

Abstract
The increasing resistance of malarial parasites to almost all available drugs calls for the identification of new compounds and the detection of novel targets. Here, we establish the antimalarial activities of risedronate, one of the most potent bisphosphonates clinically used to treat bone resorption diseases, against blood stages of Plasmodium falciparum (50% inhibitory concentration [IC50] of 20.3 ± 1.0 μM). We also suggest a mechanism of action for risedronate against the intraerythrocytic stage of P. falciparum and show that protein prenylation seems to be modulated directly by this drug. Risedronate inhibits the transfer of the farnesyl pyrophosphate group to parasite proteins, an effect not observed for the transfer of geranylgeranyl pyrophosphate. Our in vivo experiments further demonstrate that risedronate leads to an 88.9% inhibition of the rodent parasite Plasmodium berghei in mice on the seventh day of treatment; however, risedronate treatment did not result in a general increase of survival rates.
INTRODUCTION
Malaria is the leading cause of morbidity and mortality in tropical regions, with 300 to 500 million clinical cases and around 1 million deaths a year (40). Plasmodium falciparum is responsible for the most severe form of the disease. Malaria expansion in some areas has been attributed to the failure of vector control policies and, mainly, to the increase of parasite resistance to drugs commonly used for its therapy (33). This alarming scenario has accelerated research into new antimalarial drugs, focusing efforts on the study not only of conventional but also of novel targets, such as isoprenoid biosynthesis (41). Isoprenoids are abundant and diverse compounds widely distributed in nature, and many of them (e.g., retinoids, carotenoids, ubiquinones, dolichols, and prenyl groups bound to proteins) are essential components of the cellular machinery of all organisms due to their roles in a plethora of biological processes (39).
All isoprenoids are derived from a common precursor, isopentenyl pyrophosphate (IPP), and its isomer, dimethylallyl pyrophosphate (DMAPP). Farnesyl pyrophosphate synthase (FPPS) catalyzes the consecutive condensation of IPP with DMAPP and geranyl pyrophosphate (GPP) to produce farnesyl pyrophosphate (FPP). FPP biosynthesis is considered the branching point in the isoprenoid synthesis pathway, since it is the substrate for enzymes that catalyze the first committed step for the biosynthesis of diverse isoprenoids. FPP can also be condensed with an additional molecule of IPP to yield geranylgeranyl pyrophosphate (GGPP), a reaction catalyzed by geranylgeranyl pyrophosphate synthase (GGPPS) (17).
FPP and GGPP are substrates for prenyl:protein transferases (farnesyl transferase and geranylgeranyl transferase), catalyzing the posttranslational modification of proteins (6). Protein prenylation is a general pathway in eukaryotic cells and has been described for several protozoan parasites (12, 19, 26, 34, 44), including P. falciparum (5). In addition, our group has characterized farnesylated and geranylgeranylated proteins in P. falciparum and showed that prenylation can be inhibited by terpenes (30). The process of protein prenylation is a very attractive goal for the development of new drugs targeting malignant cells and protozoan parasites (4, 16).
Bisphosphonates are potent inhibitors of bone resorption and are in clinical use for the treatment and prevention of osteoporosis, Paget's disease, hypercalcemia caused by malignancy, and tumor metastasis in bone (29), and it is promising as an antibacterial, anticancer, and antiparasitic drug (10, 32, 36). In vitro and in vivo antiparasitic effects of bisphosphonates against Leishmania mexicana, Leishmania donovani, Trypanosoma cruzi, Trypanosoma brucei, Cryptosporidium parvum, Toxoplasma gondii, and P. falciparum have been established (10, 15, 24, 27, 31, 38, 42). Recently, it was demonstrated that “lipophilic” bisphosphonates are effective against Plasmodium liver stages (35). Specifically, nitrogen-containing bisphosphonates (N-BPs) have been developed to be employed in the treatment of bone diseases, targeting FPPS. N-BPs prevent the posttranslational prenylation of proteins in J774 macrophages (25) and osteoclasts in vitro by inhibiting FPPS (8, 13).
In this study, we characterized the in vitro and in vivo antiplasmodial activities of risedronate, a commercially available N-BP. Our results further confirm the inhibition of parasite protein prenylation as a possible mechanism of action for risedronate, as this drug decreased FPP and GGPP biosynthesis and inhibited FPP transfer to proteins.
MATERIALS AND METHODS
Reagents.
Percoll was purchased from GE Healthcare. [1-(n)-3H]geranylgeranyl pyrophosphate triammonium salt {[1-(n)-3H]GGPP} (16.5 Ci/mmol), [1-(n)-3H]farnesyl pyrophosphate triammonium salt {[1-(n)-3H]FPP} (16.5 Ci/mmol), and [1-14C]isopentenyl pyrophosphate triammonium salt ([14C]IPP) (55.0 Ci/mmol) were obtained from GE Healthcare. Life Technologies supplied Albumax I. Sigma-Aldrich provided isopentenyl pyrophosphate (IPP), farnesyl pyrophosphate ammonium salt (FPP), geranylgeranyl pyrophosphate ammonium salt (GGPP), farnesol (FOH), and geranylgeraniol (GGOH). Risedronate was purchased from Gerbrás Química Farmacêutica (Brazil).
Plasmodium falciparum culture.
P. falciparum clone 3D7 was cultured according to a protocol described previously by Trager and Jensen (37), where human serum was replaced by Albumax I (0.5%) (21). Parasites were grown in a 40-ml volume in an atmosphere of 5% CO2, 5% O2, and 90% N2. The cultures were initially synchronized in the ring stage (1 to 20 h after reinvasion) by two treatments with 5% (wt/vol) d-sorbitol solution in water for subsequent maintenance in culture until the differentiation to the trophozoite (20 to 30 h after reinvasion) or schizont (30 to 35 h after reinvasion) stage. Parasite development and multiplication were monitored by the microscopic evaluation of Giemsa-stained thin smears.
Inhibition tests with risedronate and rescue assays.
Risedronate was dissolved in sterile deionized water, resulting in a 25 mM stock solution. The inhibition tests were carried out with flat-bottomed microtiter plates using the following drug concentrations: 3,000, 300, 30, 3, 0.3, 0.03, and 0.003 μM. We employed a method described previously by Desjardins and coauthors (9), with some modifications, to determine risedronate 50% inhibitory concentrations (IC50s) for P. falciparum intraerythrocytic stages after 48 h of treatment. Briefly, synchronic ring-stage parasite cultures (5% hematocrit and 1% parasitemia) were exposed to increasing drug concentrations, and the parasitemia and parasite morphologies were determined with Giemsa-stained smears immediately before the start and at intervals of 24 to 96 h, instead of [3H]hypoxanthine incorporation. All tests were performed in triplicates for three independent experiments. The IC50, IC90 (± standard deviation), and 95% confidence interval (CI95%) values for growth inhibition were calculated by using Origin 8.1 software (OriginLab). For the rescue assays, FPP, GGPP, and IPP were solubilized in RPMI medium (5 mM stock solution), and different concentrations of each compound (100 nM to 1,000 nM) were then added simultaneously to synchronous P. falciparum cultures in the ring stage previously treated with 20 μM risedronate. Parasitemia was assessed every 24 h. Statistical analysis was performed by using one-way analysis of variance (ANOVA) followed by Dunnett's post hoc test (GraphPad Prism, CA). A P value of <0.05 was considered statistically significant.
Treatment with risedronate and metabolic labeling.
Asynchronous cultures of P. falciparum were treated with 15 μM risedronate for 36 h and labeled with [1-3H]GGPP (3.125 μCi/ml) or [1-3H]FPP (3.125 μCi/ml) in normal RPMI 1640 medium for the last 12 h in the presence of drug. After labeling, ring, trophozoite, and schizont forms were purified on a 40%-70%-80% discontinuous Percoll gradient (3), followed by cell lysis in a solution containing ice-cold 10 mM Tris-HCl (pH 7.2), 150 mM NaCl, 2% (vol/vol) Triton X-100, 0.2 mM phenylmethylsulfonyl fluoride (PMSF), 5 mM iodoacetamide, 1 mM N-(p-tosyl-lysine)chloromethyl ketone, and 1 μg/ml leupeptin (TEN-Triton). After incubation for 15 min at 4°C, lysates were centrifuged at 10,000 × g for 30 min. Supernatants of parasites were stored in liquid N2 for subsequent SDS-PAGE analysis. For the analysis of isoprenoids, synchronic cultures in the ring stage were treated with 15 μM risedronate for 36 h and metabolically labeled with [1-14C]IPP for the last 12 h. After labeling, schizont-stage parasites were purified on a 40%-70%-80% discontinuous Percoll gradient as described above and freeze-dried prior to lipid extraction as described elsewhere previously (30). Risedronate at 15 μM was considered the ideal drug concentration to be used in our metabolic labeling experiments, since approximately 90% of the parasite population remained viable after 36 h of treatment.
Reverse-phase thin-layer chromatography (RP-TLC).
Similar amounts of schizont-stage pellets of untreated or risedronate-treated parasites labeled with [1-14C]IPP as described above were extracted with hexane for the subsequent separation of alcohols on reverse-phase Silica Gel 60 plates (Merck) with acetone-H2O (6:1, vol/vol) (23). Plates were sprayed with En3Hance (DuPont NEN) and subjected to autoradiography for 45 days at −70°C. The standards were visualized with iodine vapor, and Rf values were determined. Hexane extracts of uninfected erythrocytes were used as a control group.
Gel electrophoresis.
SDS-PAGE was performed with 12.5% gels as described elsewhere previously (22). The same numbers of drug-treated or untreated parasites labeled with [1-3H]GGPP or [1-3H]FPP were extracted in TEN-Triton and solubilized in SDS sample buffer (50 mM Tris-HCl [pH 6.8], 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol) and applied onto each well for protein extract analyses. All gels were incubated with Amplify (Amersham), dried, and exposed to Kodak X-Omat film with intensifying screen sets at −70°C for 30 days.
Immunoprecipitation assays.
Synchronic cultures in the ring stage were treated with 15 μM risedronate for 24 h and metabolically labeled with [1-3H]FPP or [1-3H]GGPP for an additional 12 h in the presence of the drug. After labeling, schizont-stage parasites were purified on a 40%-70%-80% discontinuous Percoll gradient as described above. Pellets of untreated and treated schizont-stage parasites were resuspended in immunoprecipitation buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% [vol/vol] Triton X-100, 0.5% [wt/vol] sodium deoxycholate, 0.1% [wt/vol] SDS, 5 μg/ml protease inhibitor cocktail [0.2 mM phenylmethylsulfonyl fluoride, 1 mM benzamidine, 2 mM β-mercaptoethanol, chymostatin {5 mg/ml}, and 1 μg/ml leupeptin, antipain, and pepstatin A]) and then precleared with protein A-Sepharose beads (GE Healthcare) (28). Schizont forms were then incubated with anti-human Ras or anti-Rap1/Krev-1 monoclonal immunoglobulins (1:20 dilution; Cell Signaling Technology) for 2 h at 4°C. The antigen-antibody complex was precipitated by using 100 μl of a 10% protein A-Sepharose slurry. After five washes with phosphate-buffered saline (PBS), the bound antigen was released in SDS sample buffer and analyzed by SDS-PAGE and autoradiography (20). Densitometric analyses were performed by using Image J software (National Institutes of Health).
In vivo assays.
Each male BALB/c mouse (3 to 4 weeks old) (n = 10 to 15 per group) was injected intraperitoneally (i.p.) with 106 blood-stage Plasmodium berghei strain ANKA parasites. Our laboratory previously established this parasite burden as the 50% lethal dose 14 days after inoculation. Risedronate treatment with different concentrations was initiated in 2 h after infection on day 0 and continued for 7 days. The drug was diluted in PBS and administered i.p. at 10, 15, 20, and 25 mg/kg of body weight/day. Parasitemia levels were monitored microscopically by examining Giemsa-stained thin blood smears on days 4, 7, 11, 14, and 17 postinfection. Throughout this period, the spontaneous death of each animal was computed. The percentage of parasitemia inhibition was calculated as follows: 100 − [(mean parasitemia for the treated group/mean parasitemia for the control group) × 100] (14). For comparisons of average parasitemias at different time points, analysis of variance was performed with a post hoc Mann-Whitney test for comparisons of the means (Origin 8.1 software; OriginLab). All in vivo assays were approved by the Ethics Committee for Animal Experimentation of the Instituto de Ciências Biomédicas, University of São Paulo.
RESULTS
Inhibition of P. falciparum development after treatment with risedronate.
To test the inhibitory effect of risedronate on P. falciparum growth, parasites were cultured in the absence or presence of increasing concentrations of the drug. The growth of parasites was inhibited in a dose-dependent manner with an IC50 of 20.3 ± 1.0 μM (CI95%, 19.17 to 21.43; IC90, 38.6 ± 0.9 μM). The inhibitory effect of risedronate was reverted by the simultaneous addition of FPP or GGPP at 1 μM during 48 h of treatment (88.3% ± 9.1% and 83.5% ± 9.8% of growth, respectively). When IPP, an upstream precursor of FPP, was incubated under the same experimental conditions, the inhibitory effect of risedronate was not reversed, validating the hypothesis that FPPS is a potential target of risedronate (Fig. 1).
Fig. 1.

Effect of a 48-h risedronate treatment on the growth of P. falciparum. Parasite growth inhibition by risedronate (55.2% ± 7.1%) was partially reversed by the addition of FPP (88.3% ± 9.1%) or GGPP (83.5% ± 9.8%) to a final concentration of 1,000 nM in parasite cultures. The addition of IPP did not reverse the inhibition. Parasite growth percentages when FPP, GGPP, or IPP was added alone in parasite culture were not statistically different from those of the parasite control. One-way analysis of variance with Dunnett's multiple-comparison test was used to determine statistical differences (*) between FPP plus risedronate versus risedronate (P < 0.05) and between GGPP plus risedronate versus risedronate (P < 0.01). Results are expressed as the means of parasite growth (percent) ± standard deviations from three independent experiments.
Effect of risedronate on the biosynthesis of P. falciparum isoprenoids.
To determine the effect of risedronate on isoprenoid biosynthesis, parasites were metabolically labeled with [1-14C]IPP, and lipid extracts of schizont-stage parasites for untreated and treated parasites (15 μM risedronate for 36 h) were analyzed by RP-TLC (Fig. 2). [1-14C]IPP incorporation into isoprenoids was inhibited in risedronate-treated schizont-stage parasites, as the intensities of bands with Rf values corresponding to farnesol (FOH) and geranylgeraniol (GGOH) standards were decreased. The intensities of bands with Rf values equivalent to prenol of 8 and 9 isoprenic chains were also reduced.
Fig. 2.
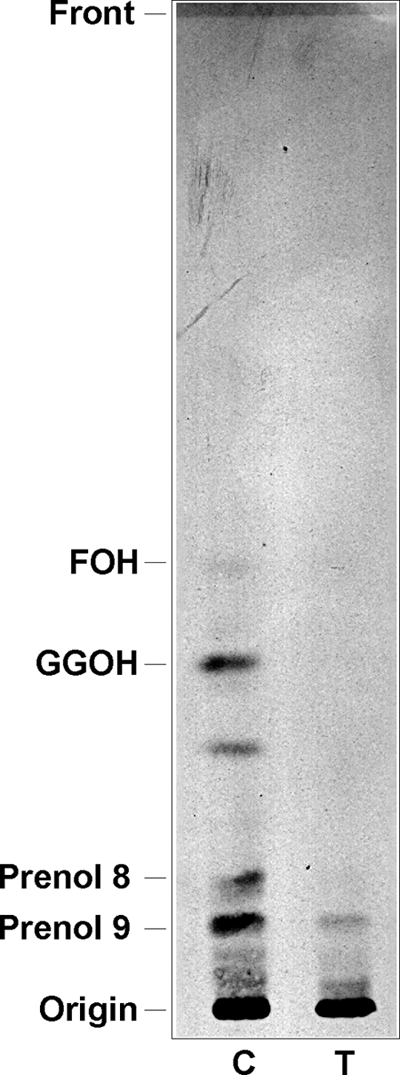
RP-TLC analysis of hexane extract schizont-stage P. falciparum parasites. Parasites were untreated and treated for 36 h with 15 μM risedronate and then metabolically labeled for the last 12 h with [1-14C]IPP. The solvent system was acetone-H2O (6:1, vol/vol). Abbreviations: C, control; T, risedronate-treated parasites; FOH, farnesol; GGOH, geranylgeraniol.
Risedronate interferes with Plasmodium protein isoprenylation.
Given that FOH and GGOH biosyntheses were clearly inhibited after risedronate treatment, one could speculate that one of the effects of risedronate is probably related to its interference with the protein isoprenylation process. To test this hypothesis, we labeled parasites with [1-3H]FPP or [1-3H]GGPP in order to analyze protein extracts by SDS-PAGE. When ring- and schizont-stage parasites were treated with 15 μM risedronate and then labeled with [1-3H]FPP, we observed significant differences in the incorporation of this precursor into proteins. The intensities of bands presenting molecular masses of around 51, 41, 27 to 29, and 7 kDa decreased. Interestingly, trophozoite-stage parasites presented almost all the farnesylated proteins inhibited (except for the 7-kDa band) after incubation with risedronate (Fig. 3A). In contrast, when parasites were treated with the drug and labeled with [1-3H]GGPP, bands with an apparent molecular mass of 27 to 29 kDa showed an increase of intensity for all stages evaluated (Fig. 3B). Noninfected red blood cells showed no incorporation of radioactivity under these conditions (data not shown). In order to evaluate whether risedronate specifically alters p21ras and p21rap in P. falciparum protein lysates, untreated and risedronate-treated schizont forms previously labeled with [1-3H]FPP or [1-3H]GGPP were immunoprecipitated with anti-p21ras or anti-p21rap antibodies, respectively. Compared to the control group, Ras-like protein levels in schizont-stage parasites were reduced approximately 2-fold after treatment with risedronate. On the other hand, an increase of 3.8-fold versus the control group was detected for a band of 21 kDa corresponding to Rap-like proteins in treated parasites (Fig. 3C). Taken together, these results reinforce that risedronate modulates the incorporation of [1-3H]FPP and [1-3H]GGPP into parasite proteins in an opposite manner.
Fig. 3.

In vitro treatment of P. falciparum cultures with risedronate and metabolic labeling with [1-3H]GGPP or [1-3H]FPP. (A and B) Parasites were untreated or treated for 36 h with 15 μM risedronate and labeled for the last 12 h with [1-3H]FPP (A) or [1-3H]GGPP (B). (C) Immunoprecipitation using anti-p21ras or anti-p21rap antibody with schizont-stage parasites metabolically labeled with [1-3H]FPP or [1-3H]GGPP, respectively. The parasites were harvested and purified by Percoll gradients, lysed, and analyzed by SDS-PAGE and fluorography. The parasite stage analyzed is indicated at the bottom of each panel (ring, trophozoite, and schizont forms). Molecular mass standards are indicated on the left (kDa). C, control; T, risedronate-treated parasites. This is a representative experiment of three independent assays.
In vivo effect of risedronate.
After determining the antiplasmodial effect of risedronate in vitro, we verified its efficacy in BALB/c mice infected with P. berghei strain ANKA. The administration of 20 and 25 mg/kg risedronate for 4 days led to decreases of parasitemia of 68.9% and 83.6%, respectively. On the seventh day of treatment the inhibitions were 63% and 88.9% with 20 and 25 mg/kg, respectively (Fig. 4A). After recovering the parasitemia, a dose-response curve was obtained for estimating the ID50 (dose causing 50% inhibition), equivalent to 17 ± 1.8 mg/kg after 7 days of treatment. Four days after the interruption of treatment (11 days postinfection), the parasitemias of the groups treated with 10, 15, 20, and 25 mg/kg/day were 15.3%, 15.9%, 15.2%, and 5.7%, respectively. Conversely, the group that received PBS presented parasitemia of 25.6%. Among the groups treated with risedronate, only the animals that received 25 mg/kg had a significant inhibition of 77.8% (see Table S1 in the supplemental material), demonstrating that even after treatment discontinuation, the parasitemia of the animals remained low in relation to that of the controls; however, parasite recrudescence was observed for all treated groups. By Kaplan-Meier survival analysis there was no difference between risedronate-treated mice and PBS-treated groups (Fig. 4B).
Fig. 4.

Effect of risedronate on mice infected with P. berghei erythrocytic-stage parasites. Mice were infected by the intraperitoneal injection of 1 × 106 Plasmodium berghei ANKA-parasitized murine erythrocytes and treated with PBS or different doses of risedronate i.p. for 7 days. (A) Parasitemia was monitored on days 4, 7, 11, 14, and 17 after infection. Each time point represents the means ± standard errors, and asterisks indicate significant differences between PBS- and risedronate-treated groups. A Mann-Whitney test was used to evaluate statistic differences between groups, and a P value of <0.05 were considered statistically significant (n = 10 per group). (B) Mortality was checked daily, and survival curves were plotted according to Kaplan-Meier analysis using GraphPad Prism 5 (GraphPad Software, CA). Differences between groups were evaluated by a log-rank test, but there were no significant differences between groups (P > 0.07; n = 15 per group).
DISCUSSION
In this report we confirmed that risedronate, a nitrogen-containing bisphosphonate (N-BP), has a potent activity against the blood stages of P. falciparum in vitro and P. berghei in vivo. The IC50 established for parasite growth is in the range obtained for the same isolate (3D7) in previous studies (27, 35). We also showed that the inhibitory effect induced by risedronate can be partially reversed by the simultaneous addition of FPP or GGPP during P. falciparum culture treatment (Fig. 1). The restoration observed after the addition of GGPP is plausible, since Couto et al. (7) previously demonstrated that P. falciparum is able to convert GGPP into FPP. In contrast, when we added IPP to the cultures, the parasites could not recover, suggesting that the inhibition of FPPS is a potential target for risedronate, which could also act by inhibiting GGPPS. Luckman et al. (25) verified the same event of restoration after coincubating J774 macrophages with alendronate and FPP or GGPP, observing a partial prevention of apoptotic events.
Our results regarding the effect of risedronate on isoprenoid biosynthesis (Fig. 2) suggest the inhibition of FPPS. The RP-TLC profile for treated parasites shows that bands with Rf values equivalent to FOH and GGOH are decreased compared to the signal from untreated parasites, leading us to speculate that risedronate inhibits enzymes involved in FPP and/or GGPP synthesis. It is known that the major target of N-BPs, as risedronate, is FPPS (11); therefore, we assume the possible role of risedronate as a potent inhibitor of the isoprenoid pathway in P. falciparum by inhibiting FPPS and, consequently, blocking protein farnesylation and geranylgeranylation. In J774 macrophages, the drug inhibited protein prenylation, including Ras protein prenylation (25).
These results are supported by the fact that several phosphonate and bisphosphonate analogues of FPP have been demonstrated to inhibit farnesyltransferase (18). Since risedronate inhibits FPP and GGPP biosynthesis, it is expected, in the case of treated and [1-3H]FPP- and [1-3H]GGPP-labeled parasites, to exhibit an increase of intensity in the bands corresponding to isoprenylated proteins. However, the profile of farnesylated proteins was decreased in intensity compared to the control group. This result could be explained by the inhibition of FPP transfer to proteins. On the other hand, the profile of geranylgeranylated proteins was increased in intensity, probably because the transfer of GGPP to proteins was not altered by risedronate. Moreover, this result shows either that risedronate inhibits P. falciparum GGPPS or that GGPP is inhibited as a result of the FPP suppression. Singh et al. (35) showed recently that lipophilic bisphosphonates have activity against a recombinant GGPPS from Plasmodium vivax. Recently, Artz et al. (1) showed that risedronate inhibits recombinant GGPPS from P. vivax using enzymatic activity assays and crystallography approaches. In T. gondii, N-BP was shown to inhibit the bifunctional enzyme FPPS/GGPPS (23). Considering that the Ras and Rab proteins are farnesylated and geranylgeranylated, respectively, the inhibition of Ras and increase of Rab after risedronate incubation (Fig. 3C) confirm our hypothesis that in P. falciparum, risedronate inhibits FPPS and the transfer of the FPP group to proteins.
Risedronate is indicated for the treatment and prevention of various bone diseases, such as osteoporosis in postmenopausal women, glucocorticoid-induced osteoporosis, and severe diseases like Paget's disease of bone (PDB). This is a chronic disorder for which treatment with oral risedronate at a dose of 17.5 mg once daily for 8 weeks significantly decreases abnormal markers associated with PDB (43). Risedronate and other bisphosphonates possess antiprotozoal activity, specifically against Chagas' disease (2) and malaria. Recently, Singh et al. (35) studied the effect of lipophilic bisphosphonates (BPH-715) blocking the prenylation of proteins, resulting in a potent activity against P. berghei liver stages in vivo. Mice treated with BPH-715 showed complete protection, without the appearance of erythrocytic-stage parasites after 28 days of observation. However, this lipophilic bisphosphonate had only a mild inhibitory effect on erythrocytic stages of the parasite. Unlike the findings of Singh et al. (35), the bisphosphonate used in our experiments was effective in intraerythrocytic stages, where mice infected with P. berghei and treated with risedronate at 25 mg/kg/day for 7 days presented an 88.9% inhibition of parasitemia. It is noteworthy that in our in vivo studies, we determined the ID50 (dose causing 50% inhibition) of risedronate sodium, which was 17 mg/kg/day for 7 days. Even with the efficacy of risedronate in reducing parasitemia in erythrocytic stages, the survival of mice was not prolonged, suggesting that during treatment there was an inhibition of the development of Plasmodium berghei intraerythrocytic forms but no cure of infected animals. In a previous experiment, the animals that received risedronate over 7 days died unexpectedly on the ninth day postinfection, suggesting a possible toxic effect of the drug when associated with P. berghei infection. Curiously, when noninfected mice were treated for longer periods (14 days), risedronate did not show toxicity, since it was not lethal to the animals. Probably, the progressive asthenia caused by infection made the animals more susceptible and was a determining factor for the lethality despite the antiplasmodial activity of risedronate. For that reason, we decided to treat the infected mice for only 7 days and monitor them regarding their parasitemia and survival rates on the following days, thereby preventing the toxic effects of risedronate through extended periods of treatment. Our studies demonstrate that treatment with sodium risedronate exhibits significant in vivo antiplasmodial activity but also indicate a requirement for more investigation into the efficacy and safety of risedronate as therapy against malaria. Also, other ways of administration and other bisphosphonate-derived drugs should be evaluated.
In conclusion, our studies showed that risedronate interferes with farnesylated and geranylgernanylated proteins in P. falciparum as a consequence of the inhibition of the biosynthesis of FPP and GGPP. We also demonstrated that risedronate inhibits the transfer of FPP to parasite proteins. Additional experiments are required to assess the specificity of risedronate in the inhibition of protein farnesyltransferase in P. falciparum. These results suggest that the antiplasmodial activities of N-BPs may be exploited as potential antimalarial candidates.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from CNPq and FAPESP (Brazil). F.M.J. is the recipient of a postgraduate fellowship from CNPq. A.Y.S. and D.C.M. receive postgraduate fellowships from FAPESP.
We thank S. Wendel (Sírio Libanês Hospital, NESTA) for providing the erythrocytes and Gerhard Wunderlich for critical reading of the manuscript.
Footnotes
Supplemental material for this article may be found at http://aac.asm.org/.
Published ahead of print on 28 February 2011.
REFERENCES
- 1. Artz J. D., et al. 17 November 2010, posting date Molecular characterization of a novel geranylgeranyl pyrophosphate synthase from Plasmodium parasites. J. Biol. Chem. doi:10.1074/jbc.M109.027235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bouzahzah B., Jelicks L. A., Morris S. A., Weiss L. M., Tanowitz H. B. 2005. Risedronate in the treatment of murine Chagas' disease. Parasitol. Res. 96:184–187 [DOI] [PubMed] [Google Scholar]
- 3. Braun-Breton C., et al. 1986. In vivo time course of synthesis and processing of major schizont membrane polypeptides in Plasmodium falciparum. Mol. Biochem. Parasitol. 20:33–43 [DOI] [PubMed] [Google Scholar]
- 4. Chakrabarti D., et al. 1998. Protein prenyl transferase activities of Plasmodium falciparum. Mol. Biochem. Parasitol. 94:175–184 [DOI] [PubMed] [Google Scholar]
- 5. Chakrabarti D., et al. 2002. Protein farnesyltransferase and protein prenylation in Plasmodium falciparum. J. Biol. Chem. 277:42066–42073 [DOI] [PubMed] [Google Scholar]
- 6. Clarke S. 1992. Protein isoprenylation and methylation at carboxyl-terminal cysteine residues. Annu. Rev. Biochem. 61:355–386 [DOI] [PubMed] [Google Scholar]
- 7. Couto A. S., Kimura E. A., Peres V. J., Uhrig M. L., Katzin A. M. 1999. Active isoprenoid pathway in the intra-erythrocytic stages of Plasmodium falciparum: presence of dolichols of 11 and 12 isoprene units. Biochem. J. 341(Pt. 3):629–637 [PMC free article] [PubMed] [Google Scholar]
- 8. Coxon F. P., et al. 2000. Protein geranylgeranylation is required for osteoclast formation, function, and survival: inhibition by bisphosphonates and GGTI-298. J. Bone Miner. Res. 15:1467–1476 [DOI] [PubMed] [Google Scholar]
- 9. Desjardins R. E., Canfield C. J., Haynes J. D., Chulay J. D. 1979. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob. Agents Chemother. 16:710–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Docampo R., Moreno S. N. 2001. Bisphosphonates as chemotherapeutic agents against trypanosomatid and apicomplexan parasites. Curr. Drug Targets Infect. Disord. 1:51–61 [DOI] [PubMed] [Google Scholar]
- 11. Dunford J. E., et al. 2008. Structure-activity relationships among the nitrogen containing bisphosphonates in clinical use and other analogues: time-dependent inhibition of human farnesyl pyrophosphate synthase. J. Med. Chem. 51:2187–2195 [DOI] [PubMed] [Google Scholar]
- 12. Field H., Blench I., Croft S., Field M. C. 1996. Characterisation of protein isoprenylation in procyclic form Trypanosoma brucei. Mol. Biochem. Parasitol. 82:67–80 [DOI] [PubMed] [Google Scholar]
- 13. Frith J. C., Monkkonen J., Auriola S., Monkkonen H., Rogers M. J. 2001. The molecular mechanism of action of the antiresorptive and antiinflammatory drug clodronate: evidence for the formation in vivo of a metabolite that inhibits bone resorption and causes osteoclast and macrophage apoptosis. Arthritis Rheum. 44:2201–2210 [DOI] [PubMed] [Google Scholar]
- 14. Gessler M. C., Nkunya M. H., Mwasumbi L. B., Heinrich M., Tanner M. 1994. Screening Tanzanian medicinal plants for antimalarial activity. Acta Trop. 56:65–77 [DOI] [PubMed] [Google Scholar]
- 15. Ghosh S., et al. 2004. Effects of bisphosphonates on the growth of Entamoeba histolytica and Plasmodium species in vitro and in vivo. J. Med. Chem. 47:175–187 [DOI] [PubMed] [Google Scholar]
- 16. Gibbs J. B., et al. 1996. Farnesyltransferase inhibitors and anti-Ras therapy. Breast Cancer Res. Treat. 38:75–83 [DOI] [PubMed] [Google Scholar]
- 17. Goldstein J. L., Brown M. S. 1990. Regulation of the mevalonate pathway. Nature 343:425–430 [DOI] [PubMed] [Google Scholar]
- 18. Holstein S. A., Cermak D. M., Wiemer D. F., Lewis K., Hohl R. J. 1998. Phosphonate and bisphosphonate analogues of farnesyl pyrophosphate as potential inhibitors of farnesyl protein transferase. Bioorg. Med. Chem. 6:687–694 [DOI] [PubMed] [Google Scholar]
- 19. Ibrahim M., Azzouz N., Gerold P., Schwarz R. T. 2001. Identification and characterisation of Toxoplasma gondii protein farnesyltransferase. Int. J. Parasitol. 31:1489–1497 [DOI] [PubMed] [Google Scholar]
- 20. Kessler S. W. 1975. Rapid isolation of antigens from cells with a staphylococcal protein A-antibody adsorbent: parameters of the interaction of antibody-antigen complexes with protein A. J. Immunol. 115:1617–1624 [PubMed] [Google Scholar]
- 21. Kimura E. A., Couto A. S., Peres V. J., Casal O. L., Katzin A. M. 1996. N-linked glycoproteins are related to schizogony of the intraerythrocytic stage in Plasmodium falciparum. J. Biol. Chem. 271:14452–14461 [DOI] [PubMed] [Google Scholar]
- 22. Laemmli U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 23. Ling Y., Li Z. H., Miranda K., Oldfield E., Moreno S. N. 2007. The farnesyl-diphosphate/geranylgeranyl-diphosphate synthase of Toxoplasma gondii is a bifunctional enzyme and a molecular target of bisphosphonates. J. Biol. Chem. 282:30804–30816 [DOI] [PubMed] [Google Scholar]
- 24. Ling Y., et al. 2005. Bisphosphonate inhibitors of Toxoplasma gondii growth: in vitro, QSAR, and in vivo investigations. J. Med. Chem. 48:3130–3140 [DOI] [PubMed] [Google Scholar]
- 25. Luckman S. P., et al. 1998. Nitrogen-containing bisphosphonates inhibit the mevalonate pathway and prevent post-translational prenylation of GTP-binding proteins, including Ras. J. Bone Miner. Res. 13:581–589 [DOI] [PubMed] [Google Scholar]
- 26. Lujan H. D., Mowatt M. R., Chen G. Z., Nash T. E. 1995. Isoprenylation of proteins in the protozoan Giardia lamblia. Mol. Biochem. Parasitol. 72:121–127 [DOI] [PubMed] [Google Scholar]
- 27. Martin M. B., et al. 2001. Bisphosphonates inhibit the growth of Trypanosoma brucei, Trypanosoma cruzi, Leishmania donovani, Toxoplasma gondii, and Plasmodium falciparum: a potential route to chemotherapy. J. Med. Chem. 44:909–916 [DOI] [PubMed] [Google Scholar]
- 28. Moura I. C., et al. 2001. Limonene arrests parasite development and inhibits isoprenylation of proteins in Plasmodium falciparum. Antimicrob. Agents Chemother. 45:2553–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rodan G. A. 1998. Mechanisms of action of bisphosphonates. Annu. Rev. Pharmacol. Toxicol. 38:375–388 [DOI] [PubMed] [Google Scholar]
- 30. Rodrigues Goulart H., et al. 2004. Terpenes arrest parasite development and inhibit biosynthesis of isoprenoids in Plasmodium falciparum. Antimicrob. Agents Chemother. 48:2502–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rodriguez N., et al. 2002. Radical cure of experimental cutaneous leishmaniasis by the bisphosphonate pamidronate. J. Infect. Dis. 186:138–140 [DOI] [PubMed] [Google Scholar]
- 32. Rohmer M., Grosdemange-Billiard C., Seemann M., Tritsch D. 2004. Isoprenoid biosynthesis as a novel target for antibacterial and antiparasitic drugs. Curr. Opin. Invest. Drugs 5:154–162 [PubMed] [Google Scholar]
- 33. Sanchez C. P., Dave A., Stein W. D., Lanzer M. 2010. Transporters as mediators of drug resistance in Plasmodium falciparum. Int. J. Parasitol. 40:1109–1118 [DOI] [PubMed] [Google Scholar]
- 34. Shen P. S., Sanford J. C., Samuelson J. 1996. Entamoeba histolytica: isoprenylation of p21ras and p21rap in vitro. Exp. Parasitol. 82:65–68 [DOI] [PubMed] [Google Scholar]
- 35. Singh A. P., et al. 2010. Lipophilic bisphosphonates are potent inhibitors of Plasmodium liver-stage growth. Antimicrob. Agents Chemother. 54:2987–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stresing V., Daubine F., Benzaid I., Monkkonen H., Clezardin P. 2007. Bisphosphonates in cancer therapy. Cancer Lett. 257:16–35 [DOI] [PubMed] [Google Scholar]
- 37. Trager W., Jensen J. B. 1976. Human malaria parasites in continuous culture. Science 193:673–675 [DOI] [PubMed] [Google Scholar]
- 38. Urbina J. A., et al. 1999. Trypanosoma cruzi contains major pyrophosphate stores, and its growth in vitro and in vivo is blocked by pyrophosphate analogs. J. Biol. Chem. 274:33609–33615 [DOI] [PubMed] [Google Scholar]
- 39. Wang K., Ohnuma S. 1999. Chain-length determination mechanism of isoprenyl diphosphate synthases and implications for molecular evolution. Trends Biochem. Sci. 24:445–451 [DOI] [PubMed] [Google Scholar]
- 40. WHO. 2008. World malaria report, vol. WHO/HTM/GMP/2008.1. World Health Organization, Geneva, Switzerland [Google Scholar]
- 41. Wiesner J., Jomaa H. 2007. Isoprenoid biosynthesis of the apicoplast as drug target. Curr. Drug Targets 8:3–13 [DOI] [PubMed] [Google Scholar]
- 42. Yardley V., et al. 2002. In vivo activities of farnesyl pyrophosphate synthase inhibitors against Leishmania donovani and Toxoplasma gondii. Antimicrob. Agents Chemother. 46:929–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yoh K., Takata S., Yoshimura N., Hashimoto J. 2010. Efficacy, tolerability, and safety of risedronate in Japanese patients with Paget's disease of bone. J. Bone Miner. Metab. 28:468–476 [DOI] [PubMed] [Google Scholar]
- 44. Yokoyama K., et al. 1998. The effects of protein farnesyltransferase inhibitors on trypanosomatids: inhibition of protein farnesylation and cell growth. Mol. Biochem. Parasitol. 94:87–97 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.