

Abstract
Herein we report the synthesis of vinyl sulfone end functionalized PEGylated polymers by reversible addition-fragmentation chain transfer (RAFT) polymerization for conjugation to proteins. Poly(ethylene glycol) methyl ether acrylate (PEGA) was polymerized in the presence of 1-phenylethyl dithiobenzoate with 2,2′-azobis(2-methylpropionitrile) as the initiator to generate well-defined polyPEGAs with number-average molecular weights (Mn) by gel permeation chromatography (GPC) of 6.7 kDa, 11.8 kDa and 16.1 kDa. Post-polymerization, the majority of polymer chains contained the dithioester functional group at the omega chain end, and the polydispersity indexes (PDI) of the polymers ranged from 1.08 to 1.24. The dithioester was subsequently reduced via aminolysis, and the resulting thiol was trapped with a divinyl sulfone in situ to produce semi-telechelic, vinyl sulfone polyPEGAs with efficiencies ranging between 85% and 99%. It was determined that the retention of vinyl sulfone was directly related to reaction time, with the maximum dithioester being transformed into a vinyl sulfone within 30 minutes. Longer reaction times resulted in slow decomposition of the vinyl sulfone end group. The resulting semi-telechelic vinyl sulfone polymers were then conjugated to a protein containing a free cysteine, bovine serum albumin (BSA). Gel electrophoresis demonstrated that the reaction was highly efficient and that conjugates of increasing size were readily prepared. After polymer attachment, the activity of the BSA was 92% of the unmodified biomolecule.
Introduction
Conjugation of proteins and peptides to polymer scaffolds is of interest for drug delivery, biotechnology, and nanotechnology applications. Compared to the unmodified biomolecules, polymer conjugates have improved pharmacokinetic properties. Conjugates can also target cancerous tissue passively due to the enhanced permeability retention (EPR) effect or actively with the addition of targeting moieties on the polymer.1, 2 The conjugation site on the therapeutic directly affects its activity.1, 3 A variety of functional groups have been employed for these conjugations including: pyridyl disulfides and maleimides for thiols, ketones and aldehydes for aminooxys, or azide-alkynes.3, 4 The aforementioned functional groups are chemospecific, and the reactions take place under mild conditions, two characteristics that are essential for generating active protein- and peptide-polymer conjugates. The maleimide group in particular reacts with thiols at mild pHs. As a result this group is commonly used for conjugations to cysteine residues in engineered peptides and proteins. A drawback of the maleimide is its instability; it is easily hydrolyzed and exhibits unwanted side reactions with amines at alkaline pH. Another functional group that chemospecifically reacts with thiols is the vinyl sulfone, and the advantage of this group is its increased stability in aqueous and alkaline conditions.4-7
Many of these chemospecific functional groups have been employed to attach monodisperse poly(ethylene oxide) (PEG) to proteins in order to generate well-defined therapeutic conjugates.2, 4 PEG conjugates have been shown to display improved pharmacokinetic properties, as mentioned above; however, a downside to the use of this polymer is that it cannot be broken down in vivo. As a result, it can accumulate in the liver and gastrointestinal tract with unknown side effects.8 Synthesizing polymers from poly(ethylene glycol) methyl ether acrylate (PEGA) generates macromolecules in which the PEG side chains are attached to the polymer backbone by ester linkages.9 These PEG chains are generally short (4 to 10 units). As a result, these branched PEGs prevent nonspecific absorption of proteins similar to linear PEG,10 and increase polymer molecular weight allowing for prolonged circulation and passive targeting.1
Reversible addition-fragmentation chain transfer (RAFT) polymerization has emerged as a controlled radical polymerization (CRP) technique that is capable of producing a wide variety of well-defined polymers including poly(PEGA).11-13 RAFT polymerization is mediated by a chain transfer agent (CTA) consisting of a thiocarbonylthio group or other suitable moiety. The thiocarbonylthio functional group can be manipulated post-polymerization to reveal a thiol by reduction with primary amines14 or sodium borohydride.15 However, in the case of the reductive transformation of methacrylate polymers, the thiol can bite back onto the polymer chain, generating a cyclic thioester.16, 17 Recently, Roth et al. showed that the thiol of poly(methyl methacrylates) could be trapped as a disulfide upon performing the aminolysis reaction in the presence of methanethiosulfonate.18 The thiocarbonylthio group can also be removed by radical coupling.19-22
A number of groups have synthesized CTAs that contain a protein reactive moiety and have subsequently shown that the polymer can be conjugated to proteins or the polymer can be modified post-polymerization to install protein reactive groups.23 However, to our knowledge vinyl sulfone end functionalized polymers have not been synthesized using controlled radical polymerization. Herein, we present for the first time a method in which generated thiols are subsequently reacted with divinyl sulfone to generate semi-telechelic vinyl sulfone polymers for conjugation to proteins (Scheme 1).
Scheme 1.

Polymer synthesis and protein conjugation (protein structure from PDB # 1E7H).
Experimental
Materials
All chemicals were purchased from Sigma-Aldrich, Fisher Scientific, Acros, and EMD and used as received unless otherwise specified. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized from acetone.
Analytical Techniques
1H NMR spectra were acquired on a Bruker Avance 500 MHz or 600 MHz DRX and processed using Topspin 1.2 NMR software. Gel permeation chromatography (GPC) was conducted on a Shimadzu HPLC system equipped with a refractive index detector RID-10A, one Polymer Laboratories PLgel guard column, and two Polymer Laboratories PLgel 5 μm mixed D columns. LiBr (0.1 M) in DMF at 40 °C was used as the eluent (flow rate: 0.80 mL/min). Calibration was performed using near-monodisperse poly(methyl methacrylate) standards from Polymer Laboratories. Chromatograms were processed using the EZStart 7.2 chromatography software. UV-Vis spectra were obtained on a Biomate 5 Thermo Spectronic UV-Vis spectrometer using quartz cells. Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was carried out using 4-15% tris(hydroxymethyl)aminomethane (TRIS)-glycine precast gradient gels (Invitrogen), and samples were dissolved in TRIS buffer containing SDS, bromophenol blue and glycerol.
Methods
Typical polymerization of PEGA
1 (50.0 mg, 0.193 mmol), AIBN (15.9 mg, 0.097 mmol), and PEGA (0.970 g, 2.14 mmol) were loaded into a Schlenk tube. Dimethylformamide (DMF) (1.24 mL) was added to give a 1 M solution of PEGA. Subsequently, the tube was subjected to three freeze-pump-thaw cycles. Immersion of the Schlenk tube into an 80 °C oil bath initiated the polymerization. After 2.75 h (80% conversion) the reaction flask was removed from the oil bath and opened to the atmosphere to give crude 2a. The polymer was purified by dialysis against 1/1 EtOAc/MeOH (MWCO 1,000 Da). Polymer conversions were calculated from the 1H NMR spectra by averaging of the integrations of the vinylic proton peaks of PEGA and the peak centered at 4.26 ppm (COOCH2) of pPEGA and PEGA in CD3CN. The molecular weight was calculated upon comparison of the average integrations of three poly(PEGA) peaks, 4.40-3.80 ppm (COOCH2), 3.75-3.35 ppm (ethylene glycol units) and 3.35-3.2 ppm (OCH3), with the α-aromatic protons of the chain end at 7.30 and 7.18 ppm. 1H NMR (CD3CN): δ 7.98 (o-CH), 7.64 (p-CH), 7.34 (m-CH), 7.30 (m-CH), 7.18 (p-CH, o-CH), 4.73 (CH(CH3)Ph), 4.40-3.80 (COOCH2), 3.75-3.35 (OCH2CH2O), 3.35-3.2 (OCH3), 2.8-2.3 (CHCH2 main chain), 1.8-1.4 (CHCH2 main chain), 1.31 (CH2CH(CH3)Ph), 1.18 (CH(CH3)Ph). Number-average molecular weight (Mn) g/mol (1H NMR): 6,000. Mn g/mol (DMF GPC): 6,700. Polydispersity index (PDI): 1.09. UV (DCE): λmax = 305 nm. CTA end-group by UV-Vis: 92%. CTA end group by 1H NMR: >99%.
UV-vis analysis of end-group
A series of increasing concentrations of 1-phenylethyl dithiobenzoate (1) were prepared in 1,2-dichloroethane (DCE) and their absorbance was measured at 305 nm (λmax(SC=SC)). A line was fit to these data points though the origin, which gave a molar absorptivity (ε) of the dithiobenzoate moiety of 15790 (L · mol−1 · cm−1) in DCE (Figure S1). Purified polymers were dissolved in DCE and their absorbance at 305 nm was measured. This procedure was repeated 5x. The absorbance at 305 nm provided the concentration of dithiobenzoate present. The concentration of polymer was calculated with the molecular weights determined from the 1H NMR spectra. The concentration of dithiobenzoate was divided by the concentration of polymer and multiplied by 100 to give the retention of dithiobenzoate on the polymer.
Typical aminolysis and in situ conjugation of 2a with divinyl sulfone to afford pPEGA-vinyl sulfone (2b)
2a (75.0 mg, 0.0125 mmol) was dissolved in MeOH (1.5 mL) in Schlenk tube one. N-Butylamine (0.050 mL, 0.510 mmol) was placed in Schlenk tube two. Divinyl sulfone (0.075 mL, 0.747 mmol) was dissolved in a 0.1 M pH 8.6 phosphate buffer (PB) containing 10 mM tris(2-carboxyethyl) phosphine hydrochloride (TCEP) and 10 mM ethylenediaminetetraacetic acid (EDTA) (1.5 mL) in Schlenk tube three. All three tubes were subjected to three freeze-pump-thaw cycles under argon. The contents of tube one were added to tube two and stirred for 10 minutes. The contents of tube three were then added to tube two. The reaction was stirred for 30 min at 23 °C, followed by dialysis in H2O, then dialysis in MeOH (MWCO 1000 Da) to afford 2b. The end-group retention was determined by comparing the integrations of the peaks at 7.30 and 7.18 ppm corresponding to the α-aromatic protons of the end-group to the integrations of the vinyl sulfone at 6.80, 6.34, and 6.25 ppm. Error for the instrument is inherently 5%. We determined that depending on the ppm values utilized for the end group retention the error was 6% for polymer 2b. Thus, we report the error as +/− 6%. 1H NMR (CD3CN): δ=7.30 (m-CH), 7.18 (p-CH, o-CH), 6.80 (geminal-CH), 6.34 (cis), 6.25 (trans), 4.40-3.80 (COOCH2), 3.75-3.35 (OCH2CH2O), 3.35-3.2 (OCH3), 2.8-2.3 (CHCH2 main chain), 1.8-1.4 (CHCH2 main chain), 1.31 (CH2CH(CH3)Ph), 1.18 (CH(CH3)Ph). Mn g/mol (1H NMR): 6,000. Mn g/mol (DMF GPC): 7,100. PDI: 1.10.
Typical conjugation of vinylsulfone pPEGA to bovine serum albumin (BSA)
10 μL of a 10 mg/mL of BSA in 0.1 M PB, pH 9.0 was added to 44 μL (1 mg/mL TCEP in 0.1 M PB, pH 9.0) and diluted to a total volume of 100 μL. This 10:1 TCEP:BSA solution was allowed to sit at 23 °C for 1 h. 10 μL of the 10:1 TCEP:BSA solution, 18 equivalents of 2b (molecular weight from GPC was used, 7,100 g/mol), and 1 μL of DMSO were combined. The reaction was allowed to proceed for 2 h at 23 °C and then analyzed by SDS-PAGE under reducing conditions.
Typical conjugation of divinyl sulfone to BSA
The same protocol was used as described for the conjugation of BSA to 2b. Briefly, 10 μL of a 10 mg/mL of BSA in 0.1 M PB, pH 9.0 was added to 44 μL (1 mg/mL TCEP in 0.1 M PB, pH 9.0) and diluted to a total volume of 100 μL. This 10:1 TCEP:BSA solution was allowed to sit at 23 °C for 1 h. 10 μL of the 10:1 TCEP:BSA solution, 18 equivalents of divinyl sulfone and 1 μL of DMSO were combined. The reaction was allowed to proceed for 2 h at 23 °C and then analyzed by SDS-PAGE under non-reducing conditions.
Typical blocking of vinyl sulfone polymer with 2-mercaptoethanol
Polymer (2b) was dissolved in 25 mM PB, pH 9.0 to a final concentration of 10 mM, 40 equivalents of 2-mercaptoethanol were added, and the reaction was allowed to proceed for two hours. The excess 2-mercaptoethanol was removed by centriprep (MWCO 50 kDa).
Typical purification of BSA and BSA conjugates
After allowing the conjugation to proceed for 2 h at 23 °C, the reaction was quenched by addition of 40 equivalents of 2-mercaptoethanol. The mixture was diluted to 25 mM PB concentration and loaded onto a 1.0 mL HiTrap Q FF (anion exchange column). The column was washed with 5 column volumes of 25 mM PB pH 8.0 to remove unreacted polymer, 2-mercaptoethanol, and DMSO. The conjugate was eluted from the column with 0.4 M NaCl, 25 mM PB, pH 8.0. Purified conjugates were analyzed by SDS-PAGE, and the removal of unconjugated polymer was confirmed by iodine staining.
Typical activity assay of BSA and BSA-pPEGA conjugate
Assessment of BSA and BSA conjugate esterase activity was performed following a literature procedure with slight modification.24-27 Briefly, the following samples were diluted to 1.0 mM (total volume 9 μL) with 0.4 M NaCl, 25 mM PB, pH 8.0 after column purification: BSA, BSA-vinyl sulfone, BSA-pPEGA (2b), and BSA + polymer quenched with 2-mercaptoethanol. Then 1.0 μL of 10 mM 4-nitrophenyl acetate dissolved in acetonitrile was added. Esterase activity was monitored by measuring the absorbance of the 4-nitrophenolate anion at 405 nm after 30 min. For BSA, BSA-vinyl sulfone, BSA + blocked polymer measurements were preformed in triplicate on two different batches purified as described above. For the BSA-pPEGA conjugates, measurements were performed in triplicate on four batches purified as described above. Statistical analysis was carried out using the Student’s t-test.
Results and Discussion
Polymer synthesis
Dithiobenzoate magnesium was synthesized by a Grignard reaction between bromobenzene and carbon disulfide. Addition of (1-chloroethyl)benzene to the mixture afforded CTA 1 in 81% yield (see ESI for details). Other routes have been reported that generate 1 in 75% to 88% yield.28, 29 RAFT polymerization of PEGA was mediated by 1 in DMF at 80 °C using [1]:[0.5]:[11] of 1:AIBN:PEGA (Scheme 1). The polymerization was terminated after 2.75 h (80% conversion). The crude reaction mixture was then dialyzed against 1:1 MeOH:EtOAc to afford 2a. Analysis by GPC against monodisperse polymethyl methacrylate (PMMA) standards provided a Mn of 6.7 kDa and a PDI of 1.09. The molecular weight was calculated from the 1H NMR spectrum by comparison of the aromatic protons of the end groups to the methylene protons adjacent to the ester on the polymer (6.0 kDa), and found to be within 10% of the molecular weight determined from GPC using PMMA standards. This molecular weight was higher than the targeted molecular weight of 4.2 kDa. The difference between the targeted and obtained molecular weight may be due to the relatively high polymerization temperature used; however, the PDI was 1.09 indicating a well-defined polymer.
Dithioester chain end retention was critical for further elaboration. The dithiobenzoate group exhibits a strong UV absorbance at approximately 305 nm due to π to π* transition and a weak visible absorbance at 530 nm due to n to π* transitions that provides the distinct color.30 Post-polymerization UV-vis can be performed to assess the retention of the dithiobenzoate moiety. Using the 1H NMR molecular weight (6.0 kDa), a series of solutions of 2a were analyzed in DCE at 305 nm. This analysis indicated that 92% of the polymer chains contained the dithiobenzoate functional group. However, this value was most likely an underestimate because the electronics of the C=S bond of the CTA changed post-polymerization. The R-group of the CTA was a styrene unit, while the R-group of the polymer was the ester of the pPEGA. As the adjacent carbon to the thiocarbonylthio (R-group) becomes more electron deficient the molar absorptivity decreases. This was confirmed by UV-vis analysis of dithiobenzoate CTAs containing R-groups consisting of styrene, amide, and ester (data not shown). Comparison of the α-group (styrene) to the ω-group (dithiobenzoate) of 2a by 1H NMR indicated >99% retention post polymerization. This confirmed that the majority of the chains contained the dithiobenzoate moiety, demonstrating that well-defined polymers with high retention of the reactive dithioester group were prepared for further elaboration.
Synthesis of semi-telechelic polymers
The dithioester group is labile and susceptible to nucleophilic attack. When primary amines are employed, generation of a free thiol is observed. As a result, transformation of the dithiobenzoate group to vinyl sulfone was explored via the thiol generated by aminolysis. The conversion was performed in two steps in situ (Scheme 3). For the initial studies, a polymer with a Mn (GPC) of 7,400, PDI of 1.08, and chain end retention of 92% by UV-Vis and 98% by 1H NMR was employed. This polymer was dissolved in MeOH and the oxygen was removed by freeze-pump-thawing to minimize disulfide formation. The solution was then added to oxygen-free butylamine and stirred for 10 min, after which the red color from the dithiobenzoate disappeared. Subsequently, oxygen-free, pH 8.6 phosphate buffer containing TCEP and vinyl sulfone was added, and the reaction was allowed to proceed for 8 h. The mixture was dialyzed to remove the excess divinyl sulfone.
Scheme 3.

Synthesis of semitelechelic vinyl sulfone pPEGA
The percent conversion to the vinyl sulfone end group was analyzed by 1H NMR and the polymer by GPC. Comparison of the aromatic protons (α-group) to the vinyl protons (ω-group) indicated that 90% ± 6% of the chains contained the vinyl sulfone (Figure 1). The GPC chromatogram showed a Mn of 8.2 kDa and a PDI of 1.09; the chromatogram contained a slight high molecular weight shoulder indicative of chain-chain coupling (Figure S2). This could have resulted from polymer coupling through the divinyl sulfone or the free thiol of one polymer attacking an ester unit on another polymer chain. It was unlikely that the shoulder corresponded to disulfide formation between two chains because TCEP was present during the conjugation.
Figure 1.

1H NMR in acetonitrile-D3 of Mn 7,400 g/mol (GPC) polymer before and after reaction with vinyl sulfone.
Although the retention of dithioester prior to the reaction was >98%, only 90% of the transformed polymer chains contained a vinyl sulfone. We envisioned that there could be several reasons for this. First, it is known that thiol terminated methacrylates back bite onto the polymer backbone.16, 31 This nucleophilic attack usually occurs on the second monomer unit from the thiol forming a stable five-membered thioester. The thiol terminated poly(PEGA) may be susceptible to the same reaction. Another possibility was oxidation of thiol to the sulfonate, which would preclude reaction with the vinyl sulfone. Finally, it was feasible that the excess butylamine or another polymer could react with a chain already terminated with a vinyl sulfone. The latter would also result in the observed high molecular weight shoulder in the GPC chromatogram. To gain insight into what was occurring, the reaction was monitored by 1H NMR.
A solution of 2a was added to butylamine and stirred for 10 min. After addition of the aqueous solution containing vinyl sulfone to reduced 2a, aliquots were removed at 30 min, 1 h, 2.5 h, and 12 h and purified as described above. 1H NMR analysis (Figure 2) indicated that greater than 99% ± 6% of the polymer chains contained vinyl sulfones at 30 min (2b) and 1 h. Even after 2.5 h 94% ± 6% of the polymers contained vinyl sulfones, while at 12 h 85% ± 6% of the chains contained vinyl sulfones. The integration of the methylene protons adjacent to the ester peak 4.15 ppm remained the same within error. This suggested that the thiol was effectively generated and reacted quickly with the added vinyl sulfone, while the polymer itself remained unchanged. Again a small high molecular weight shoulder and slight increase in the molecular weight was observed for 2b (Figure 3). Likely this was a result of reaction of a few of the newly formed vinyl sulfone polymer chains with thiol-terminated polymer. The gradual disappearance of the vinyl sulfone group over time suggested degradation likely by the slow addition of the excess butyl amine. However, even after 12 h, greater than 85% of the polymer chains contained the desired functionality. This demonstrated the remarkable selectivity of the vinyl sulfone group in the presence of primary amines, which is important for site selective conjugation to free cysteines in the presence of lysine residues.
Figure 2.

1H NMR in acetonitrile-D3 over time while trapping thiol with divinyl sulfone.
Figure 3.
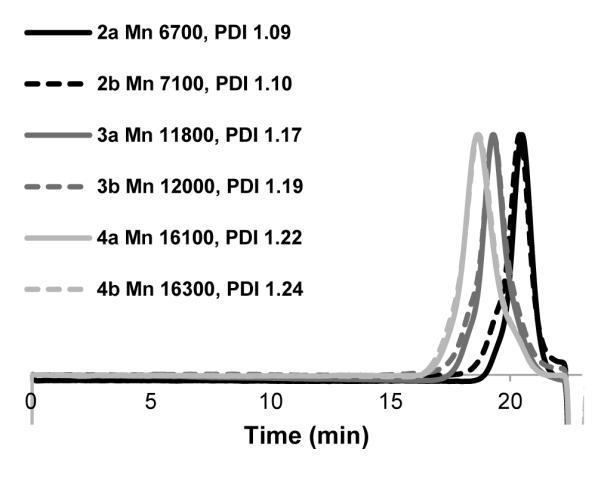
GPC chromatograms of 2a, 2b, 3a, 3b, 4a, and 4b normalized and analyzed by comparison to monodisperse PMMA standards.
To investigate if the reaction was possible with a bismaleimide, 1,8-bis-maleimide diethylene glycol was conjugated following the same procedure. In contrast to the former reaction, where the vinyl sulfone group had not hydrolyzed to an appreciable extent even at 12 h, only 16.5% and 7.5% of the polymer chains contained maleimide after 1 h and 12 h, respectively (Figure S7). Maleimides are known to hydrolyze quickly in neutral to alkaline environments,4 and this data was consistent with literature. This result further demonstrated the enhanced stability of the vinyl sulfone, which will be useful for storage and bioconjugation of this polymer.
Synthesis of higher molecular weight vinyl sulfone polymers
Next, longer chains were pursued in order to generate polymers of useful size for therapeutic applications. The subsequent polymers were synthesized using ratios of [1]:[0.5]:[37] and [1]:[0.5]:[74], and the polymerizations were stopped at 2.75 h (82% conversion) and 2.75 h (78% conversion) to generate 3a and 4a, respectively, after dialysis. The targeted molecular weight for 3a was 14 kDa, and GPC indicated a Mn of 11.8 kDa and a PDI of 1.17 (Figure 3). Similar to 2a the theoretical and GPC molecular weights do not correlate. Retention of the dithiobenzoate post-polymerization for 3a was determined to be 88% by comparison of the R and Z-group by 1H NMR. The targeted molecular weight for 4a was 26.5 kDa, and GPC indicated a Mn of 16.1 kDa and a PDI of 1.22 (Figure 3). Retention of the dithiobenzoate post-polymerization was determined to be 89% by comparison of the R and Z-groups by 1H NMR. These results indicated that loss of control occurs when accessing higher molecular weights, because the PDIs increased and the retention of the dithioester decreased. We have previously observed this with RAFT polymerization of PEGA.32
Aminolysis and in situ conjugation to divinyl sulfone using a 30 min incubation time were pursued for 3a and 4a to afford 3b and 4b, respectively. GPC analysis indicated a Mn of 12.0 kDa and PDI of 1.19 for 3b and 16.3 kDa and PDI of 1.24 for 4b (Figure 3). The same 1H NMR analysis of 3b and 4b was performed as for 2b. The integrations of the aromatic protons of the α-group were compared to the vinyl sulfone peaks to give 93% ± 6% and 85% ± 6% retention of vinyl sulfone, respectively. This result demonstrated that the dithioesters that were available were readily transformed to vinyl sulfones.
Conjugation of semi-telechelic polymers to a protein
Conjugations of bovine serum albumin (BSA) to the semi-telechelic vinyl sulfone polymers (2b, 3b, and 4b) were pursued. BSA is a convenient model system because it is commercial available and contains a cysteine that does not participate in a disulfide bond (Cys-34) with a molecular mass of ~66 kDa. Although BSA contains a free cysteine, it is known that this thiol is partially oxidized. We had previously demonstrated that when a ratio of 10:1 TCEP:BSA was used, it was possible to obtain 3 free thiols.33 Thus the BSA was reduced in this manner, and conjugations were performed in 0.1 M phosphate buffer pH 9.0 with 10% dimethylsulfoxide (DMSO) using 20 equivalents of polymer to protein and 10:1 TCEP:BSA.
The conjugations were allowed to proceed for 2 h at 23 °C and then analyzed by SDS-PAGE under reducing conditions. Lanes 3, 4, and 5 showed the formation of a high molecular weight species at ~72 kDa, ~150 kDa, and ~200 kDa; respectively (Figure 4). Since three thiols were available for conjugation, it was possible to obtain one to three sites of labeling. The different molecular weight polymers provided distinctly different molecular weight conjugates by SDS-PAGE. This demonstrated the ability to target different molecular weight protein-polymer conjugates with these vinyl sulfone-terminated pPEGAs, which is important for tuning the pharmacokinetic properties of polymer therapeutics. Furthermore, the conjugations were highly efficient as evidenced by the minimal amount of unmodified protein present after the reaction. As a control, BSA with Cys34 pre-blocked with maleimide was incubated with 2b and no shift was observed (Figure S8).
Figure 4.
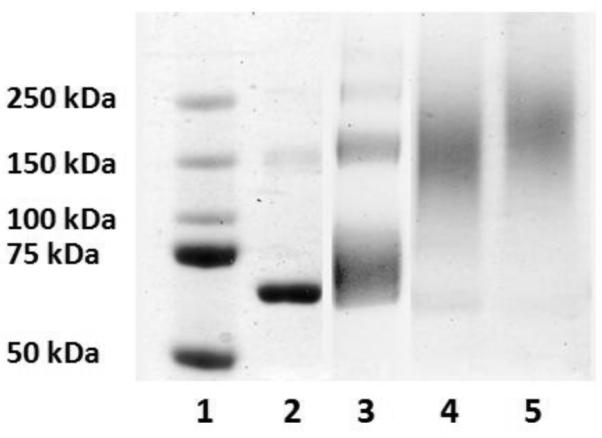
SDS-PAGE of the conjugation of 2b, 3b, and 4b to BSA (reducing conditions). Lane 1) protein marker, 2) BSA, 3) 2b + BSA, 4) 3b + BSA, and 5) 4b + BSA.
To confirm that the integrity of the protein was maintained, activity studies were pursued. It known that BSA, as well as human serum albumin, possesses esterase activity.24-27 Additionally, if the secondary and tertiary structures of serum albumins are altered, the proteins are no longer active. Conjugation of BSA to 2b was undertaken as described above, and the reaction was quenched with 2-mercaptoethanol. Unmodified BSA was also subjected to the same reaction and purification conditions, minus the polymer. Conjugation was confirmed by SDS-PAGE (Figure S8). Analysis of the esterase activity of the unmodified BSA and BSA-pPEGA (2b) conjugate were performed with 4-nitrophenyl acetate (NPA) by measuring the appearance of 4-nitrophenylate at 405 nm. Two additional controls in which vinyl sulfone-pPEGA blocked with 2-mercaptoethanol was added to unmodified BSA and BSA conjugated to divinyl sulfone were also analyzed.
It is known that BSA is rapidly acetylated by NPA.27 Our results indicated that the BSA-pPEGA (2b) conjugate maintained 92 % activity compared to unmodified BSA (Figure 5). These results are similar to those previously obtained for BSA-N-isopropylacrylamide conjugates.24, 25 The controls had high activity (94-95%), yet were statistically different than both the unmodified BSA and BSA-polymer conjugate. The origin of this reduction in activity indicated that the presence of substrate (either divinyl sulfone or non-reactive polymer) during the reaction had a small deleterious effect on protein activity, which likely contributed to the 8% reduction in conjugate activity observed. Together these data demonstrate that attachment of polymer chains to the BSA does not inhibit esterase activity of BSA to an appreciable extent. This suggests that this particular polymer may be useful to prepare PEGylated therapeutic conjugates with retention of activity.
Figure 5.
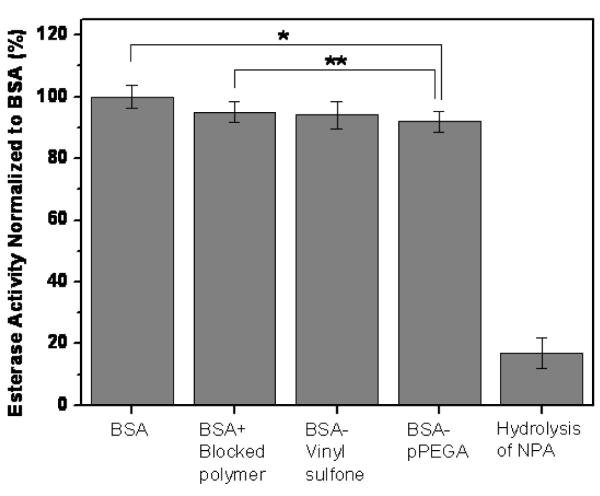
Esterase activity of BSA-pPEGA (2b) conjugate compared to unmodified BSA and BSA incubated with pre-blocked-vinyl sulfone-pPEGA. Error bars indicate the standard deviation of six replicates, except for BSA-pPEGA, which was 12 replicates. *, **Denote statistical deviation from unmodified BSA (p < 0.05).
Conclusion
RAFT polymerization was employed to synthesize well-defined polyPEGAs with vinyl sulfone chain ends. Access of various molecular weights was demonstrated. Aminolysis of these polymers enabled trapping of the subsequent thiol with divinyl sulfone to generate semi-telechelic vinyl sulfone polymers capable of reacting with free cysteines. We demonstrated that the transformation occurred rapidly and efficiently, while preserving the integrity of the polymer. The polymers were then effectively used to synthesize protein-polymer conjugates. Esterase activity studies revealed that the BSA-polymer conjugate retained 92% of BSA activity. We anticipate that this technique will allow for protein-polymer conjugates with properties tailored to their desired application. Furthermore, this polymer functionalization technique should be useful to conjugate proteins, DNA, peptides, and other biomolecules, without complication due to hydrolysis of chain ends. In addition, the vinyl sulfone chain end would be an excellent partner in thiol-ene reactions for a wide variety of polymer materials applications.
Supplementary Material
Scheme 2.

Synthesis of pPEGA.
Table 1.
Table of polymers.
| entry | Conversion (%) |
[1]:AIBN:PEGA | Mn (theory) |
Mn (GPC) |
PDI | end group (%)a |
|---|---|---|---|---|---|---|
| 2a | 80 | 1:0.5:11 | 4200 | 6700 | 1.09 | Dithiobenzoate (>99) |
| 2b | - | - | 4200 | 7100 | 1.10 | Vinyl Sulfone (>99) |
| 3a | 82 | 1:0.5:37 | 14000 | 11800 | 1.17 | Dithiobenzoate (88) |
| 3b | - | - | 14000 | 12000 | 1.19 | Vinyl Sulfone (93) |
| 4a | 78 | 1:0.5:74 | 26500 | 16100 | 1.22 | Dithiobenzoate (89) |
| 4b | - | - | 26500 | 16300 | 1.24 | Vinyl Sulfone (85) |
Calculated by comparison of the R to Z-group by 1H NMR.
Acknowledgements
This research was funded by the National Science Foundation (CHE-0809832). GNG thanks the Christopher S. Foote Graduate Research Fellowship in Organic Chemistry and the NIH Biotechnology Training Grant. SNSA thanks the NIH sponsored Chemistry and Biology Interface (CBI) Training Program. NMM thanks the UC LEADS program. HDM thanks the Alfred P. Sloan Foundation for additional funding. The authors appreciate Dr. Sung Hye Kim for her assistance with the statistical analysis.
Footnotes
Supporting Information Supporting information is available free of charge via the Internet at http://pubs.acs.org. CTA synthesis, protocol for conjugation of the bis-maleimide, and UV-vis analysis of the CTA, GPC spectra, 1H NMR spectra, and SDS PAGE gel images not provided in the text.
References
- (1).Duncan R. Nat. Rev. Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- (2).Veronese FM, Harris JM. Adv. Drug Delivery Rev. 2008;60:1–2. doi: 10.1016/j.addr.2007.04.018. [DOI] [PubMed] [Google Scholar]
- (3).Heredia KL, Maynard HD. Org. Biomol. Chem. 2007;5:45–53. doi: 10.1039/b612355d. [DOI] [PubMed] [Google Scholar]
- (4).Hermanson GT. Bioconjugate Techniques. 1st ed Elvsevier Science; San Diego: 1996. p. 785. [Google Scholar]
- (5).Bae JW, Lee E, Park KM, Park KD. Macromolecules. 2009;42:3437–3442. [Google Scholar]
- (6).Ding ZL, Long CJ, Hayashi Y, Bulmus EV, Hoffman AS, Stayton PS. Bioconjugate Chem. 1999;10:395–400. doi: 10.1021/bc980108s. [DOI] [PubMed] [Google Scholar]
- (7).Shimoboji T, Ding ZL, Stayton PS, Hoffman AS. Bioconjugate Chem. 2001;12:314–319. doi: 10.1021/bc000107b. [DOI] [PubMed] [Google Scholar]
- (8).Murakami Y, Tabata Y, Ikada Y. Drug Delivery. 1997;4:23–31. [Google Scholar]
- (9).Han DK, Hubbell JA. Macromolecules. 1997;30:6077–6083. [Google Scholar]
- (10).Satomi T, Nagasaki Y, Kobayashi H, Otsuka H, Kataoka K. Langmuir. 2007;23:6698–6703. doi: 10.1021/la0624384. [DOI] [PubMed] [Google Scholar]
- (11).Lowe AB, McCormick CL. Prog. Polym. Sci. 2007;32:283–357. [Google Scholar]
- (12).Moad G. Aust. J. Chem. 2006;59:661–662. [Google Scholar]
- (13).Han DH, Pan CY. J. Polym. Sci., Part A: Polym. Chem. 2008;46:341–352. [Google Scholar]
- (14).Qiu XP, Winnik FM. Macromol. Rapid Commun. 2006;27:1648–1653. [Google Scholar]
- (15).Narain R, Gonzales M, Hoffman AS, Stayton PS, Krishnan KM. Langmuir. 2007;23:6299–6304. doi: 10.1021/la700268g. [DOI] [PubMed] [Google Scholar]
- (16).Xu JT, He JP, Fan DQ, Wang XJ, Yang YL. Macromolecules. 2006;39:8616–8624. [Google Scholar]
- (17).Gruendling T, Pickford R, Guilhaus M, Barner-Kowollik C. J. Polym. Sci., Part A: Polym. Chem. 2008;46:7447–7461. [Google Scholar]
- (18).Roth PJ, Kessler D, Zentel R, Theato P. Macromolecules. 2008;41:8316–8319. [Google Scholar]
- (19).Perrier S, Takolpuckdee P, Mars CA. Macromolecules. 2005;38:2033–2036. [Google Scholar]
- (20).Heredia KL, Grover GN, Tao L, Maynard HD. Macromolecules. 2009;42:2360–2367. doi: 10.1021/ma8022712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HD. Chem. Commun. 2009:2148–2150. doi: 10.1039/b822799c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Tao L, Kaddis CS, Loo RRO, Grover GN, Loo JA, Maynard HD. Macromolecules. 2009 doi: 10.1021/ma901540p. DOI: 10.1021/ma901540p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gauthier MA, Klok HA. Chem. Commun. 2008:2591–2611. doi: 10.1039/b719689j. [DOI] [PubMed] [Google Scholar]
- (24).Boyer C, Bulmus V, Liu JQ, Davis TP, Stenzel MH, Barner-Kowollik C. J. Am. Chem. Soc. 2007;129:7145–7154. doi: 10.1021/ja070956a. [DOI] [PubMed] [Google Scholar]
- (25).De P, Li M, Gondi SR, Sumerlin BS. J. Am. Chem. Soc. 2008;130:11288–11289. doi: 10.1021/ja804495v. [DOI] [PubMed] [Google Scholar]
- (26).Means GE, Bender ML. Biochemistry. 1975;14:4989–4994. doi: 10.1021/bi00693a031. [DOI] [PubMed] [Google Scholar]
- (27).Tildon JT, Ogilvie JW. J. Biol. Chem. 1972;247:1265–1271. [PubMed] [Google Scholar]
- (28).Kanagasabapathy S, Sudalai A, Benicewicz BC. Tetrahedron Lett. 2001;42:3791–3794. [Google Scholar]
- (29).Sudalai A, Kanagasabapathy S, Benicewicz BC. Org. Lett. 2000;2:3213–3216. doi: 10.1021/ol006407q. [DOI] [PubMed] [Google Scholar]
- (30).Kitchin AD, Velate S, Chen M, Ghiggino KP, Smith TA, Steer RP. Photoch. Photobio. Sci. 2007;6:853–856. doi: 10.1039/b702811c. [DOI] [PubMed] [Google Scholar]
- (31).Lima V, Jiang XL, Brokken-Zijp J, Schoenmakers PJ, Klumperman B, Van Der Linde R. J. Polym. Sci., Part A: Polym. Chem. 2005;43:959–973. [Google Scholar]
- (32).Bays E, Tao L, Chang CW, Maynard HD. Biomacromolecules. 2009;10:1777–1781. doi: 10.1021/bm9001987. [DOI] [PubMed] [Google Scholar]
- (33).Heredia KL, Bontempo D, Ly T, Byers JT, Halstenberg S, Maynard HD. J. Am. Chem. Soc. 2005;127:16955–16960. doi: 10.1021/ja054482w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.