

Abstract
Increased microbial drug resistance has generated a global requirement for new anti-infective agents. As part of an effort to develop new, low-molecular-mass peptide antibiotics, we used a rationale-based minimalist approach to design short, nonhemolytic, potent, and broad-spectrum antibiotic peptides with increased serum stability. These peptides were designed to attain an amphipathic structure in helical conformations. VS1 was used as the lead compound, and its properties were compared with three series of derivates obtained by (i) N-terminal amino acid addition, (ii) systematic Trp substitution, and (iii) peptide dendrimerization. The Trp substitution approach underlined the optimized sequence of VS2 in terms of potency, faster membrane permeation, and cost-effectiveness. VS2 (a variant of VS1 with two Trp substitutions) was found to exhibit good antimicrobial activity against both the Gram-negative Escherichia coli and the Gram-positive bacterium Staphylococcus aureus. It was also found to have noncytolytic activity and the ability to permeate and depolarize the bacterial membrane. Lysis of the bacterial cell wall and inner membrane by the peptide was confirmed by transmission electron microscopy. A combination of small size, the presence of unnatural amino acids, high antimicrobial activity, insignificant hemolysis, and proteolytic resistance provides fundamental information for the de novo design of an antimicrobial peptide useful for the management of infectious disease.
INTRODUCTION
The accelerated emergence of pathogenic bacteria resistant to conventional antibiotics is a major threat today (1). Therefore, the development of a new class of broad-spectrum antibiotics is an urgent need. Cationic antimicrobial peptides (AMPs) could be one of the best possible alternatives (31). AMPs are an important component of the natural defenses of most living organisms against invading pathogens (12). Naturally occurring AMPs are generally 12 to 50 amino acids in length and are folded into several structural groups, including helix, sheet, extended, and looped structures (43). Although they show marked variability in length, amino acid composition, and structure, a majority of them share two common features and functionally important characteristics, namely, a net positive charge that facilitates interaction with negatively charged microbial surfaces and the ability to form an amphipathic secondary structure that permits incorporation into microbial membranes (38). Although the exact mechanism of action of AMPs is still not completely understood, it has been well established that AMPs interact with the cell membrane of the susceptible microorganism, where either their accumulation in the membrane causes increased permeability and loss of barrier function or they enter the membrane to access cytoplasmic targets (4). Development of resistance against peptides that act on the microbial membrane is unlikely, as it would require substantial alterations in the lipid composition of the cell membranes of the microorganisms (28).
However, most natural AMPs are large, have low potency, and show toxicity to host cells (21). Moreover, due to their peptidic nature, they suffer from poor bioavailability and poor proteolytic stability (25). These features have significantly hampered their pharmaceutical development as therapeutic agents. Short designer AMPs with increased half-lives offer excellent templates for future antibiotic drug design.
Different approaches are being followed in efforts to increase the effectiveness of AMPs, including alteration of sequences, inclusion of unnatural or d-amino acids or beta-amino acids, cyclization of peptides and peptoid mimics, and synthesis of multivalent constructs of short peptides (9, 18, 27, 33, 37, 40). Short designer AMPs that are less likely to induce resistance and that minimize damage to host cells or tissues appear to be the most promising candidates.
In the present work, we focused on the use of a nonproteinogenic amino acid, α,β-dihydrophenylalanine (ΔPhe), in designing relatively short antimicrobial peptides. The presence of more than one ΔPhe in peptides has been shown to constrain the peptide in a 310 or α-helical conformation, along with providing enhanced resistance to enzymatic degradation compared to their phenylalanine-containing counterparts (23, 29). We used a de novo-designed prototype undecapeptide peptide (VS1) incorporating ΔPhe as a lead in an optimization strategy to design three sets of peptides with the same basic motif, where ΔPhe is placed at two residue spaces. In order to identify the most promising, cost-effective antibiotic peptide as a drug candidate, the designed peptides were compared in terms of (i) spectrum of activity, (ii) specificity, (iii) microbicidal properties, and (iv) membrane permeabilization.
MATERIALS AND METHODS
Materials.
Amino acid derivatives and resin for peptide synthesis were obtained from Nova Biochem; diisopropylcarbodiimide (DIPCDI), piperidine, dimethyl formamide (DMF), dichloromethane (DCM), hydroxybenzotriazole (HOBt), isobutylchloroformate (IBCF), trifluoroacetic acid (TFA), triisopropyl silane (TIS), dl-threo-β-phenylserine, sodium hydroxide, citric acid, 3-[4,5-dimethylthiazol-2-yl]-2,5diphenyltetrazolium bromide (MTT), DiSC3-5 (3,3′-dipropylthiadicarbocyanine) iodide, dimethyl sulfoxide (DMSO), bovine serum albumin (BSA), and N-methyl morpholine (NMM) were from Sigma-Aldrich (St. Louis, MO); sodium chloride, acetic anhydride, and tetrahydrofuran (THF) were from Qualigens (Mumbai, India); ethyl acetate, diethyl ether, sodium acetate, and sodium sulfate were from Merck (Mumbai, India); silica gel thin-layer chromatography (TLC) plates (60F-254) were from Merck (Germany); acetic acid was from SD Fine Chem Ltd. (Mumbai, India); acetonitrile was from Burdick and Jackson (Muskegon, MI). and RPMI 1640 and fetal bovine serum (FBS) were from Invitrogen (Carlsbad, CA).
Preparation of Fmoc-X-dl-threo-β-phenylserine.
Fmoc-X-dl-threo-β-phenylserine (where Fmoc is 9-fluorenylmethoxy carbonyl and X is Lys [Boc], Trp [Boc], or Ala) was synthesized by a method of salt coupling using mixed anhydride. Fmoc amino acid (15 mmol) dissolved in 15 ml of sodium-refluxed and distilled THF was activated at −15°C for 10 min with IBCF and NMM (15 mmol each). A solution of 15 mmol of dl-threo-β-phenylserine made in 1 equivalent of NaOH (15 ml) was added to the above-mentioned mixed anhydride, and the reaction mixture was stirred at room temperature overnight. Following evaporation of THF, citric acid was added to the aqueous solution to reach a solution of pH 2.0. The precipitate obtained was dissolved in 100 ml ethyl acetate and transferred to a separating funnel. Following the removal of the lower aqueous layer, the ethyl acetate layer was washed extensively with water to remove the citric acid. The complete removal of citric acid was confirmed by measuring the pH. The ethyl acetate layer was further washed with brine and allowed to pass through a bed of anhydrous sodium sulfate. Evaporation of ethyl acetate on a rotary evaporator resulted in solid dipeptide acids.
Preparation of Fmoc-X-ΔPhe azalactone.
Fmoc-X-dl-threo-β-phenylserine was mixed with recrystallized anhydrous sodium acetate (obtained by fusing the salt and allowing it to cool in a desiccator) in freshly distilled acetic anhydride and stirred overnight. The thick slurry obtained was mixed with ice and stirred at 8 to 10°C. Following trituration, the yellow dipeptide azalactone was filtered on a sinter funnel and dried to constant weight. The authenticity and purity of the azalactones were assessed by TLC, mass spectroscopy, and UV-visible spectroscopy.
Peptide synthesis.
Peptides were synthesized as C-terminal amides using standard Fmoc chemistry on rink amide MBHA (4-methylbenzhydrylamine hydrochloride salt) resin in the manual mode, with DIPCDI and HOBt as coupling agents. Fmoc-Lys (Fmoc)-OH was used to make a branching core for the synthesis of the lysine-branched dimer VSD1. Piperidine treatment of the lysine derivative immobilized on the resin gave rise to two amino groups (α and ε), allowing the synthesis of two identical peptide chains, as shown in Table 1. The synthesis of the dendrimer VSD1 was accomplished on a K-K2 core generated by coupling of Fmoc-Lys (Fmoc)-OH to the two amino groups of the lysine resin synthesized as described above. The side chain protection used was Boc (Lys or Trp). Couplings were carried out using DMF at a 4-fold molar excess at a final concentration of ∼500 mM. Removal of Fmoc was carried out using 20% piperidine in DMF. Both the coupling of amino acids and the Fmoc deprotection were monitored by the Kaiser test (16). ΔPhe was introduced into peptides as an Fmoc-X-ΔPhe azalactone (where X is Lys [Boc], Trp [Boc], or Ala) dipeptide block (23), which was allowed to couple overnight in DMF. At the completion of assembly of the peptides, following Fmoc removal, the amino termini were acetylated using 20% acetic anhydride in DCM. After acetylation of the peptides, the resin was washed extensively with DMF, DCM, and methanol and dried in a desiccator under vacuum.
Table 1.
Amino acid sequences of designed peptides
| Peptide | Amino acid sequence | Length (residues) | Charge | ΔPhe | Trp | MW (calculated) | MW (observed) |
|---|---|---|---|---|---|---|---|
| VS1 | Ac-K-A-ΔF-W-K-ΔF-V-K-ΔF-V-K-NH | 11 | +4 | 3 | 1 | 1,466.8 | 1,468 |
| Series I (analogs with variable length) | |||||||
| VSL1 | Ac-ΔF-K-A-ΔF-W-K-ΔF-V-K-ΔF-V-K-NH2 | 12 | +4 | 4 | 1 | 1,607.4 | 1,608 |
| VSL2 | Ac-A-ΔF -K-A-ΔF-W-K-ΔF-V-K-ΔF-V-K-NH2 | 13 | +4 | 4 | 1 | 1,679.8 | 1,679 |
| VSL3 | Ac-K-A-ΔF-K-A-ΔF-W-K-ΔF-V-K-ΔF-V-K-NH2 | 14 | +5 | 4 | 1 | 1,807.3 | 1,808.2 |
| Series II (analogs with different Trp contents) | |||||||
| VS2 | Ac-K-W-ΔF-W-K-ΔF-V-K-ΔF-V-K-NH2 | 11 | +4 | 3 | 2 | 1,577.6 | 1,575.1 |
| VS3 | Ac-K-W-ΔF-W-K-ΔF-W-K-ΔF-V-K-NH2 | 11 | +4 | 3 | 3 | 1,665.7 | 1,667 |
| VS4 | Ac-K-W-ΔF-W-K-ΔF-W-K-ΔF-W-K-NH2 | 11 | +4 | 3 | 4 | 1,752.4 | 1,754.7 |
| Series III (branched dimeric analog) | |||||||
| VSD1 | Ac-K-A-ΔF-W-K-ΔF-V-K-ΔF-V-K-NH\K | 23 | +8 | 6 | 2 | 3,001 | 3,000.6 |
| Ac-K-A-ΔF-W-K-ΔF-V-K-ΔF-V-K-NH/ |
Synthesis of FITC-labeled peptides.
To the free amino terminus of peptide VS2 on resins, Fmoc-ε-aminohexanoic (Ahx) acid-OH was coupled using HOBt and DIPCDI in DMF. Fmoc was removed by treatment with 20% piperidine-DMF. The resin was washed with DMF and equilibrated in pyridine-DMF-DCM (12:7:5 [vol/vol/vol]). A 1.1 equivalent of fluorescein isothiocyanate (FITC) in pyridine-DMF-DCM (12:7:5 [vol/vol/vol]) was added to the resin and allowed to couple overnight. An orange color on the resin and a negative Kaiser test indicated coupling.
Cleavage of the peptides from resin.
Peptides were cleaved by stirring the resin in a cleavage mixture (95% TFA, 2.5% water, and 2.5% TIS) for 2 h at room temperature. The suspension was filtered using a sinter funnel, the TFA was rotary evaporated, and the peptide was precipitated by adding cold dry ether. The ether was filtered through a sinter funnel, and the peptide on the funnel was dissolved in 10% acetic acid and lyophilized.
Peptide purification and mass spectrometry.
Crude peptides were purified by reverse-phase high-performance liquid chromatography (RPHPLC) on a Deltapac C18 column (15-μm inside diameter [i.d.], 300 by 19 mm) using an acetonitrile-water linear gradient of 5 to 65% acetonitrile (0.1% TFA)-water (0.1% TFA) with a flow rate of 5 ml/min for 60 min, with detection at 214 and 280 nm. The purified peptides were reinjected into an analytical reversed-phase C18 column (Phenomenex; C18; 5-μm i.d; 250 by 4.6 mm) using an acetonitrile-water linear gradient of 5 to 65% acetonitrile (0.1% TFA)-water (0.1% TFA) at a flow rate of 1 ml/min over 60 min and were found to be 98% pure. The identity of the purified (98%) peptides was confirmed by electrospray ionization mass spectrometry at the International Centre for Genetic Engineering and Biotechnology (ICGEB), New Delhi, India.
Solubility measurements.
Water was added to the purified peptide powders to attain complete dissolution, and the concentration of the peptide in the spun supernatant (13,000 rpm; 10 min) was determined by measurement of the absorbance at 280 nm. The extinction coefficient (ε) for α,β-didehydrophenylalanine (ΔPhe) is 19,000 M−1 cm−1 (23) and for tryptophan is 5,050 M−1 cm−1.
Antibiotic susceptibility testing.
MICs against the Gram-negative bacterium Escherichia coli ML35p and the Gram-positive bacterium Staphylococcus aureus (ATCC 700699) were determined according to a modified MIC method for cationic antimicrobial peptides (41). Bacterial cells grown overnight were diluted in Mueller-Hinton (MH) broth to a density of 105 CFU/ml. One hundred microliters of this culture was aliquoted into the wells of a 96-well flat-bottom microtiter plate (Costar), and 11 μl of stocks of each peptide (in 0.2% BSA and 0.01% acetic acid) was added. This mixture was incubated at 37°C in a rotary shaker incubator (Kuhner, Switzerland) set at 200 rpm. After 18 h of incubation, the optical density at 600 nm (OD600) was measured using a microtiter plate reader (Versa Max tunable; Molecular Devices, Sunnyvale, CA). The MIC is defined as the lowest concentration of a drug that inhibits the measurable growth of an organism after overnight incubation. Peptide concentrations were determined spectrophotometrically at 280 nm (ε280, 19,000 M−1 cm−1 for ΔPhe and 5,050 M−1 cm−1 for tryptophan). Each experiment was done in triplicate and was repeated at least twice.
Hemolytic-activity testing.
Human blood in 10% citrate phosphate dextrose was obtained from the Rotary Blood Bank, New Delhi, India. Red blood cells (RBCs) were harvested by spinning (1,000 × g; 5 min; room temperature). They were washed three to five times with phosphate-buffered saline (PBS). The packed cell volume obtained was used to make a 0.8% (vol/vol) suspension in PBS; 100 μl of this RBC suspension was transferred to each well of a 96-well microtiter plate and mixed with 100 μl of peptide solution at twice the desired concentration. The microtiter plate was incubated (37°C; 60 min) and centrifuged (1,000 × g; 5 min; room temperature). The supernatant (100 μl) was transferred to new wells, and the OD414 was measured with a microtiter plate reader (Versa Max tunable; Molecular Devices, Sunnyvale, CA) to monitor RBC lysis. Cells incubated with PBS alone served as the negative control, and RBCs lysed using 0.1% Triton X-100 were used to measure 100% lysis (positive control).
Mammalian-cell cytotoxicity.
The cytotoxicity of the antibiotic peptides was determined using an MTT assay against HeLa cells. Briefly, cells (5 × 104/well) were cultured at 37°C overnight in RPMI 1640 containing 10% fetal bovine serum in 96-well microtiter plates. The next day, peptides (prepared in RPMI 1640) at 1× and 5× MIC were added to the cells and incubated for 18 h at 37°C. Ten percent DMSO was taken as the positive control, and untreated cells served as the negative control; 20 μl of MTT solution (5 mg/ml) in PBS was added, and the cells were incubated (37°C; 3 to 4 h). Supernatant (120 μl) was removed, DMSO (100 μl) was added, and the resulting suspension was mixed to dissolve the formazan crystals formed by MTT reduction. The ratio of the OD570 for treated cells to the OD570 for untreated cells was used to calculate percent viability.
Proteolytic stability of peptides to trypsin and chymotrypsin.
The proteolytic stability of the peptide VS1 was compared with that of its phenylalanine analog (Ac-KAFWKFVKFVK-NH2) using reversed-phase HPLC. Peptide VS1 and trypsin/chymotrypsin, taken at a ratio of 100:0.5 (mol/mol) in 0.1 M ammonium bicarbonate, 0.1 mM CaCl2, pH 8.3, were incubated in a rotary shaker incubator (37°C; 200 rpm; 30 min). An aliquot was injected into a reverse-phase analytical C18 column (Phenomenex; C18; 5-μm i.d.; 250 by 4.6 mm), using an acetonitrile-water linear gradient of 5 to 65% acetonitrile (0.1% TFA)-water (0.1% TFA) at a flow rate of 1 ml/min over 65 min. An identically treated control peptide (phenylalanine analog) sample was also injected into a reverse-phase analytical C18 column using an acetonitrile-water linear gradient of 5 to 65% acetonitrile (0.1% TFA)-water (0.1% TFA) at a flow rate of 1 ml/min over 65 min. For the variable time-dependent assay, peptides were incubated with enzyme for 0, 1.5, and 2.5 h, respectively.
Outer membrane permeabilization (NPN) assay.
The membrane permeabilization activity of the peptides was determined by the 1-N-phenylnaphthylamine (NPN) assay of Loh et al. (20). In overview, an overnight culture of E. coli ML35p was diluted in MH broth and grown to an OD600 of 0.5 to 0.6. The cells were harvested, washed, and resuspended in the same volume of buffer (5 mM HEPES [pH 7.2], 5 mM glucose). For the NPN assay, 2 ml of cells and 10 μM NPN were mixed, and the fluorescence was measured with a fluorescence spectrophotometer (excitation wavelength, 350 nm; emission wavelength, 420 nm). The increase in fluorescence due to partitioning of NPN into the outer membrane (OM) was measured by the addition of various concentrations of peptide (0.25 μM to 2 μM). Polymyxin B (PMB) was taken as a positive control. All experiments were performed three times, and the trends that were observed were reproducible.
Inner membrane permeabilization assay (PI and Syto9 uptake-based assay).
Overnight-grown E. coli ML 35p cells were subcultured to an OD600 of 0.35. The cells were harvested (4,000 rpm; 10 min; 4°C), washed, and resuspended in buffer (5 mM glucose in 10 mM sodium phosphate buffer, pH 7.5) to 108 CFU/ml. Fifteen microliters of E. coli suspension (108 CFU/ml) was incubated (37°C; 200 rpm) in 135 μl of buffer (5 mM glucose, 5 mM HEPES, pH 7.2) containing peptides at 1× MIC for 90 min. The samples were then incubated with propidium iodide (PI) (2.7 μM) and Syto9 (6 μM) for 15 min. A smear was made, heat fixed, and visualized under a Nikon fluorescence microscope. Cells without peptide treatment served as a control for the experiment.
FACS-based analysis for PI uptake.
Overnight-grown cells were subcultured to an OD600 of 0.35. The cells were harvested (4,000 rpm; 10 min; 4°C), washed, and resuspended in buffer (5 mM glucose, 5 mM HEPES, pH 7.2) to 10 CFU/ml. Twenty-five microliters of E. coli suspension (108 CFU/ml) was incubated in 480 μl of buffer containing peptide and 5 μl of PI (1 mg/ml) for 15 min at room temperature and then subjected to fluorescence-activated cell sorter (FACS) analysis; 25 μl of cells suspended in 470 μl of buffer with and without PI served as controls.
Membrane depolarization assay (DiSC3-5) assay.
The depolarization of the cytoplasmic membrane of E. coli ML35p by the peptides was determined by a modified version of the method of Wu et al. (42) using the membrane potential-sensitive cyanine dye DiSC3-5. In this experiment, E. coli cells, in mid-exponential phase (OD600, 0.35), were collected by centrifugation, washed once with buffer (5 mM HEPES, pH 7.2, 5 mM glucose), and resuspended to an OD600 of 0.05. The cells were incubated with 1 μM DiSC3-5 for 2 h for maximal uptake of the dye, after which 100 mM KCl was added to equilibrate the cytoplasmic and external potassium ion concentrations. The cells were mixed with the desired concentration of peptide, and the fluorescence was monitored at an excitation wavelength of 622 nm and an emission wavelength of 655 nm. Dye released with the addition of 1% DMSO was monitored as a control.
Cellular uptake of peptide studied by confocal microscopy.
E. coli ML35p cells grown overnight were subcultured to an OD600 of 0.35 (1 × 108 CFU/ml). The cells were harvested by centrifugation (4,000 rpm for 10 min), washed, and resuspended in 5 mM glucose-10 mM sodium phosphate buffer (pH 7.5) to yield 108 CFU/ml; 100 μl of E. coli suspension (108 CFU/ml) was mixed with 5 μl of FITC-labeled peptide VS2 aqueous stock solution to yield a final peptide concentration of 5 μM (1× MIC) and incubated (37°C; 200 rpm) for 30 min in a Kuhner rotary shaker incubator. The cells were harvested by centrifugation (4,000 rpm for 10 min) and washed twice with 5 mM glucose in 10 mM sodium phosphate buffer (pH 7.5). A smear was made on a poly-l-lysine-coated slide. For confocal microscopy, confocal laser scanning (Radiance 2100; Bio-Rad) under a Nikon microscope (objective plane Apo 60×/1.4 oil) was used to observe the slide. The excitation wavelength for FITC was 494 nm (argon laser), and fluorescence was detected through an HQ 515/30 emission filter (high-quality band-pass). Image processing was conducted with Lazer Sharp (Bio-Rad), and Photoshop 6.0 (Adobe Systems, San Jose, CA) was used for the final image assembly.
Examination of bacterial-membrane damage by electron microscopy. (i) Scanning electron microscopy (SEM).
Overnight-grown cells were subcultured to an OD600 of 0.35. The cells were harvested (4,000 rpm; 10 min; 4°C), washed, and resuspended in HEPES buffer, pH 7.2, to obtain 106 CFU/ml. Fifteen microliters of E. coli suspension (106 CFU/ml) was incubated for 30 min and 60 min in 135 μl of 5 mM HEPES buffer, pH 7.2, containing the peptide at its MIC (5 μM). The cells were spun down (4,000 rpm; 4°C; 10 min) and washed three times in 0.1 M phosphate buffer, pH 7.4. The cells were fixed in 2.5% glutaraldehyde buffered with 0.1 M phosphate buffer, pH 7.4, at 4°C for 2 h. They were further washed in 0.1 M phosphate buffer, postfixed with 1% OSO4, and dehydrated with graded ethanol. The sample was dried with hexamethyldisilizane (HMDS) and coated with gold (15 nm). Observations were made on a Zeiss EV040 scanning microscope at the Jawaharlal Nehru University, New Delhi, India.
(ii) Transmission electron microscopy (TEM).
A sample containing E. coli (1 × 106 CFU/ml) in Mueller-Hinton medium was incubated for 1 h with peptide (VS2). The peptide was taken at its MIC value (5 μM). After being centrifuged at 3,000 × g for 10 min, the pellets were washed with 0.1 M phosphate buffer and resuspended in the same buffer. E. coli cells were fixed with 2% glutaraldehyde in 0.1 M phosphate buffer for 1 h at room temperature (20°C). The cells were washed with 0.1 M phosphate buffer (pH 7.2) and postfixed with 1% OSO4 in 0.1 M phosphate buffer for 1 h at 4°C. For ultrastructure study, samples were dehydrated with graded acetone, cleared with toluene, infiltrated with a toluene and araldite mixture at room temperature and then finally in pure araldite at 50°C, and embedded in an Eppendorff tube (1.5 ml) with a pure araldite mixture at 60°. Ultrathin-section cutting was done with an ultramicrotome (Ultramicotome Lecia EM UC6). Sections were taken on a 3.05-mm-diameter 200-mesh copper grid and stained with uranyl acetate and lead acetate. The grids were examined under a transmission electron microscope (JEOL 2100F).
RESULTS
Rationale-based design of prototype peptide antibiotic.
Ab initio and de novo designing resulted in a short, cationic, amphipathic undecapeptide, VS1 (Ac-K-A-ΔF-W-K-ΔF-V-K-ΔF-V-K-CONH2), where Ac is an acetyl group. In order to allow initial electrostatic interaction with the negatively charged bacterial membrane, VS1 harbored four lysine residues on its hydrophilic face. An excessive charge has been shown to have a deleterious effect on activity (because it prevents structuring), and the optimal charge for maximal antimicrobial activity has been shown to be +4 (10). Our idea to keep the +4 charge was derived from such studies. Since, valine is the amino acid most commonly found in natural, as well as synthetic, antimicrobial peptides, two valine residues were incorporated on the hydrophobic face to maintain amphipathicity (3). A single alanine at the N-terminal hydrophobic face was incorporated to reduce peptide hydrophobicity while maintaining its helicity. Abiotic residues (ΔPhe) were placed 2 residues apart. In peptides, such placements of ΔPhe have been shown to induce a helical conformation (22). Taking a cue from the membrane-active properties of Trp, a single Trp was also incorporated in the prototype peptide VS1. The helical-wheel representation in Fig. 1A shows the segregation of polar and apolar faces of a 310 helix.
Fig. 1.

(A) De novo design of antimicrobial peptides: prototypic, amphipathic, cationic undecapeptide sequence in 310 helical-wheel configuration. (B) Chemical structure of ΔPhe.
Systematic modifications in the prototype peptide sequence.
Using the basic template of VS1 (11 residues; +4), systematic variations in the sequence resulted in three sets of peptides. The first set included three peptides, VSL1 (12 residues; +4), VSL2 (13 residues; +4), and VSL3 (14 residues; +5). These peptides were identical to VS1 up to 11 residues from the C terminus and differed only in their N-terminal extensions. The second set of peptides were Trp analogs of VS1: VS2 (two Trp), VS3 (three Trp), and VS4 (four Trp). They were identical to VS1 in length, charge, and number of ΔPhe residues and differed only in their Trp contents. The third approach resulted in a single peptide dimer of VS1; a dendrimer, VSD1, consisted of two VS1 peptide sequences on a core lysine residue. Thus, the branched dimer had a net charge of +8 (+4 per chain). The physicochemical properties, such as length, charge, and number of ΔPhe and Trp residues, of the designed peptides are provided in Table 1.
Effects of increasing length, Trp substitution, and dendrimerization of VS1 on antibiotic activity and cell selectivity. (i) Antibacterial activity.
The antibacterial activities of VS1 and its analogs against E. coli ML35p and S. aureus are shown in Table 2. The prototype peptide VS1 showed moderate potency (MIC for E. coli, 25 μM), a narrow spectrum of activity (MIC for S. aureus, >50 μM), and no hemolysis of RBCs or toxicity to HeLa cells. An increase in the chain length of VS1 resulted in a 5-fold increase in potency against E. coli (MIC, 5 μM for VSL1, VSL2, and VSL3); however, there was no augmentation in activity against the Gram-positive bacterium S. aureus. Moreover, in VSL3, even the combined effect of additional positive charge (+5) and increased length of the peptide was unable to broaden the spectrum of antibiotic activity. However, in the second series of peptides, sequential inclusion of Trp residues in VS1 resulted in substantial enhancement of the antimicrobial activity of the peptides. Replacement of Ala by Trp in VS1 led to VS2, which exhibited a 5-fold increase in activity against E. coli (MIC, 5 μM). This replacement also resulted in moderate activity against S. aureus (MIC, 50 μM). VS3, where Ala and Val in the VS1 sequence were replaced with two Trp residues, was equipotent with VS2 against E. coli but showed enhanced activity against S. aureus (MIC, 25 μM). VS4 (four Trp substitutions in VS1) was also equipotent with VS2 and VS3 against the Gram-negative E. coli (MIC, 5 μM) but showed further enhancement in activity against the Gram-positive S. aureus (MIC, 10 μM). The dendrimer VSD1 showed excellent antimicrobial activity against both E. coli (MIC, 1 μM) and S. aureus (MIC, 5 μM).
Table 2.
MICs of peptide analogs against Gram-negative (E. coli ML35p) and Gram-positive (S. aureus 700699) bacteria
| Peptidea | MIC (μM)b |
|
|---|---|---|
| E. coli ML35p | S. aureus (ATCC 700699) | |
| VS1 | 25 | >50 |
| Series I | ||
| VSL1 | 5 | 50 |
| VSL2 | 5 | >50 |
| VSL3 | 5 | >50 |
| Series II | ||
| VS2 | 5 | 50 |
| VS3 | 5 | 25 |
| VS4 | 5 | 10 |
| Series III | ||
| VSD1 | 1 | 5 |
| Rifampin | 5 | 1 |
| Kanamycin | 4 | 7.5 |
Rifampin and kanamycin were taken as reference antibiotics.
MICs are shown as the geometric means of three sets of determinations.
(ii) Cell selectivity (hemolysis and toxicity to HeLa cells).
None of the designed peptides were hemolytic or cytotoxic at their MICs, except VS3 (negligible hemolysis at 5 μM, the MIC for E. coli), VS4 (∼60% hemolysis at 5 μM, the MIC for E. coli), and VSD1 (∼10% at 1 μM, the MIC for E. coli). At these concentrations, none of the peptides showed any toxicity to HeLa cells. However, we observed that at 5× MIC values, which are much higher than physiologically relevant concentrations, the peptides VSL2, VSL3, VS2, and VS3 showed some degree of hemolysis and cytotoxicity. VSL2 was ∼15% hemolytic and 50% toxic to HeLa cells, whereas VSL3 was ∼13% hemolytic and 55% toxic at 5× MIC values. VS2 showed 12% hemolysis and 30% cytotoxicity, and VS3 showed 19% hemolysis and 40% toxicity to HeLa cells at 5× MIC values. VSL1 was exceptional; it was nonhemolytic and cytotoxic at the MIC but showed 75% hemolysis and 60% cytotoxicity at 5× MIC values (Fig. 2A and B). These results indicated that out of all the peptides, VS2 provided the right balance between antimicrobial activity and hemolytic activity and was chosen for further studies.
Fig. 2.
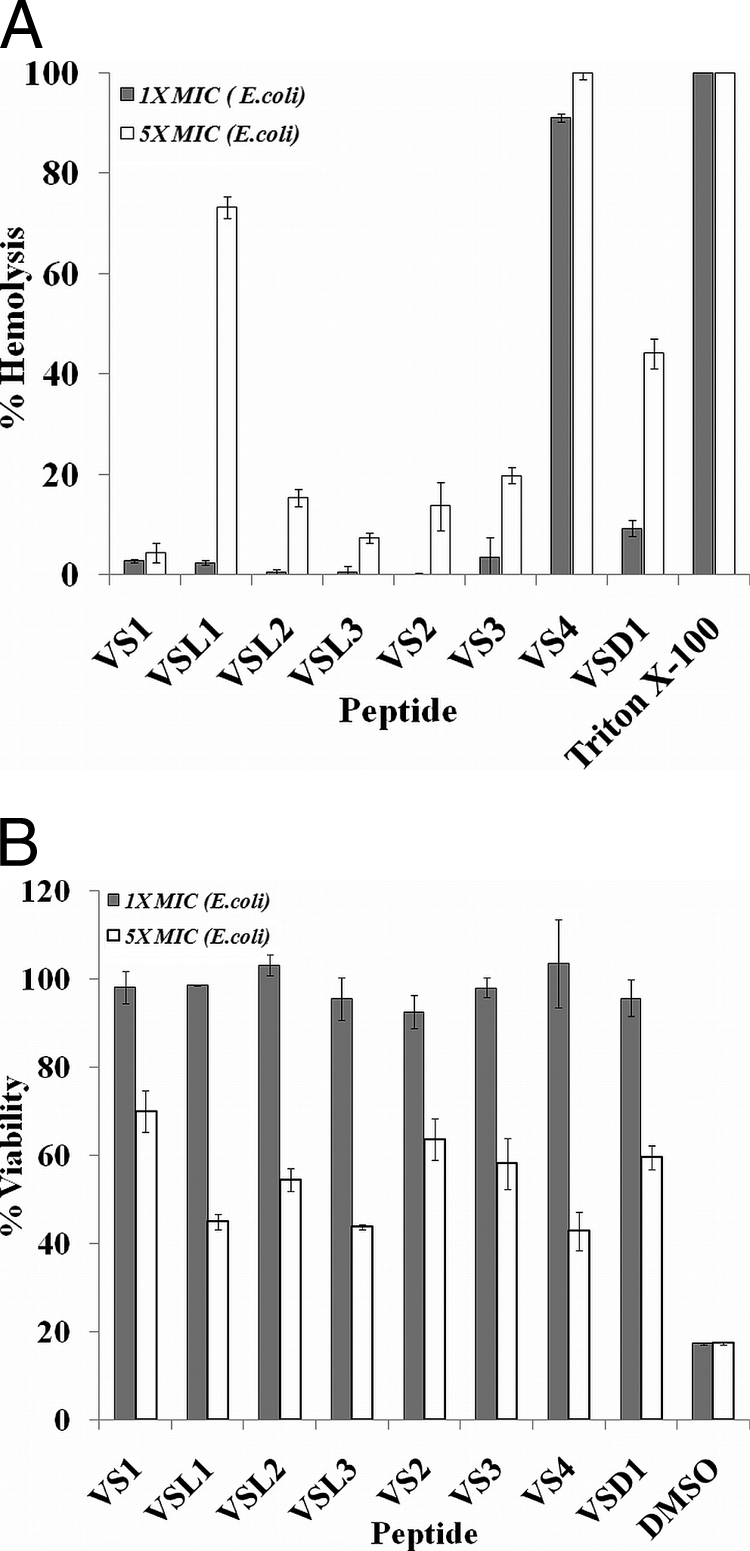
(A) Percent hemolysis caused by peptides. Peptide antibiotics at MIC and 5× MIC (E. coli) were incubated with 0.4% RBCs in PBS. The results are expressed as percent hemolysis. RBCs incubated with 0.1% Triton X-100 were considered to be 100% lysed. Standard deviations from three observations are plotted. (B) Mammalian-cell cytotoxicity (MTT assay) of peptide antibiotics at MIC and 5× MIC (E. coli) against HeLa cell lines. Test samples were incubated with cells for 24 h in RPMI 1640. Untreated cells served as a negative control. The ratio of the OD570 for peptide-treated cells to the OD570 for untreated cells was used to calculate the percent viability of the cells. Standard deviations from three observations are plotted.
Circular-dichroism-based structures of peptides.
Circular-dichroism (CD) studies showed that all the peptides acquired helical structures in a membrane-mimetic environment of 20 mM SDS (the critical micelle concentration for SDS is 8 mM) (Fig. 3A). All the peptides exhibited an excitonic couplet at 267 nm (+) and 298 nm (−), which is the signature for the formation of a right-handed 310 helix in ΔPhe-containing peptides (23). However, different degrees of helicity were observed for the different peptides, and a particular trend was observed in each series of peptides, as shown in Fig. 3B. In the first series, increasing the lengths of the peptides resulted in increases in their helicities (measured in terms of the intensity of the +267-nm band), with the only exception being VSL2, which showed no increase in helicity with increasing length. However, an interesting and reverse trend was observed in the second series of peptides, where the helical intensity (the intensity of the +267-nm band) decreased with increasing Trp content. VSD1, from the third set, showed the highest molar ellipticity value of the helix-associated excitonic couplet. The helicity in VSD1 resulted from the combined helicities of two helical peptides (VS1).
Fig. 3.

(A and B) Circular-dichroism-based conformational analysis of designed peptides (25 μM each) in 20 mM SDS-10 mM sodium phosphate buffer, pH 7.5. (B) Histogram showing mean residue ellipticity (MRE) values for the peptides (for the 267-nm band).
Proteolytic stability of peptides.
Peptide stability is one of the most important parameters in the development of peptide therapeutics (35). In order to determine the relative stability of the ΔPhe-containing prototype peptide VS1, its analog containing phenylalanine (Phe) instead of ΔPhe was synthesized, and both were treated with trypsin/chymotrypsin under identical conditions. Analysis by RPHPLC showed that after 1 h, the saturated (Phe) analog (peak at 38 min) disappeared, giving rise to two small peaks at 14 min and 25 min. On the other hand, VS1 containing ΔPhe was found to be stable to proteolytic degradation; there were no observed changes in the retention time (39 min) and peak intensity of VS1 after 2.5 h, as shown in Fig. 4.
Fig. 4.

(A and B) HPLC profile of VS1 at 0 and 2.5 h of incubation with trypsin/chymotrypsin. (C and D) HPLC profile of Phe analog at 0 and 2.5 h of incubation with trypsin/chymotrypsin.
Increased bacterial-membrane permeabilization and membrane depolarization caused by peptides. (i) Outer membrane permeabilization assay (N-napthylphenyl amine assay).
The abilities of peptides to destabilize the outer membrane were judged by the permeabilization of the outer membrane. NPN is a hydrophobic fluorescent molecule that fluoresces strongly in a hydrophobic environment, like the interior of a membrane, and weakly in an aqueous environment (20). Normally, the outer membrane of a bacterial cell is impermeable to NPN. However, permeabilization of the outer membrane by cationic peptides allows NPN to enter the hydrophobic environment of the membrane, resulting in enhanced fluorescence of the NPN. Figure 5 shows that the cationic peptides were able to permeabilize the bacterial membrane at concentrations of 0.25 μM to 2 μM, which is well below their MICs. VS4 and VSD1 showed maximum increase in NPN fluorescence at 2 μM. Indeed, for VS4 and VSD1 at 2 μM concentration, the increase in fluorescence was much higher than for polymyxin B at the same concentration (2 μM), which was taken as the positive control. PMB was taken as a control because it is a bactericidal antibiotic that kills almost all Gram-negative bacteria at rather low concentrations. Moreover, it has been shown to permeate the OM of E. coli, changing the packing order of lipopolysaccharides (LPS) and increasing the permeability of the OM to a variety of molecules, including its own uptake (self-promoted uptake) (6). In addition, it is a decapeptide antibiotic with a net positive charge (+5) and a molecular mass of 1,200 Da (comparable to the sizes and charges of the designed peptides). Outer membrane permeabilization by these peptides was remarkable, as only very few peptides are able to permeabilize membranes at such low concentrations (2).
Fig. 5.

Outer membrane permeabilization (NPN assay) by peptide. Polymyxin B was taken as a positive control. A dose-dependent increase in the fluorescence intensity was observed upon addition of peptides at concentrations much lower (0.25 to 2 μM) than their MICs.
(ii) Cytoplasmic membrane permeation (PI and Syto9 uptake by cells).
Florescence microscopic studies on cytoplasmic membrane permeabilization by the designed peptides is shown in Fig. 6A. PI is a membrane-impermeable red fluorophore that enters only those cells that have disrupted cell membranes and shows enhanced fluorescence upon binding to DNA. The fluorescence emission maximum for DNA-bound PI is about 615 to 620 nm when excited by a 488-nm laser. Thus, cells with disrupted membranes appear red when excited by the PI filter, whereas the cells with intact membranes do not show any fluorescence when excited using a PI filter. On the other hand, Syto9 is a cell-permeant nucleic acid stain. Its absorption and emission maxima are 485 to 486 nm and 498 to 501 nm, respectively. It has permeability to virtually all cell membranes (living or dead), including bacterial and mammalian cells. By differential staining of cells with Syto9 and PI, we were able to compare cells with disrupted membranes to the total number of cells. Comparison of PI and Syto9 fluorescence shows that peptides were able to permeabilize almost all bacterial cell membranes at their respective MICs after 90 min. Figure 6A shows that there was no significant difference between the Syto9 (green) and PI (red) fluorescence levels in the peptide-treated cells. However, cells without any peptide treatment showed only Syto9 fluorescence and no PI florescence. Since these cells have intact membranes, PI was unable to permeate the cells.
Fig. 6.

(A) Fluorescence microscopy of peptide-induced permeability of E. coli ML35p cells visualized by Syto9 and PI staining at 90 min. After 90 min, all the peptide-treated cells had increased membrane permeability, as seen by PI (red) fluorescence, except control cells without peptide treatment. On the other hand, Syto9 stained all the bacterial cells (live, dead, and membrane permeabilized) nonspecifically to give green fluorescence. Peptides were taken at their respective MICs. (B) Kinetics of PI uptake as studied by FACS for four peptides, VS1, VSL1, VS2, and VSD1, belonging to different series. The peptides were taken at their respective MICs. Cells without any peptide served as a control.
(iii) A FACS-based PI uptake assay was used to study the kinetics of inner membrane permeabilization by four selected peptides (VS1, VS2, VSL1, and VSD1). As shown in Fig. 6B, VS1 and VSL1 were very slow in inducing membrane permeabilization. At their respective MICs (25 μM and 5 μM), VS1 and VSL1 were able to permeabilize only ∼10% of E. coli cells, whereas VS2 showed very fast kinetics of inner membrane permeabilization. At its MIC, it permeabilized ∼96% of cells in 10 min. However, VSD1 was even faster in action: they permeabilized ∼98% of cells in 5 min, but were hemolytic.
(iv) Membrane depolarization (DiSC3-5) assay.
The dye DiSC3-5 (3,3′-dipropylthiadicarbocyanine iodide) is known to be distributed between bacterial cells and the surrounding medium, depending on the membrane potential gradient. Once inside the membrane, the dye aggregates and self-quenches. With the addition of a membrane-permeabilizing agent, the dye is released, and the increase in fluorescence can be monitored over time. Figure 7 shows disruption of the bacterial membrane potential by VS2 and DMSO (positive control). DMSO increases the membrane permeability and exerts a marked inhibitory effect on a wide range of bacteria and fungi. The graph clearly demonstrates that, similar to DMSO, VS2 also efficiently disturbed the potential gradient across the bacterial membrane in a dose-dependent manner.
Fig. 7.

E. coli membrane depolarization caused by the VS2 peptide. (▵) Disruption of the potential gradient across the membrane was assessed by the increase in the fluorescence of the potential-sensitive dye diSC3-5. DMSO (10%) (□) served as a positive control.
(v) Confocal microscopic analysis of uptake of FITC-labeled VS2 peptide.
Z-sectioned confocal images of cells treated with FITC-VS2 for 30 min showed the presence of the peptide in the intracellular milieu of E. coli (Fig. 8). This was an interesting observation, as it suggested that although the primary target for the activity of VS2 was the bacterial membrane, it might also have some intracellular targets, and the bacterial killing by VS2 might be the result of the combined effects of the two independent activities.
Fig. 8.

(A) Confocal images of cells incubated with FITC-labeled VS2 for 30 min. (B) Z section of an individual E. coli mL35p cell showing intracellular presence of peptide.
(vi) Examination of bacterial lysis by electron microscopy.
The effect of VS2 on the morphology of peptide-treated E. coli was investigated by scanning electron microscopy. A change in the cell morphology was observed. Increased roughness of the cell wall was observed after 30 min of incubation, while cell shrinkage was observed after 60 min of incubation with the peptides (Fig, 9, left).
Fig. 9.

SEM and TEM micrographs of E. coli cells treated with VS2 at its MIC (5 μM) for 30 min and 60 min. (A) Untreated cells with smooth, intact surfaces. (B) Peptide-induced breakage and roughness in the cell wall after 30 min. (C) Increased damage to the bacterial cell wall in the form of cracks after 60 min. Cell shrinkage due to loss of turgor was also observed. Similarly, TEM micrographs of ultrathin sections of negatively stained, untreated E. coli cells showed a normal cell shape with an undamaged structure of inner membrane and intact outer membrane (A′). (B′) Peptide-induced breakage in the cell wall and cell membrane after 30 min. Leakage of the inner mass of the cell was also visible. (C′) Complete damage to the cell wall and inner membrane and lysis of the bacterial cell after 60 min. Arrows indicate the sites of cell membrane damage on E. coli cells.
In TEM micrographs, VS2 appeared to have the most severe effect on the bacterial cell wall and cell membrane. Cell wall breakage and variability in wall thickness was observed after 30 min. Separation of the cytoplasmic membrane from the cell wall was also observed. More severe effects were observed after 60 min, when bacterial cell lysis due to membrane damage was clearly visible. E. coli cells without any peptide served as a control (Fig, 9, right).
DISCUSSION
The aim of our work was to identify candidates for developing novel potent and cost-effective antibiotics, starting with a short cationic AMP as a template and then systematically engineering its structure to enhance the degree and spectrum of activity without imparting excessive hemolytic activity to the peptide. Here, we describe our experimental results related to the effectiveness of the three series of peptides that were designed.
The key element in the design of the lead template 11-residue peptide were three ΔPhe residues, four lysine residues, and one tryptophan, along with valine and alanine residues. The three ΔPhe residues were separated by two amino acid residues, an arrangement that induces 310 helical structures in peptides (22). Four lysine residues in such an arrangement were expected to form a polar face, while ΔPhe residues align themselves to form a hydrophobic face, resulting in an amphipathic helical structure. A tryptophan residue was included in the design because of its known membrane-active properties and association with antibacterial activity in peptides like indolicidin and tritrypticin (24, 36). Peptides varying from 10 to 12 residues have shown significant antimicrobial activity (15). Previous reports from our laboratory have demonstrated antimicrobial activity of ΔPhe-containing decapeptides (8).
Although increased length is often associated with enhanced activity, there are no set rules that correlate length with the activity of antimicrobial peptides. We decided to synthesize three more peptides, VSL1, VSL2, and VSL3, containing 12, 13, and 14 residues, respectively. We found that increase in length did not result in any significant enhancement of antibacterial activity. Next, we sequentially increased the Trp content of VS1, resulting in VS2, VS3, and VS4, with two, three, and four Trp residues, respectively. Thus, VS4 was made up of only ΔPhe, Lys, and Trp residues. We observed remarkable changes in the properties of these Trp-containing peptides. The presence of two Trp residues in VS2 enhanced its activity against E. coli (MIC, 5 μM) and S. aureus (MIC, 50 μM). Addition of another Trp in VS3 did not alter its activity against E. coli (MIC, 5 μM) but enhanced its potency against S. aureus (MIC, 25 μM). VS4, with four Trp residues, was the most potent antibacterial peptide. However, the results of hemolysis experiments showed that VS4 was most hemolytic at the 5× MIC values. High Trp content in the antimicrobial peptides is known to be associated with enhanced hemolysis, making them unsuitable for use as antimicrobial agents (7). These results indicate that the presence of two Trp residues in VS2 provided the right balance of charge, hydrophobicity, and appropriate membrane properties, making it a reasonably potent and broad-spectrum antimicrobial peptide. Dendrimerization has been used as a strategy to potentiate the activity of antimicrobial peptides, as it allows multivalent binding (due to doubling of the charge) to the bacterial membrane and generates a higher local concentration that results in membrane destabilization (19). In support of this, our results showed that a branched dimeric peptide, VSD1, exhibited excellent activity against E. coli (MIC, 1 μM) and methicillin-resistant S. aureus (MIC, 5 μM). Dewan et al., in their study on ΔPhe-containing peptides, have shown that, along with increased antimicrobial properties, branched dimers show faster kill kinetics and increased serum stability compared to the linear dimeric analogs (8). Duplication of the vital antimicrobial structure in VSD1 resulted in very high antibacterial activity but simultaneously compromised its cell selectivity. Although it showed very low hemolysis (∼7%) at its MIC, it was considerably hemolytic (∼40%) at 5× MIC values.
Structure-activity relationship studies on helical antibacterial peptides have shown that most of the α-helical peptides are unstructured in aqueous solution and acquire a helical structure in the presence of a lipid-like environment (5). Peptide chain lengths have been associated with increased potency of helical peptides, as long helices can better span the bacterial lipid membrane (32). All ΔPhe-containing peptides showed some degree of helicity in SDS. For the first series of peptides, where the length of the template was successively increased by 1 residue, it was observed that VSL1 (12 residues) showed an increase in helicity and 5-fold enhancement in activity against E. coli. VSL2 (13 residues), however, was equipotent to VSL1 and showed lower helicity than VSL1. In VSL3, with increased length and higher helical content, there was no gain in antimicrobial activity. A gradual decrease in helicity was observed on incorporation of 2, 3, and 4 Trp residues. However, helicity taken as a parameter did not correlate with the activities of peptides of both series. The results indicate that the helicity of a similarly structured peptide alone may not correlate with activity. There are studies in the literature that cite a similar lack of clear structure-function correlation for AMPs (13). In such studies, the activity of AMPs is shown to be derived from a correct balance of charge, hydrophobicity, solubility, and amphipathicity, collectively called the “interfacial activity” (30). It has been shown that peptide composition is also an important parameter for the antimicrobial activity of short designer peptides (34).
The cell membrane is vital for any microorganism and is the primary target for most antibacterial peptides (39). However, the lethal action of a peptide could result from membrane disruption alone or from translocation through the membrane to a target receptor inside the cell, or it can be a concerted effect of both activities (14). We determined the ability of these peptides to permeabilize the cytoplasmic membrane, which is the killing mechanism of a large number of AMPs (11). In a PI uptake assay, we observed that all the peptides were able to permeabilize the bacterial membrane in 90 min. However, the results for the kinetics of membrane permeabilization by selected peptides showed that they differed in these kinetics. VS1 and VSL3 were slow in their action on the inner membrane of E. coli, as they were able to permeabilize only 10% of cells in 60 min. However, VS2 and VSD1 efficiently permeabilized the outer membrane of E. coli at their MICs in 10 min of incubation. Also, in a DiSC3-5 assay, VS2 was able to initiate membrane depolarization immediately at 0.5× its MIC, which is remarkable, as some AMPs do not depolarize the membrane until they reach concentrations as high as 4 to 10 times the MIC (42). However, the presence of a FITC-labeled VS2 peptide inside the cytoplasm of bacteria shows that the peptide translocates itself into the cell, where it might act on some intracellular targets, as well. Our finding that these peptides were able to permeabilize and depolarize the bacterial cell membrane at concentrations much lower than the MIC suggests that membrane permeabilization is not the sole cause of bacterial death and that the designed peptides might have a multimodal mechanism of action (17, 26). SEM images of VS2-treated E. coli cells showed changes in the cell wall morphology. Peptide-induced cell wall breakage was clearly visible, and an increase in the coarseness of the cell surface compared to the control was also observed. TEM images of the VS2-treated cells also confirmed the cell wall and inner membrane damage leading to cell lysis, thereby providing additional evidence of a putative multimodal action of the peptides. These observations suggest that the antibacterial activity of VS2 might be a multistep process involving initial permeabilization and depolarization of the bacterial membrane, leading to its destabilization, and then action on intracellular components of cell, ultimately leading to cell death.
Our study with a set of designed peptides demonstrated that synthetic peptides, such as VS2, containing a helicogenic residue, ΔPhe, that exhibited a broad spectrum of activity showed increased protease resistance and faster killing kinetics and could be suitable candidates for further development.
Conclusion.
Short abiotic antimicrobial peptides are promising candidates for overcoming the critical and accelerating problem of bacterial resistance to currently utilized antibiotics. To provide a road map for the development of improved second-generation therapeutics, we investigated a rationale-based approach for optimizing the peptide sequence to significantly enhance its antimicrobial activity. Increase in length did not dramatically alter the antimicrobial activity of peptides, suggesting that a length of 11 to 12 residues is optimum for activity. The resultant study adds significantly to the field, providing a lead peptide, VS2, with high potency, a broad spectrum of activity, enhanced proteolytic resistance, and faster membrane permeabilization kinetics, based on which novel potent antimicrobials could be produced.
ACKNOWLEDGMENTS
We thank the Council of Scientific and Industrial Research, India, for financial assistance. We acknowledge core funding from ICGEB, New Delhi, India.
We are grateful to Dinkar Sahal, ICGEB, New Delhi, India, for providing strains of E. coli and S. aureus. We acknowledge Rashmi Shrivastva from the Immunology Group, ICGEB, for help in mass spectrometry and Ruchita Pal from the Advanced Instrumentation Research Facility (AIRF), JNU, for providing instrumental support for electron microscopy. We also thank Manjula Kalia for her help in taking confocal images. We thank anonymous reviewers for their constructive criticism that has enriched our work.
Footnotes
Published ahead of print on 14 February 2011.
REFERENCES
- 1. Andersson D. I., Hughes D. 2010. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 8:260–271 [DOI] [PubMed] [Google Scholar]
- 2. Arnusch C. J., et al. 2007. Enhanced membrane pore formation by multimeric/oligomeric antimicrobial peptides. Biochemistry 46:13437–13442 [DOI] [PubMed] [Google Scholar]
- 3. Bíliková K., et al. 2002. Apisimin, a new serine-valine-rich peptide from honeybee (Apis mellifera L.) royal jelly: purification and molecular characterization. FEBS Lett. 528:125–129 [DOI] [PubMed] [Google Scholar]
- 4. Brogden K. A. 2005. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 3:238–250 [DOI] [PubMed] [Google Scholar]
- 5. Dathe M., Wieprecht T. 1999. Structural features of helical antimicrobial peptides: their potential to modulate activity on model membranes and biological cells. Biochim. Biophys. Acta 1462:71–87 [DOI] [PubMed] [Google Scholar]
- 6. Daugelavicius R., Bakiene E., Bamford D. H. 2000. Stages of polymyxin B interaction with the Escherichia coli cell envelope. Antimicrob. Agents Chemother. 44:2969–2978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deslouches B., et al. 2005. De novo generation of cationic antimicrobial peptides: influence of length and tryptophan substitution on antimicrobial activity. Antimicrob. Agents Chemother. 49:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dewan P. C., Anantharaman A., Chauhan V. S., Sahal D. 2009. Antimicrobial action of prototypic amphipathic cationic decapeptides and their branched dimers. Biochemistry 48:5642–5657 [DOI] [PubMed] [Google Scholar]
- 9. Fernandez-Lopez S., et al. 2001. Antibacterial agents based on the cyclic D,L-alpha-peptide architecture. Nature 412:452–455 [DOI] [PubMed] [Google Scholar]
- 10. Giangaspero A., Sandri L., Tossi A. 2001. Amphipathic alpha helical antimicrobial peptides. Eur. J. Biochem. 268:5589–5600 [DOI] [PubMed] [Google Scholar]
- 11. Hancock R. E., Rozek A. 2002. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 206:143–149 [DOI] [PubMed] [Google Scholar]
- 12. Hancock R. E., Sahl H. G. 2006. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24:1551–1557 [DOI] [PubMed] [Google Scholar]
- 13. Haug B. E., Stensen W., Stiberg T., Svendsen J. S. 2004. Bulky nonproteinogenic amino acids permit the design of very small and effective cationic antibacterial peptides. J. Med. Chem. 47:4159–4162 [DOI] [PubMed] [Google Scholar]
- 14. Haukland H. H., Ulvatne H., Sandvik K., Vorland L. H. 2001. The antimicrobial peptides lactoferricin B and magainin 2 cross over the bacterial cytoplasmic membrane and reside in the cytoplasm. FEBS Lett. 508:389–393 [DOI] [PubMed] [Google Scholar]
- 15. Javadpour M. M., et al. 1996. De novo antimicrobial peptides with low mammalian cell toxicity. J. Med. Chem. 39:3107–3113 [DOI] [PubMed] [Google Scholar]
- 16. Kaiser E., Colescott R. L., Bossinger C. D., Cook P. I. 1970. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 34:595–598 [DOI] [PubMed] [Google Scholar]
- 17. Kragol G., et al. 2001. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 40:3016–3026 [DOI] [PubMed] [Google Scholar]
- 18. Liu D., DeGrado W. F. 2001. De novo design, synthesis, and characterization of antimicrobial beta-peptides. J. Am. Chem. Soc. 123:7553–7559 [DOI] [PubMed] [Google Scholar]
- 19. Liu Z., et al. 2006. Multivalent antimicrobial peptides from a reactive polymer scaffold. J. Med. Chem. 49:3436–3439 [DOI] [PubMed] [Google Scholar]
- 20. Loh B., Grant C., Hancock R. E. 1984. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 26:546–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Marr A. K., Gooderham W. J., Hancock R. E. 2006. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr. Opin. Pharmacol. 6:468–472 [DOI] [PubMed] [Google Scholar]
- 22. Mathur P., Jagannathan N. R., Chauhan V. S. 2007. Alpha, beta-dehydrophenylalanine containing cecropin-melittin hybrid peptides: conformation and activity. J. Pept. Sci. 13:253–262 [DOI] [PubMed] [Google Scholar]
- 23. Mathur P., Ramakumar S., Chauhan V. S. 2004. Peptide design using alpha,beta-dehydro amino acids: from beta-turns to helical hairpins. Biopolymers 76:150–161 [DOI] [PubMed] [Google Scholar]
- 24. Nagpal S., Gupta V., Kaur K. J., Salunke D. M. 1999. Structure-function analysis of tritrypticin, an antibacterial peptide of innate immune origin. J. Biol. Chem. 274:23296–23304 [DOI] [PubMed] [Google Scholar]
- 25. Nguyen L. T., et al. 2010. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One 5:e12684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Park C. B., Kim H. S., Kim S. C. 1998. Mechanism of action of the antimicrobial peptide buforin II: buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun. 244:253–257 [DOI] [PubMed] [Google Scholar]
- 27. Patch J. A., Barron A. E. 2002. Mimicry of bioactive peptides via non-natural, sequence-specific peptidomimetic oligomers. Curr. Opin. Chem. Biol. 6:872–877 [DOI] [PubMed] [Google Scholar]
- 28. Peschel A., Sahl H. G. 2006. The co-evolution of host cationic antimicrobial peptides and microbial resistance. Nat. Rev. Microbiol. 4:529–536 [DOI] [PubMed] [Google Scholar]
- 29. Ramagopal U. A., Ramakumar S., Sahal D., Chauhan V. S. 2001. De novo design and characterization of an apolar helical hairpin peptide at atomic resolution: compaction mediated by weak interactions. Proc. Natl. Acad. Sci. U. S. A. 98:870–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rathinakumar R., Wimley W. C. 2008. Biomolecular engineering by combinatorial design and high-throughput screening: small, soluble peptides that permeabilize membranes. J. Am. Chem. Soc. 130:9849–9858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reddy K. V., Yedery R. D., Aranha C. 2004. Antimicrobial peptides: premises and promises. Int. J. Antimicrob. Agents 24:536–547 [DOI] [PubMed] [Google Scholar]
- 32. Ringstad L., Schmidtchen A., Malmsten M. 2006. Effect of peptide length on the interaction between consensus peptides and DOPC/DOPA bilayers. Langmuir 22:5042–5050 [DOI] [PubMed] [Google Scholar]
- 33. Rotem S., Mor A. 2009. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 1788:1582–1592 [DOI] [PubMed] [Google Scholar]
- 34. Strøm M. B., et al. 2003. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem. 46:1567–1570 [DOI] [PubMed] [Google Scholar]
- 35. Strömstedt A. A., Pasupuleti M., Schmidtchen A., Malmsten M. 2009. Evaluation of strategies for improving proteolytic resistance of antimicrobial peptides by using variants of EFK17, an internal segment of LL-37. Antimicrob. Agents Chemother. 53:593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Subbalakshmi C., Bikshapathy E., Sitaram N., Nagaraj R. 2000. Antibacterial and hemolytic activities of single tryptophan analogs of indolicidin. Biochem. Biophys. Res. Commun. 274:714–716 [DOI] [PubMed] [Google Scholar]
- 37. Tam J. P., Lu Y. A., Yang J. L. 2002. Antimicrobial dendrimeric peptides. Eur. J. Biochem. 269:923–932 [DOI] [PubMed] [Google Scholar]
- 38. Tossi A., Sandri L., Giangaspero A. 2000. Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 55:4–30 [DOI] [PubMed] [Google Scholar]
- 39. Vaara M. 1992. Agents, that increase the permeability of the outer membrane. Microbiol. Rev. 56:395–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wade D., et al. 1990. All-D amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. U. S. A. 87:4761–4765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wayne P. A. 1999. Methods for determining bactericidal activity of antimicrobial agents; approved guideline. Document M26-A. Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 42. Wu M., Maier E., Benz R., Hancock R. E. 1999. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry 38:7235–7242 [DOI] [PubMed] [Google Scholar]
- 43. Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415:389–395 [DOI] [PubMed] [Google Scholar]