCR6 and CCL20: partners in intestinal immunity and lymphorganogenesis. Ann. N. Y. Acad. Sci. 1072:52–61 [DOI] [PubMed] [Google Scholar]
- 49. Yamamoto M., et al. 2009. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 206:2747–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.