

Abstract
Among Gram-negative bacteria, resistance to β-lactams is mediated primarily by β-lactamases (EC 3.2.6.5), periplasmic enzymes that inactivate β-lactam antibiotics. Substitutions at critical amino acid positions in the class A β-lactamase families result in enzymes that can hydrolyze extended-spectrum cephalosporins, thus demonstrating an “extended-spectrum” β-lactamase (ESBL) phenotype. Using SHV ESBLs with substitutions in the Ω loop (R164H and R164S) as target enzymes to understand this enhanced biochemical capability and to serve as a basis for novel β-lactamase inhibitor development, we determined the spectra of activity and crystal structures of these variants. We also studied the inactivation of the R164H and R164S mutants with tazobactam and SA2-13, a unique β-lactamase inhibitor that undergoes a distinctive reaction chemistry in the active site. We noted that the reduced Ki values for the R164H and R164S mutants with SA2-13 are comparable to those with tazobactam (submicromolar). The apo enzyme crystal structures of the R164H and R164S SHV variants revealed an ordered Ω loop architecture that became disordered when SA2-13 was bound. Important structural alterations that result from the binding of SA2-13 explain the enhanced susceptibility of these ESBL enzymes to this inhibitor and highlight ligand-dependent Ω loop flexibility as a mechanism for accommodating and hydrolyzing β-lactam substrates.
INTRODUCTION
The most common β-lactamases found in Escherichia coli and Klebsiella pneumoniae are the class A enzymes TEM, SHV, and CTX-M (4, 23). Point mutations in the blaTEM and blaSHV genes result in single-amino-acid substitutions that broaden the substrate spectrum of the common TEM and SHV family enzymes to include extended-spectrum cephalosporin antibiotics like ceftazidime or cefotaxime (9). These altered β-lactamases are referred to as “extended-spectrum” β-lactamases (ESBLs), and this property is referred to as the “ESBL phenotype” (23). The amino acid substitutions responsible for this phenotype in both TEM and SHV enzymes are located in three areas of the class A β-lactamase, the Ω loop (Ambler residues 164 to 179), the B3 β-strand, and residue 104 (2).
The Ω loop borders the active site on one side and includes the catalytic residue Glu166, which, in addition to aiding in acylation, primes a water molecule needed in the regeneration of the enzyme via the deacylation of the enzyme-substrate complex at Ser70 (2, 16). In wild-type (wt) TEM and SHV, two salt bridges, between residues Arg164 and Asp179 and between residues Arg164 and Glu171, stabilize the loop in place, thereby ensuring the presence of the catalytic Glu166 in the active site. Naturally occurring variants with single-amino-acid changes disrupting the salt bridge at residues 164 to 179, including D179N (SHV-8) in the SHV family and R164H (TEM-29) and R164S (TEM-12) in the closely related TEM β-lactamases, have been isolated in the clinic and were shown to have ESBL phenotypes (18, 24). Such ESBL activity is hypothesized to result in the increased flexibility of residues in the Ω loop due to the elimination of the R164-D179 salt bridge, thus allowing the active site to accommodate the larger R1 groups of cephalosporin antibiotics. Furthermore, among these ESBL substitutions found in variants of TEM-1 and TEM-2 (the latter is considered a separate “parent” enzyme to the TEM family, differing from TEM-1 by 1 amino acid but with equivalent biochemical properties) (23), the most common amino acid changes are from Arg164 to Ser or His (10). Additional ESBLs arise from an alteration of D179; these have been observed in the clinic for both TEM (D179E) and SHV (D179N, D179S, D179G, and D179A) β-lactamases (1, 6, 15, 24). The finding that a substitution at either of these positions results in the ESBL phenotype suggests that merely disrupting this interaction sufficiently destabilizes the Ω loop to allow larger cephalosporin substrates access to the catalytic center. This destabilization of the Ω loop was previously predicted for SHV-1 (14) and a closely related variant (3) and was observed for a distantly related β-lactamase (13). Furthermore, the crystal structure of TEM-64 (E104K/R164K/M182T variant) in complex with a boronic acid transition-state analog revealed a shift of some residues of the Ω loop, with N170 having moved substantially (26). However, TEM-64 has two active-site substitutions that confer the ESBL phenotype, E104K and R164K, and is also complexed with an inhibitor, which thus limits interpretations regarding what precisely contributes to the observed conformational changes. To our knowledge, experimental structural studies have not been reported for single R164 or D179 mutants of TEM or SHV in both the absence and presence of a substrate or inhibitor.
To investigate if increased Ω loop flexibility results from substitutions in this R164-D179 salt bridge in SHV, we engineered and crystallized the R164H and R164S variant forms of SHV-1 and determined their atomic structures to 1.8 and 1.34 Å, respectively. To further probe whether ligand binding affects the Ω loop conformation, we soaked crystals of these SHV-1 R164 mutants with SA2-13, a rationally designed inhibitor of SHV-1 β-lactamase (22) (Fig. 1). The structures of both R164H and R164S SHV-1 β-lactamase variants in complex with SA2-13, at 1.65 and 1.8 Å resolutions, respectively, revealed a remarkable ligand-induced disordering of the Ω loop, thereby providing new insight into the dynamic nature of the Ω loop in R164 ESBL β-lactamases.
Fig. 1.
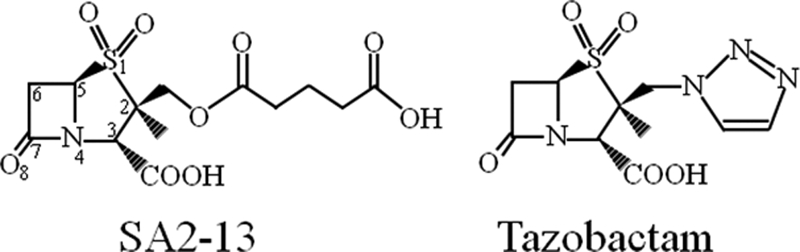
Chemical structures of SA2-13 and tazobactam.
MATERIALS AND METHODS
Inhibitors.
The synthesis of SA2-13 was performed by the laboratory of John Buynak (22). Tazobactam was a kind gift of Wyeth Pharmaceuticals, previously located in Pearl River, NY.
Mutagenesis.
Single-base-pair substitutions of the blaSHV-1 gene were made by using the QuikChange II site-directed mutagenesis kit (Stratagene) on the pBC SK(−) vector (Stratagene), as previously described (14). Mutations were confirmed by standard nucleotide sequencing reactions. Sequence-confirmed plasmids harboring the mutated blaSHV genes were transformed into E. coli DH10B chemically competent cells (Invitrogen) for protein expression.
Expression and purification.
The two altered enzymes R164H and R164S were expressed in E. coli DH10B cells and purified as previously described for other SHV-1 variants (21), with the addition of two purification steps for the R164 mutants. First, after periplasmic fractionation, the crude extract was loaded onto a hand-poured Q Sepharose FF column (GE Healthcare), and the active β-lactamase-containing flowthrough was retained and purified by preparative isoelectric focusing (pIEF), with the latter step performed as previously described (12, 21). The eluent from the active bands of the pIEF gel, as determined by using nitrocefin strips, was loaded onto a Mono Q column (GE Healthcare) in 20 mM Tris (pH 8.5) and eluted by a 0 to 250 mM sodium chloride gradient. Samples were prepared for crystallization by gel filtration in 20 mM HEPES (pH 8.5) on a Superdex 75 10/300 GL column (GE Healthcare).
Crystallization and soaking.
Single crystals of the SHV R164H and R164S variants were obtained by vapor diffusion at room temperature in sitting drops. The drop compositions were similar to those under wt SHV-1 crystallization conditions (16). For each drop, 2 μl of 5 mg/ml mutant protein in 2 mM HEPES (pH 7.0) was mixed with 0.5 μl of Cymal-6 detergent (Hampton Research) and combined with 2.5 μl of reservoir solution. Reservoir solutions contained 21 to 30% polyethylene glycol 6000 (PEG 6000) and 0.1 M HEPES buffer at pH 6.6 to 7.6 for all crystals discussed here, except for the R164H:SA2-13 complex, whose reservoir consisted of 27% PEG 10000 and 0.1 M HEPES at pH 7.3.
Ligand soaking experiments with 50 mM SA2-13 were performed at room temperature for 30 to 60 min in solutions of mother liquor (30 min for the R164S variant and 60 min for the R164H variant). The crystals were cryoprotected by being briefly passed through synthetic mother liquor supplemented with 10% (+/−)-2-methyl-2,4-pentanediol (MPD), as well as 50 mM SA2-13 for the ligand-soaked crystals, and immediately flash-frozen in liquid N2.
X-ray diffraction data were collected at the National Synchrotron Light Source, beamline X29. The data were processed with HKL2000 (20) (Table 1). The crystal space group was P212121, and crystals were isomorphous to wild-type SHV-1 crystals, so the structures were solved by isomorphous replacement (Protein Data Bank [PDB] accession number 1VM1). Crystallographic refinement was carried out by using REFMAC (27), and COOT (8) was used for the graphical fitting of the electron density maps. ProDRG was used to create initial ligand coordinate and connectivity restraint files used for fitting ligands into the electron density maps (25). Structural figures were made by using PyMOL (http://www.pymol.org/).
Table 1.
Crystallographic data collection and refinement statistics for the four crystal structures
| Statistica | Value for variantb |
|||
|---|---|---|---|---|
| R164H apo | R164H:SA2-13 | R164S apo | R164S:SA2-13 | |
| Data collection | ||||
| Space group | P212121 | P212121 | P212121 | P212121 |
| Unit cell lengths (Å) | 48.7, 55.6, 84.3 | 49.4, 55.5, 82.9 | 48.8, 55.6, 83.8 | 49.5, 55.2, 83.5 |
| Wavelength (Å) | 1.1000 | 1.1000 | 1.1000 | 1.1000 |
| Resolution (Å) | 50–1.80 (1.86–1.80) | 50–1.65 (1.71–1.65) | 50–1.34 (1.39–1.34) | 50–1.80 (1.86–1.80) |
| Mean redundancy (SD) | 3.6 (3.6) | 4.4 (3.5) | 3.5 (3.2) | 3.5 (2.9) |
| No. of unique reflections | 19,477 | 26,947 | 48,908 | 19,684 |
| 〈I〉/σ〈(I)〉c | 14.5 (2.0) | 16.1 (3.93) | 20.5 (3.24) | 15.0 (2.74) |
| Mean Rmerge (%) (SD) | 9.3 (39.7) | 7.5 (22.6) | 5.6 (23.4) | 7.7 (23.0) |
| Mean completeness (%) (SD) | 88.9 (70.3) | 95.6 (70.3) | 99.0 (95.4) | 95.1 (67.7) |
| Refinement | ||||
| Resolution range (Å) | 25.1–1.80 | 46.1–1.65 | 31.8–1.341 | 42.6–1.802 |
| R factor (%) | 17.24 | 16.2 | 16.33 | 14.7 |
| R free (%) | 22.84 | 21.1 | 18.99 | 19.2 |
| RMSD from ideality | ||||
| Bond length (Å) | 0.011 | 0.010 | 0.009 | 0.012 |
| Bond angle (°) | 1.29 | 1.33 | 1.32 | 1.36 |
| Avg B factor (Å2) | 27.9 | 19.4 | 15.4 | 18.8 |
| Protein | 27.8 | 17.6 | 12.8 | 17.1 |
| Inhibitor | 24.9 | 26.2 | ||
| Other ligands | 50.1 | 34.9 | ||
| Water molecules | 36.4 | 32.7 | 27.9 | 31.7 |
RMSD, root mean square deviation.
Data in parentheses are for the highest-resolution shell.
The ratio between average spot intensity and average uncertainty in the intensity measurements.
Kinetics.
Kinetic measurements of Ki and kinact for the inactivation of wild-type SHV-1 and the R164 mutants with SA2-13 were carried out as previously reported (11, 22).
Protein structure accession numbers.
Coordinates have been deposited in the Protein Data Bank under accession numbers 3OPH for R164S apo, 3OPL for R164H apo, 3OPP for R164S:SA2-13, and 3OPR for R164H:SA2-13.
RESULTS
Kinetics of inhibition.
The kinetic analysis results for the R164 variants show that the Ki of SA2-13 for the R164H and R164S β-lactamases is lower than that of wt SHV-1 (Table 2). A somewhat similar trend was seen with tazobactam, although the decrease in Ki was not as great when R164 was substituted (Table 2). The R164 substitutions did not seem to affect the kinact parameter, as this rate constant remains relatively uniform in comparisons of the wild type and the R164 variant when inhibited by either SA2-13 or tazobactam (Fig. 1 and Table 2).
Table 2.
Kinetic parameters of inhibition of SHV-1 and R164 variants
| Variant | Ki (μM) | kinact (s−1) | kinact/Ki (μM−1 s−1) |
|---|---|---|---|
| SA2-13 | |||
| SHV-1 | 1.70 ± 0.10 | 0.060 ± 0.006 | 0.035 ± 0.002 |
| R164S | 0.53 ± 0.02 | 0.068 ± 0.006 | 0.13 ± 0.01 |
| R164H | 0.26 ± 0.01 | 0.068 ± 0.006 | 0.26 ± 0.02 |
| Tazobactam | |||
| SHV-1 | 0.221 ± 0.015 | 0.100 ± 0.006 | 0.50 ± 0.05 |
| R164S | 0.098 ± 0.009 | 0.070 ± 0.007 | 0.71 ± 0.09 |
| R164H | 0.130 ± 0.006 | 0.080 ± 0.008 | 0.62 ± 0.07 |
R164 variant structures.
Crystal structures were determined for the R164H and R164S variant enzymes in both apo and SA2-13-bound forms (data collection and refinement statistics are listed in Table 1). For the inhibitor-bound structures, inhibitor soaking times for the R164H and R164S crystals were 60 and 30 min, respectively.
The four structures contained residues 26 to 292, except residues 165 to 171 in the R164H:SA2-13 liganded structure and residues 166 to 171 in the R164S:SA2-13 liganded structure, which were too disordered to be modeled (Fig. 2). Each of these crystal structures includes two Cymal-6 detergent molecules, with one being partially ordered; this is common for SHV-1 structures, as they contain one, and often a second, partially ordered, Cymal-6 molecule (19, 21). In both of the apo structures, the more complete Cymal-6 molecule is present in a different conformation from that of the wild-type structure, and the distal sugar moiety is disordered. The second Cymal-6 molecule is largely disordered and has been truncated in all four structures. The R164H and R164S apo structures also each contain a HEPES buffer molecule (identified by its terminal sulfonate group) noncovalently bound in the active site (Fig. 2A and C). The presence of HEPES in the active site of SHV-1 was observed previously and served as the “chemical rationale” of the fragment-based design of SA2-13 (22).
Fig. 2.

Electron density of bound ligands and Ω loops of R164 mutant structures. (A and C) R164H (A) and R164S (C) apo structures showing an |Fo|-|Fc| electron density map contoured at 2.5 σ (magenta) exhibit strong density for the HEPES sulfonate groups. The well-ordered Ω loops (residues 160 to 179) are indicated by 2|Fo-Fc| maps (blue), contoured at 1.0 σ. (B and D) Structures of R164H (B) and R164S (D) in complex with SA2-13 reveal well-ordered SA2-13 ligands and weak or absent density for the Ω loops. Map contour levels are the same as in panels A and C.
The R164H and R164S apo structures contain 199 and 341 water molecules, respectively, and the R164H:SA2-13 and R164S:SA2-13 structures include 244 and 246 water molecules, respectively. Both inhibitor-bound structures contain one hydrolyzed SA2-13 molecule with the terminal (C-7) carbon covalently bound to the active site at S70; the substitutions at residue 164 are also distinguishable in the electron density maps (Fig. 2B and D). The overall fold of the four mutant enzymes follows closely that of wild-type SHV (Fig. 3A). In the unliganded structures, the R164 substitutions cause disruptions of both the R164-D179 and R164-E171 salt bridges, yet the overall conformation and overall mobility of the Ω loop are hardly affected (Fig. 3). The main effect of the R164 mutations in the apo R164H and R164S structures is an increased mobility of a few residues, in particular the side chain at E171, which had an increase in the B factor of ∼25 Å2 compared to the wt SHV-1 structure; this side chain has shifted as well due to the absence of the R164 interaction. Additionally, part of the Ω loop, including residues E166 and N170, appeared to have shifted inwards about 0.4 Å toward the active site in the mutant structures compared to the wild-type SHV-1 structure (Fig. 3), but these shifts were probably close to the coordinate error.
Fig. 3.

Superpositions of wt SHV-1 with apo R164H and R164S structures. (A) Cartoon representations of superimposed wt SHV-1 (gray) and the R164H (blue) and R164S (magenta) variants indicating their similar overall structures. Key Ω loop and active-site residues of wt SHV-1 are highlighted in yellow, and side chains for the residues at position 164 are shown for comparison. (B) Close-up view of the Ω loop. Interactions involving the R/S/H164 side chain are indicated by dashed lines. Key active-site residue main chain (MC) atoms and side chains are labeled.
Ligand conformation.
The omit electron density maps from both inhibitor-bound structures displayed strong electron density for all the important functional groups of the SA2-13 inhibitor, which was present in the trans-enamine conformation in each case (Fig. 2). The inhibitor electron density is somewhat less continuous in the R164S-bound structure than in the R164H-bound structure, mainly in the polymethylene linker between the sulfone and terminal carboxyl groups, perhaps due to the 2-fold difference in soaking time and, therefore, possibly lower active-site occupancy.
The overall ligand conformation and ligand-protein interactions in the R164-substituted β-lactamases were similar to those of the wt SHV-1:SA2-13 complex (22), except for the loss of interactions between SA2-13 and the disordered residues of the Ω loop (Fig. 2 and 3). SA2-13 adopts a loop-like conformation in which one end is covalently attached to the protein at S70 and the other end fits into the carboxyl binding pocket, with the middle section of the compound protruding from the active site. The carboxyl binding pocket harbors the carboxyl linker addition of SA2-13 via interactions with K234, T235, and S130 similar to those of the wt SHV-1:SA2-13 structure (Fig. 4). In addition, the original β-lactam carboxyl moiety is interacting with N132 via a hydrogen bond, as was also the case for the wild-type SHV-1 SA2-13 complex (Fig. 4). As in wt SHV-1, the carbonyl oxygen atom of SA2-13 interacts with the backbone nitrogen atoms of A237 and S70 that form the oxyanion hole, and hydrophobic atoms of SA2-13 interact with V216 and Y105 via van der Waals interactions (Fig. 2 to 4). The only interaction not observed for the R164H and R164S structures is that between SA2-13 and N170, which is present only in the wild-type SHV-1 complex, as N170 is disordered in the variant acyl-enzymes (22). Also, likely as a consequence of the absence of an ordered Ω loop, SA2-13 shifted slightly in both mutant structures compared to the structure of wt SHV-1 (Fig. 4). This translation toward the space formerly occupied by the Ω loop results in shifts of the sulfone group of SA2-13 by more than 0.5 Å and of one of the oxygens of the original β-lactam carboxyl group by about 0.5 Å (Fig. 4). This small shift of SA2-13 in the R164 variants also allows SA2-13 to have a trans-enamine bond torsion angle closer to ideality; this SA2-13 torsion angle in each of the mutant structures is 180°, compared to a more distorted torsion angle of 166° in the wild-type SHV-1 complex (Fig. 4).
Fig. 4.

Superposition of SA2-13-bound wt and R164 mutant structures. The SA2-13-bound structures of wt SHV (gray) and the R164H (blue) and R164S (magenta) variants were superimposed and are shown as a stick model. Hydrogen bond interactions between SA2-13 and wt SHV-1 are indicated by black dashed lines (for clarity, the corresponding interactions in the mutant structures are not shown). Active-site residues are labeled. The Ω loop region that is ordered in the wt SHV-1 SA2-13 complex but disordered in the R164 mutant SA2-13 complex is highlighted with a red dashed circle.
DISCUSSION
We observed that the disruption of the R164-D179 and R164-E171 salt bridges in SHV by the replacement of R164 by histidine or serine results in an Ω loop that is ordered in the uninhibited (apo) structure yet becomes disordered upon the binding of SA2-13. Although the notion of increased Ω loop flexibility for R164 and D179 mutants was previously predicted for SHV-1 (14) and observed for crystal structures of more distantly related β-lactamases (one with a D179 substitution [13] and one class C β-lactamase with a 3-residue Ω loop insertion [5]), we show that this disorder can occur in a ligand-dependent fashion. This adds an additional level of complexity to the understanding of β-lactamases and their variants, particularly ESBLs. We note that a ligand-dependent shift was also observed previously for an ESBL phenotype CTX-M β-lactamase as the distance between residues 170 and 240 increased 0.8 Å upon ligand binding (7). These R164 changes in SHV-1 do not disrupt the overall fold of the enzyme or of the Ω loop in the absence of an inhibitor, thus allowing the variant apo enzymes to retain the basic wt active-site configuration. Interestingly, SA2-13 induces Ω loop flexibility in the R164 ESBL enzymes, while in wt SHV the Ω loop remains ordered upon SA2-13 binding (22). A possible explanation for this SA2-13-dependent loop disorder was obtained via a close inspection of this wt SA2-13 complex structure, which shows a potential medium-range repulsive electrostatic interaction of SA2-13 in the active site: a 3.9-Å distance between the carboxyl groups of SA2-13 and E166. Although the wt Ω loop appears unaffected, the electrostatic repulsion between these two negatively charged carboxyl groups is likely the culprit responsible for displacing the Ω loops of these SHV enzymes harboring destabilizing R164 substitutions. Perhaps the small inward shift of E166 in the apo mutant structures, as discussed above, might have further enhanced this electrostatic repulsion. After repulsion between the SA2-13 and E166 carboxyl groups causes the disorder exhibited in the Ω loop, the inhibitor experiences less repulsive force from the Ω loop side than with the Ω loop residues in place, resulting in a shift of the inhibitor toward the disordered Ω loop. This in turn helps to ensure that the disordered Ω loop residues remain displaced so long as the inhibitor is present in a stable conformation. Such disorder-causing behavior may also be time dependent in the context of a crystal lattice: consistent with the differences in the continuity of the SA2-13 electron density, fewer residues are disordered in the R164S liganded structure with a soaking time of 30 min, whereas the crystal that produced the more disordered R164H liganded structure was soaked for 60 min.
Significance of disruption of R164/D179 interactions on inhibitor susceptibility and ESBL phenotype.
The R164 side chain is part of a structurally important salt bridge network including residues D179 and E171. A substitution of residue 179 was also clinically observed via its ESBL phenotype, pointing to an important role for this salt bridge interaction (24). Detailed enzymatic and microbiological testing of D179 and R164 mutants in both SHV-1 (14) and TEM-1 (17) indicated that there are differences when each of these residues is replaced. D179 is located deeper in the Ω loop than R164, forming hydrogen bonds with the backbone nitrogens of R164 and D163, in addition to the salt bridge with the Nε and NH1 atoms of R164 discussed above (Fig. 3). Due to this buried location and the increased number of interactions compared to those with R164, we postulate that when D179 is replaced, the Ω loop could be disordered even in the uncomplexed state, perhaps similarly to what was observed previously for a D179 variant of a distantly related PC1 β-lactamase (13). Experiments are under way to investigate this hypothesis.
As was shown in a previous study, the relatively improved kinetic parameters for SA2-13 might be attributed to a decreased off-rate (kreact), in turn perhaps compounded by its ability to destabilize the Ω loop, thereby displacing E166, which is important for deacylation. We previously observed that the structure-based linked-fragment design of SA2-13 resulted in the stabilization of the trans-enamine intermediate in wt SHV-1 and that the recovery of the enzyme via deacylation is decreased by about 10-fold within the first 30 min compared to the parent compound tazobactam (22). Therefore, the enhanced ability of SA2-13 to form a stable deacylation-resistant trans-enamine intermediate and its capability of displacing the E166-containing Ω loop, with E166 being critical for acylation and deacylation, are likely both contributing to why R164 substitutions have a greater positive effect on the affinity for SA2-13 than for tazobactam (Table 2). It should, however, be kept in mind that the kinetic differences between SA2-13 and tazobactam are somewhat limited and that future investigations regarding the deacylation rates might pinpoint additional interesting differences.
The ordered or disordered state adopted by the Ω loop in ESBL variants can be ligand dependent, as described in this paper, but likely also depends on the specific type of ESBL substitution (as postulated above for R164 and D179 changes). Since the Ω loop contains an important residue needed for efficient deacylation, E166, differences in ligands and/or mutation types could lead to interesting differences in the kinetics of substrate hydrolysis and/or the inhibition of ESBL variants. Regarding inhibition differences between R164 and D179 mutations, the D179 variant of SHV-1 in E. coli DH10B is more readily inhibited (lower MIC) by both clavulanic acid and tazobactam (14) than the R164 mutants. This difference could, however, also be due in part to the somewhat lower expression level of the D179N β-lactamase. A D179 mutant of SHV was also found to be much less catalytically active toward ampicillin than the R164S mutant, as its kcat was about 100-fold lower (17). That same study found not only that the R164S variant weakly hydrolyzes ceftazidime but also that this mutant hydrolyzes cefotaxime about 2 orders of magnitude more weakly than does wild-type SHV-1, perhaps providing a rationale for why such R164 variants have not been observed clinically. Furthermore, we note that there was also a major difference in microbiological activity between the R164 and D179 SHV mutants when different-sized substrates were compared (14). Compared to R164H/S, the D179N β-lactamase expressed in E. coli yields larger relative MIC increases with increasing substrate size, indicating that the D179 β-lactamase hydrolyzes larger β-lactams more readily than the R164 variants (14). Overall, these results suggest that the D179 variants likely have their Ω loop disordered most of the time and “pay a price” in terms of efficiency at hydrolyzing small substrates, as the deacylation-critical E166 is likely seldom in the right position for efficient deacylation. However, for larger substrates that do not fit into the active site of SHV-1 with its Ω loop in the wild-type conformation, having the Ω loop disordered most of the time may be beneficial. Clearly, the D179 mutant can productively accommodate the larger substrates, the binding of which likely would have otherwise become a bottleneck toward their efficient hydrolysis. Note that even for the ESBL D179 variant with the putatively more disordered Ω loop, this loop must still occasionally return to the wt position after acylation to facilitate E166-mediated deacylation in order to explain why the MIC values for the D179 mutant are substantially higher than those for the deacylation-deficient E166A mutant (14).
In the course of our studies, we were mindful that the R164 variants of SHV have not yet been observed in the clinic. There are two likely reasons for this: (i) variants at the D179 position in SHV may be “fitter” and can outcompete R164 variants, as was shown previously under certain conditions (17), and (ii) a clinical isolate bearing this substitution may be present but has not yet been detected. Despite the absence of a “clinical correlate,” the disruption of the Ω loop at the R164-D179 salt bridge in SHV is an important and novel structural mechanism by which class A β-lactamases can evolve the ESBL phenotype (as shown by the D179 variants SHV-6, -8, and -24). Our novel finding of the ligand-induced flexibility of the Ω loop in the R164 SHV variants is unique and reveals an additional pathway that β-lactamases can utilize to arrive at the ESBL phenotype.
In summary, our studies reveal for the first time, via crystal structures of R164 mutants of the SHV-1 β-lactamase, a ligand-dependent flexibility of the Ω loop after the disruption of the R164-D179 salt bridge. There is thus an apparent “trade-off” for R164 ESBL variants between accommodating larger substrates (such as ceftazidime, which has a 2.5-fold-increased kcat for R164 compared to wild-type SHV-1 [17]), likely via increased Ω loop disorder, and substrate turnover, with the latter being directly related to having a more ordered Ω loop and, specifically, E166 side chain. Our observations also suggest that the Ω loop of SHV also undergoes major conformational changes during the catalytic cycle between the apo and bound forms. Further design of β-lactamase inhibitors and β-lactams could take advantage of the observed “tradeoff” consequences of the increased Ω loop disorder of certain ESBL variants to enhance acyl-intermediate stabilization and/or Michaelis-Menten complex accommodation.
ACKNOWLEDGMENTS
J.D.B. is supported by Robert A. Welch Foundation grant N-0871. F.V.D.A. is supported by the National Institutes of Health (grant R01 AI062968). The Veterans Affairs Merit Review Program, Geriatric Research Education and Clinical Care (GRECC), and the National Institutes of Health (grant RO1 AI063517-01) supported R.A.B.
We thank beamline personnel at NSLS X-29 for help with data collection.
Footnotes
Published ahead of print on 28 February 2011.
REFERENCES
- 1. Arlet G., Rouveau M., Philippon A. 1997. Substitution of alanine for aspartate at position 179 in the SHV-6 extended-spectrum β-lactamase. FEMS Microbiol. Lett. 152:163–167 [DOI] [PubMed] [Google Scholar]
- 2. Babic M., Hujer A. M., Bonomo R. A. 2006. What's new in antibiotic resistance? Focus on β-lactamases. Drug Resist. Updat. 9:142–156 [DOI] [PubMed] [Google Scholar]
- 3. Bonomo R. A., Rudin S. D., Shlaes D. M. 1997. OHIO-1 β-lactamase mutants: Asp179Gly mutation confers resistance to ceftazidime. FEMS Microbiol. Lett. 152:275–278 [DOI] [PubMed] [Google Scholar]
- 4. Bush K., Jacoby G. 2003. Resistant TEM and SHV β-lactamases. The Lahey Clinic, Burlington, MA: http://www.lahey.org/studies/ [Google Scholar]
- 5. Crichlow G. V., et al. 1999. Structure of the extended-spectrum class C β-lactamase of Enterobacter cloacae GC1, a natural mutant with a tandem tripeptide insertion. Biochemistry 38:10256–10261 [DOI] [PubMed] [Google Scholar]
- 6. De Champs C., et al. 2004. Frequency and diversity of class A extended-spectrum β-lactamases in hospitals of the Auvergne, France: a 2 year prospective study. J. Antimicrob. Chemother. 54:634–639 [DOI] [PubMed] [Google Scholar]
- 7. Delmas J., et al. 2010. Structural insights into substrate recognition and product expulsion in CTX-M enzymes. J. Mol. Biol. 400:108–120 [DOI] [PubMed] [Google Scholar]
- 8. Emsley P., Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 9. Fisher J. F., Meroueh S. O., Mobashery S. 2005. Bacterial resistance to β-lactam antibiotics: compelling opportunism, compelling opportunity. Chem. Rev. 105:395–424 [DOI] [PubMed] [Google Scholar]
- 10. Giakkoupi P., Tzelepi E., Tassios P. T., Legakis N. J., Tzouvelekis L. S. 2000. Detrimental effect of the combination of R164S with G238S in TEM-1 β-lactamase on the extended-spectrum activity conferred by each single mutation. J. Antimicrob. Chemother. 45:101–104 [DOI] [PubMed] [Google Scholar]
- 11. Helfand M. S., et al. 2003. Understanding resistance to β-lactams and β-lactamase inhibitors in the SHV β-lactamase: lessons from the mutagenesis of Ser130. J. Biol. Chem. 278:52724–52729 [DOI] [PubMed] [Google Scholar]
- 12. Helfand M. S., Hujer A. M., Sonnichsen F. D., Bonomo R. A. 2002. Unexpected advanced generation cephalosporinase activity of the M69F variant of SHV β-lactamase. J. Biol. Chem. 277:47719–47723 [DOI] [PubMed] [Google Scholar]
- 13. Herzberg O., Kapadia G., Blanco B., Smith T. S., Coulson A. 1991. Structural basis for the inactivation of the P54 mutant of β-lactamase from Staphylococcus aureus PC1. Biochemistry 30:9503–9509 [DOI] [PubMed] [Google Scholar]
- 14. Hujer A. M., Hujer K. M., Bonomo R. A. 2001. Mutagenesis of amino acid residues in the SHV-1 β-lactamase: the premier role of Gly238Ser in penicillin and cephalosporin resistance. Biochim. Biophys. Acta 1547:37–50 [DOI] [PubMed] [Google Scholar]
- 15. Kurokawa H., et al. 2000. A new SHV-derived extended-spectrum β-lactamase (SHV-24) that hydrolyzes ceftazidime through a single-amino-acid substitution (D179G) in the omega-loop. Antimicrob. Agents Chemother. 44:1725–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuzin A. P., et al. 1999. Structure of the SHV-1 β-lactamase. Biochemistry 38:5720–5727 [DOI] [PubMed] [Google Scholar]
- 17. Majiduddin F. K., Palzkill T. 2003. An analysis of why highly similar enzymes evolve differently. Genetics 163:457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matagne A., Lamotte-Brasseur J., Frere J. M. 1998. Catalytic properties of class A β-lactamases: efficiency and diversity. Biochem. J. 330(Pt. 2):581–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nukaga M., et al. 2003. Inhibition of class A and class C β-lactamases by penems: crystallographic structures of a novel 1,4-thiazepine intermediate. Biochemistry 42:13152–13159 [DOI] [PubMed] [Google Scholar]
- 20. Otwinowski Z., Minor W. 1997. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276:307–326 [DOI] [PubMed] [Google Scholar]
- 21. Padayatti P. S., et al. 2004. Tazobactam forms a stoichiometric trans-enamine intermediate in the E166A variant of SHV-1 β-lactamase: 1.63 Å crystal structure. Biochemistry 43:843–848 [DOI] [PubMed] [Google Scholar]
- 22. Padayatti P. S., et al. 2006. Rational design of a β-lactamase inhibitor achieved via stabilization of the trans-enamine intermediate: 1.28 A crystal structure of wt SHV-1 complex with a penam sulfone. J. Am. Chem. Soc. 128:13235–13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Paterson D. L., Bonomo R. A. 2005. Extended-spectrum β-lactamases: a clinical update. Clin. Microbiol. Rev. 18:657–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rasheed J. K., et al. 1997. Evolution of extended-spectrum β-lactam resistance (SHV-8) in a strain of Escherichia coli during multiple episodes of bacteremia. Antimicrob. Agents Chemother. 41:647–653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schuttelkopf A. W., van Aalten D. M. 2004. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60:1355–1363 [DOI] [PubMed] [Google Scholar]
- 26. Wang X., Minasov G., Shoichet B. K. 2002. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J. Mol. Biol. 320:85–95 [DOI] [PubMed] [Google Scholar]
- 27. Winn M. D., Murshudov G. N., Papiz M. Z. 2003. Macromolecular TLS refinement in REFMAC at moderate resolutions. Methods Enzymol. 374:300–321 [DOI] [PubMed] [Google Scholar]