

Abstract
Mitochondrial dysfunction is one of the possible mechanisms by which azole resistance can occur in Candida glabrata. Cells with mitochondrial DNA deficiency (so-called “petite mutants”) upregulate ATP binding cassette (ABC) transporter genes and thus display increased resistance to azoles. Isolation of such C. glabrata mutants from patients receiving antifungal therapy or prophylaxis has been rarely reported. In this study, we characterized two sequential and related C. glabrata isolates recovered from the same patient undergoing azole therapy. The first isolate (BPY40) was azole susceptible (fluconazole MIC, 4 μg/ml), and the second (BPY41) was azole resistant (fluconazole MIC, >256 μg/ml). BPY41 exhibited mitochondrial dysfunction and upregulation of the ABC transporter genes C. glabrata CDR1 (CgCDR1), CgCDR2, and CgSNQ2. We next assessed whether mitochondrial dysfunction conferred a selective advantage during host infection by testing the virulence of BPY40 and BPY41 in mice. Surprisingly, even with in vitro growth deficiency compared to BPY40, BPY41 was more virulent (as judged by mortality and fungal tissue burden) than BPY40 in both systemic and vaginal murine infection models. The increased virulence of the petite mutant correlated with a drastic gain of fitness in mice compared to that of its parental isolate. To understand this unexpected feature, genome-wide changes in gene expression driven by the petite mutation were analyzed by use of microarrays during in vitro growth. Enrichment of specific biological processes (oxido-reductive metabolism and the stress response) was observed in BPY41, all of which was consistent with mitochondrial dysfunction. Finally, some genes involved in cell wall remodelling were upregulated in BPY41 compared to BPY40, which may partially explain the enhanced virulence of BPY41. In conclusion, this study shows for the first time that mitochondrial dysfunction selected in vivo under azole therapy, even if strongly affecting in vitro growth characteristics, can confer a selective advantage under host conditions, allowing the C. glabrata mutant to be more virulent than wild-type isolates.
INTRODUCTION
Candida spp. are the most common opportunistic fungal pathogens found in humans. Candida albicans is the most prevalent fungal pathogen in mucosal and systemic fungal infections. In addition to C. albicans, Candida glabrata is now emerging as an important agent of both mucosal and bloodstream infections. The prevalence of C. glabrata has increased in the last decade, and this species now ranks as the second or third most frequently isolated Candida species among all reported cases of candidiasis (30). C. glabrata infections are treated with various antifungal agents, including azoles, polyenes, and candins. However, C. glabrata differs from C. albicans in its intrinsic low azole susceptibility. Moreover, C. glabrata can develop azole resistance at a relatively high frequency (33). The major mechanism involved in azole resistance is based on the overexpression of ATP binding cassette (ABC) transporters (34). At least three transporters, encoded by C. glabrata CDR1 (CgCDR1), CgCDR2, and CgSNQ2, participate in the development of azole resistance (33, 34, 38). The upregulation of these transporters is due to gain-of-function (GOF) mutations in the transcriptional regulator encoded by CgPDR1 (40, 42). We recently showed the existence of a large variety of GOF mutations isolated from a collection of clinical isolates (14). Surprisingly, GOF mutations in CgPDR1 also conferred a selective advantage to these azole-resistant isolates in animal models, since they resulted in enhanced virulence and fitness compared to those of wild-type strains (14).
C. glabrata belongs to the group of hemiascomycetous yeast species, to which Saccharomyces cerevisiae also belongs (13). C. glabrata is haploid, nondimorphic, and nonmating, although it possesses cryptic mating loci (4, 25). C. glabrata shares with S. cerevisiae the ability to undergo loss of mitochondrial functions (33). These events are due to loss or deletion within the mitochondrial genome. The resulting phenotype is an absence of growth on nonfermentable carbon sources (such as glycerol) and a growth deficiency in media supplemented with glucose, which is also known as a “petite” phenotype (5, 6). Remarkably, loss of mitochondrial function in C. glabrata is associated with acquisition of azole resistance (33). Such mutants can be obtained in vitro in azole-containing media at high frequencies. Some reports document the recovery of petite mutants directly from patient samples (3). Azole resistance in these cases is also caused by the upregulation of ABC transporters mediated by CgPDR1; however, it is not dependent on the presence of GOF mutations in CgPDR1 (14). The exact regulatory mechanisms behind the upregulation of ABC transporters in petite mutants are still largely unresolved. Since we documented in a previous study that azole resistance could also influence the virulence of C. glabrata, the occurrence of azole resistance through mitochondrial dysfunction led us to address the relevance of this phenotype for virulence. In this report we show that a clinical C. glabrata petite mutant exhibiting growth deficiencies in vitro still exhibited higher virulence and fitness in murine infection models than its azole-susceptible and respiration-competent parent.
MATERIALS AND METHODS
Strains and growth media.
The C. glabrata strains used in this study are listed in Table 1. Yeasts were grown in complete YEPD medium (1% Bacto peptone [Difco Laboratories, Basel, Switzerland]), 0.5% yeast extract [Difco], and 2% glucose [Fluka, Buchs, Switzerland]). To prepare inocula for experimental infections, yeasts were grown in YEPD medium. When yeasts were grown on solid medium, 2% agar (Difco) was added.
Table 1.
Azole susceptibilities of the C. glabrata isolates investigated
| Isolate | Site of isolation | MIC (μg ml−1)a |
Reference | ||
|---|---|---|---|---|---|
| FLC | ITC | VOR | |||
| BPY40 | Blood | 4 | 0.25 | 0.13 | 28 |
| BPY41 | Blood | >256 | >16 | 4 | 28 |
| DSY4339 | —b | >256 | >16 | 4 | This study |
FLC, fluconazole; ITC, itraconazole; VOR, voriconazole. The MICs of these antifungal agents were determined by the broth microdilution method as described in NCCLS document M27-A3 (8).
In vitro-generated isolate.
Isolation of C. glabrata mitochondrial mutants.
The mitochondrial mutant DSY4339 was produced in vitro by exposure of BPY40 to ethidium bromide as previously described (33). Briefly, strain BPY40 was grown overnight in YEPD and diluted 100-fold in 2 ml of YEPD containing 20 μg/ml ethidium bromide. The culture was grown overnight at 30°C, and an aliquot was plated on YEPD agar to isolate single colonies. Colonies were then selected and tested for their growth phenotypes on glycerol (YEPG)- and glucose (YEPD)-containing media. Colonies growing on glucose but not on glycerol medium were analyzed for the presence of mitochondrial DNA by staining with SYTO18 (Molecular Probes) as described previously (14).
Mouse survival and tissue burdens.
Female BALB/c mice (20 to 25 g) were initially purchased from Harlan Italy S.r.l (San Pietro al Natisone, Udine, Italy) and inbred in-house. The mice were housed in filter-top cages with free access to food and water and were used for all in vivo experiments with the approval of the institutional Animal Use Committee (Università Cattolica del Sacro Cuore, Rome, Italy). To establish systemic C. glabrata infection, mice were injected with saline suspensions of the C. glabrata strains (each in a volume of 200 μl) into their lateral tail veins.
In virulence studies, a group of 10 immunosuppressed mice was established for each yeast strain, as described earlier (20), with inocula based on 50% lethal doses (LD50s) (see Table 2). Mice were rendered neutropenic by intraperitoneal administration of cyclophosphamide (200 mg kg−1 of body weight per day) at 3 days before challenge and on the day of infection. For the LD50 determination, mice were made neutropenic (14) and then injected with serially 5-fold-diluted cell suspensions ranging from 5 × 108 to 1 × 106 cells per mouse. Each cell suspension was injected into 10 mice. LD50s were calculated by the Knudsen-Curtis method (21). Based on these results, neutropenic mice were injected with 5 × 107 CFU (which was twice the LD50 of BPY41) of each investigated strain. Mice were monitored by twice-daily inspections, with disease severity grades based on weight loss and estimation of mobility by the capacity of the mouse to twist when gripped by the tail. When one criterion was met (≥30% weight loss or severe reduction of mobility), the mice were sacrificed by use of CO2 inhalation.
Table 2.
LD50s of the C. glabrata isolates investigated
| Isolate | LD50 (CFU) in: |
|
|---|---|---|
| Immunocompetent model | Immunosuppressed model | |
| BPY40 | 7.2 × 107 | 6.8 × 107 |
| BPY41 | 3.45 × 107 | 2.3 × 107 |
| DSY4339 | 2.81 × 108 | 1.65 × 108 |
For tissue burden experiments, immunocompetent mice were inoculated with 4 × 107 CFU (20). After 7 days, mice were sacrificed by CO2 inhalation and target organs (liver, spleen, and kidney) were excised aseptically, weighted individually, and homogenized in sterile saline by using a Stomacher 80 device (Pbi International, Milan, Italy) for 120 s at high speed. Organ homogenates were diluted and plated onto YPD. Colonies were counted after 2 days of incubation at 30°C, and the numbers of CFU g−1 of organ were calculated.
CFU counts were analyzed with nonparametric Wilcoxon rank sum tests, while mean survival times were compared among groups by using the log rank test. A P value of less than 0.05 was considered significant.
Rat vaginal mucosa infection.
For the experimental vaginal infections, a previously described rat vaginal model was used (9, 10). Briefly, groups of six oophorectomized female Wistar rats (80 to 100 g; Charles River Calco, Italy) were injected subcutaneously with 0.5 mg of estradiol benzoate (Estradiolo; Amsa Farmaceutici S.r.l., Rome, Italy). Six days after the first estradiol dose, the animals were inoculated intravaginally with 107 yeast cells of each C. glabrata strain in 0.1 ml of saline solution. The inoculum was dispensed into the vaginal cavity through a syringe equipped with a multipurpose calibrated tip (Combitip; Pbi International, Milan, Italy). The number of cells in the vaginal fluid was counted by culturing 100 μl of vaginal samples using a calibrated plastic loop (Disponoic; Pbi International, Milan, Italy). The vaginal samples were obtained by washing the vaginal cavity four times with 100 μl of sterile saline solution and gentle aspiration. CFU were counted after incubation at 28°C for 48 h on Sabouraud agar containing chloramphenicol (50 μg ml−1) as described previously (10). The rat was considered colonized when at least 1 CFU was present in the vaginal lavage fluid.
Measurement of BPY40 and BPY41 fitness in vitro and in vivo.
Strains BPY40 and BPY41 were grown overnight in YEPD and diluted to a density of 5 × 106 cells ml−1. Equal volumes of each culture were mixed together, and cultures were grown under constant agitation at 30°C for 24 h. Growth of BPY40, BPY41, and the coculture was determined at 2, 4, 8, and 24 h by measuring the absorbance at 540 nm and by plating diluted samples of the cultures onto YEPD agar plates. Since BPY41 is able to grow on high concentrations of fluconazole, while BPY40 is not, the proportions of the two strains in the coculture were determined by replicating colonies onto YEPD agar containing 30 μg ml−1 of fluconazole. After incubation at 30°C for 48 h, colonies on YEPD plates and on plates containing fluconazole were counted.
For in vivo fitness assays, cultures of strains BPY40 and BPY41 were diluted to a density of 4 × 107 CFU, and these suspensions were used to infect three groups of immunocompetent mice (four per group). One group of mice was infected with BPY40, another group was infected with BPY41, and the third group was infected with both strains mixed at a ratio of 1:1. At 2, 4, and 7 days postinfection, mice were sacrificed and kidneys homogenates were obtained (see above). Diluted samples from these homogenates were plated onto YEPD. Colonies grown after 2 days of incubation at 30°C were replicated onto YEPD plates containing fluconazole (30 μg ml−1) to determine the proportions of the two strains as described above.
Construction of C. glabrata microarrays.
The nucleotide sequences of the 5,283 C. glabrata open reading frame (ORFs) and the mitochondrial genome were downloaded from the Génolevure Consortium (http://www.genolevures.org/). Following the Agilent eArray Design guidelines, two separate probe sets for each ORF were designed, each consisting of 60-base oligonucleotides. The probe selection was performed using the GE Probe Design Tool (Agilent Technologies). Probes were filtered according to their base composition and distribution, cross-hybridization potential, and melting temperature to yield two probes sets, each representing 5,210 nuclear and 6 mitochondrial ORFs. These probes cover more than 98% of the nuclear genome and represent six of the eight mitochondrial protein-encoding genes. For quality control and normalization purposes, 103 C. glabrata probes were selected randomly and spotted 20 times throughout each array in addition to standard Agilent controls, including spike controls for intra- and interarray normalizations. C. glabrata custom arrays were manufactured by in situ synthesis in the 8 × 15K format by Agilent Technologies.
cRNA synthesis, one-color labeling, and C. glabrata array hybridization.
Sample preparation was performed for three biological triplicates with separate cultures of BPY40 and BPY41. Total RNA was extracted from log-phase cultures in liquid YEPD medium at 30°C as previously described (33). Briefly, after centrifugation of 5 ml culture (corresponding to 108 cells), the yeast cell pellet was mixed with 0.3 g of glass beads, 300 μl of RNA extraction buffer (0.1 M Tris-HCl at pH 7.5, 0.1 M LiCl, 10 mM EDTA, 0.5% SDS), and 300 μl of phenol-chloroform-isoamyl alcohol (24:24:1). After 1 min of vortexing in a bead beater (Fastprep-24 instrument; MP Biomedicals Switzerland, Zürich, Switzerland), the aqueous phase was reextracted with phenol-chloroform-isoamyl alcohol, and RNA was precipitated with 600 μl of ethanol at −20°C for 1 h. The RNA pellet was resuspended in 50 μl of diethyl pyrocarbonate-treated H2O. The integrity of the input template RNA was determined prior to labeling/amplification, using the Agilent RNA 6000 Nano LabChip kit and 2100 bioanalyzer (Agilent Technologies). Agilent's One-Color Quick Amp labeling kit (Agilent Technologies) was used to generate fluorescent cRNA according to the manufacturer's instructions. Briefly, 1 μg of total RNA from each sample was used, to which a spike mix and T7 promoter primers were added, both of which are provided by the manufacturer. cDNA synthesis was promoted by Moloney murine leukemia virus (MMLV) reverse transcriptase (RT) in the presence of deoxynucleoside triphosphates (dNTPs) and RNaseOUT. Next, cRNA was produced from this first reaction with T7 RNA polymerase, which simultaneously amplifies target material and incorporates cyanine 3 (Cy3)-labeled CTP. The obtained labeled cRNAs were purified with the RNeasy minikit (Qiagen, Hilden, Germany) and quantified using a NanoDrop ND-1000 UV-visible spectrophotometer. A total of 600 ng of Cy3-labeled cRNAs from each sample in a volume of 40 μl was fragmented and hybridized for 17 h at 65°C to each array using the Gene Expression Hybridization kit (Agilent Technologies) according to the manufacturer's instructions. We used a Microarray Hybridization Chamber kit (Agilent Technologies) and a gasket slide with a format of eight microarrays per slide for sample hybridization to separate each sample in specific subarrays of the 8 × 15K format.
Microarray data analysis.
Slides were washed and processed according to the Agilent 60-mer Oligo Microarray Processing protocol and scanned on an Agilent microarray scanner G2565BA (Agilent Technologies). Data were extracted from the images with Feature Extraction (FE) software (Agilent Technologies). Each slide was processed with spike quality controls to establish the dynamic range of signals which fitted a r2 value of 1.0. FE software flags outlier features (0.03% of signals for BPY40 and BYP41 cRNA hybridizations) and detects and removes spatial gradients and local backgrounds. Data were normalized using a combined rank consistency filtering with LOWESS intensity normalization.
The gene expression values obtained with FE software were imported into GeneSpring 10.0.2 software (Agilent Technologies) for preprocessing and data analysis. For interarray comparisons, a linear scaling of the data was performed using the 75th percentile signal value of all noncontrol probes on the microarray to normalize one-color signal values. The expression of each gene was normalized by its median expression across all samples. Genes were included in the final data set if their expression changed by at least 2-fold between strain BPY41 and BPY40 in at least two independent experiments. A corrected P value of <0.05 was chosen as the cutoff for significance.
Use of bioinformatic tools.
The analysis of the consensus pattern on C. glabrata promoters (−800 to −1) was performed using the Regulatory Sequence Analysis Tools (RSAT) (http://rsat.ulb.ac.be/rsat/index.html) and implemented with the pattern discovery tool (oligo-analysis). The settings were those supplied by default by the tool provider. The position-specific scoring matrix (PSSM) consensus matrices were converted using statistical parameters to consensus patterns and viewed via Weblogo (37).
qRT-PCR.
Total RNA was extracted from log-phase cultures with an RNeasy Protect minikit (Qiagen, Hilden, Germany) by a process involving mechanical disruption of the cells with glass beads and an RNase-free DNase treatment step as previously described (35). Expression of the CgCDR1, CgCDR2, and CgSNQ2 genes was quantitatively assessed with quantitative real-time RT-PCR (qRT-PCR) in an i-Cycler iQ system (Bio-Rad). All primers and probes (35) were designed with Beacon Designer 2 (version 2.06) software (Premier Biosoft International, Palo Alto, CA) and synthesized by MWG Biotech (Ebersberg, Germany). qRT-PCRs were carried out as previously described (35). Each reaction was run in triplicate on three separate occasions. For relative quantification of the target genes, each set of primer pairs and the TaqMan probes were used in combination with the primers and probe specific for the CgACT1 reference gene in separate reactions (38). TaqMan probes were necessary for quantification of CgCDR1, CgCDR2, and CgSNQ2 expression to avoid unspecific signals originating from similar genes of the ABC transporter family. CgACT1 has been used as a housekeeping gene for normalization purposes in previous studies (14) and by other investigators (2, 26). Changes (n-fold) in gene expression relative to that of BPY40 (azole-susceptible control isolate) were determined from CgACT1-normalized expression levels. A 2-fold increase in the expression level of each gene was arbitrarily considered significant (38). CgPDR1 and CgYPS expression levels were determined by qRT-PCR without the use of TaqMan probes in a StepOne real-time PCR system (Applied Biosystems) (for practical purposes) using the Mesa Blue qPCR Mastermix Plus for Sybr assay (Eurogentec). Each reaction was run in triplicate on three separate occasions. The primers used for CgPDR1, CgACT1, and CgYPS quantification were previously described (14, 20). CgPDR1 and CgYPS expression levels were normalized by CgACT1 expression.
Microarray data accession numbers.
Microarray data are available at the NCBI GEO microarray repository. The GEO accession number for the C. glabrata Agilent array is GPL10713, and the accession number for the data is GSE23826.
RESULTS
Characteristics of BPY41, a clinical petite mutant isolate.
BPY41 was described by Posteraro et al. (28) and was collected with other C. glabrata isolates obtained during a hospital survey between 2000 and 2003. BPY41 had an azole-susceptible parent (BPY40) isolated before initiation of treatment. As published by Posteraro et al. (28), the time elapsed between isolation of BPY40 and BPY41 was about 6 months, and the cumulative fluconazole dose was 14 g. BPY40 and BPY41, described as isolates T0 and T1 in the original publication (28), respectively, belong to a series of isolates that were obtained from two different body sites (blood and vagina) at different times. The fungemic isolates BPY40 and BPY41 were indistinguishable from each other and also from the vaginal isolates by genotyping methods, including the use of Cg6/Cg12 probes and multilocus sequence typing (MLST) (28). Antifungal MIC values for BPY40 and BPY41, obtained from the work of Posteraro et al. (28), are listed in Table 1 and indicate that BPY41 exhibits cross-resistance to all azoles tested (fluconazole, itraconazole, and voriconazole). CgCDR1, CgCDR2, CgSNQ2, and CgPDR1 are overexpressed in BPY41, which is consistent with the azole resistance phenotype (14, 35). However, no CgPDR1 mutation was detected in BPY41 (14). In contrast to BPY40, BPY41 exhibited typical features of a petite phenotype, including the absence of growth in glycerol-containing medium and absence of mitochondrial staining with SYTO18 (14). As is typical of mitochondrial dysfunction, oxygen consumption measured by oxygraphy was strongly reduced in BPY41 compared to BPY40 (see Fig. S1 in the supplemental material).
Comparison of BPY40 with BPY41 for virulence and fitness in mouse infection models.
In a previous study, we showed that azole resistance mediated by CgPDR1 GOF mutations in C. glabrata correlates with increased fitness in vivo and enhanced virulence compared to those of azole-susceptible matched isolates (14). The petite phenotype of BPY41, even if it affects in vitro growth in media containing glucose, may also result in enhanced virulence since it is associated with azole resistance. To test this hypothesis, we inoculated mice intravenously with BPY40 and BPY41 and recorded survival and tissue burden over time. Surprisingly, BPY41 showed significantly increased virulence compared to the azole-susceptible isolate BPY40 (Fig. 1). Determination of tissue burdens at day 7 postinfection confirmed that CFU counts in tissues (kidneys, spleen, and liver) were significantly higher in mice infected with BPY41 than in those infected with BPY40 (Fig. 2). This observation is in disagreement with a previous study showing that laboratory-derived petite mutants were less virulent than their parental respiration-competent strain (7). We therefore included in this analysis another mitochondrion-deficient isolate which was generated from BPY40 in vitro under laboratory conditions. This isolate, DSY4339, had the typical features of mitochondrial dysfunction, i.e., absence of growth in nonfermentable carbon sources and decreased oxygen consumption compared to BPY40 (see Fig. S1 in the supplemental material). Consistent with the absence of mitochondrial structure staining by SYTO18 as described for BPY41 (data not shown) (14), no mitochondrial DNA could be recovered from DSY4339 or from BYP41 (see Fig. S2 in the supplemental material). In contrast to the case for BPY41, the virulence of DSY4339 was not significantly different from that of BPY40. These data are further supported by the 10-fold-higher LD50 of DSY4339 than of BPY41 in immunocompetent and immunodeficient mouse models of infection (Table 2). Tissue burdens were also the lowest for DSY4339, which is consistent with the low virulence of this strain (Fig. 2). These results therefore suggest that mitochondrial dysfunction can positively or negatively affect virulence, depending on the origin. However, we cannot exclude the possibility that the in vitro- or in vivo-generated mitochondrial mutants also contain nuclear mutations that decrease or increase their virulence, respectively. The occurrence of such isolates in the context of a patient is under host-selective pressure, which was not the case in the laboratory-derived isolate DSY4339, and this may explain the virulence difference between the two isolates.
Fig. 1.

Survival curves for neutropenic mice infected with BPY40 and mitochondrial mutants BPY41 and DSY4339. Statistical differences were determined using the Mantel-Cox log rank test (Prism 5.0) by comparing survival curves of mice infected with BPY40 and with other strains as indicated. Asterisks indicate statistically significant differences (**, P < 0.01; NS: not significant).
Fig. 2.

Fungal tissue burdens in immunocompetent BALB/c mice infected intravenously with 4 × 107 viable cells of C. glabrata strains. Mice were sacrificed at day 7 postinfection, and results represent values recorded separately for each of the 10 mice. Geometric means are indicated by horizontal bars, and asterisks indicate statistically significant differences (*, P < 0.05; **, P < 0.01; ***, P < 0.001). NS, not significant (P > 0.05). The origin of each strain is indicated. Prism 5.0 was used for statistical analysis, and data were processed with nonparametric Wilcoxon rank sum tests. Comparisons are indicated in the figure and associate selected data points.
Although BPY40 and BPY41 were isolated from blood, the investigated patient had almost simultaneously been infected in the vagina by C. glabrata isolates that were genotypically identical to the fungemic ones (19). Therefore, we tested virulence of the two strains in a rat model of vaginal infection. As observed in Fig. 3, BPY40 was cleared from vaginal mucosal surfaces faster than BPY41. This indicates that BPY41 could colonize mucosal surfaces more efficiently than BPY40. Compared with the azole-susceptible isolate, increased colonization by BPY41 may cause more damage on mucosal surfaces and therefore increased virulence. Our current data cannot confirm this hypothesis. Taken together, the above-mentioned results therefore suggest that azole resistance, even if resulting in loss of mitochondrial function, can contribute in specific strains to enhanced virulence in different animal models. However, the underlying conditions that accompanied the mitochondrial defects play a critical role in this phenotype.
Fig. 3.

Vaginal rat model of infection. BPY40 and BPY41 were inoculated intravaginally at 107 yeast cells in 0.1 ml of saline solution for each C. glabrata strain tested. Each isolate was inoculated in five animals. The inoculum was dispensed into the vaginal cavity, and the number of cells in the vaginal fluid was counted. The rat was considered colonized when at least 1 CFU was present in the vaginal lavage fluid. CFU are means of three measurements. Asterisks indicate a statistically significant difference between CFU counts from each infected animal (***, P < 0.001).
We next attempted to address whether enhanced virulence of BPY41 was associated with a gain in fitness in the systemic animal model. BPY40 and BPY41 were therefore injected simultaneously in mice and also cultivated in vitro in glucose-containing medium. The evolution of the population ratios of the two strains was recorded over time under both conditions (Fig. 4A and B). Remarkably, even though the population of BPY41 decreased compared to that of BPY40 over time in vitro (BPY41 made up approximately 20% of the total population [Fig. 4A]), this tendency was reversed in mice. At 7 days after infection, the percentage of BPY41 in the in vivo population increased from 50% to approximately 80%, thus indicating a 4-fold difference of BPY41 relative to the in vitro population (Fig. 4B). These results demonstrate that BPY41 gained fitness in mice even though respiratory capacities were strongly compromised in this strain background.
Fig. 4.
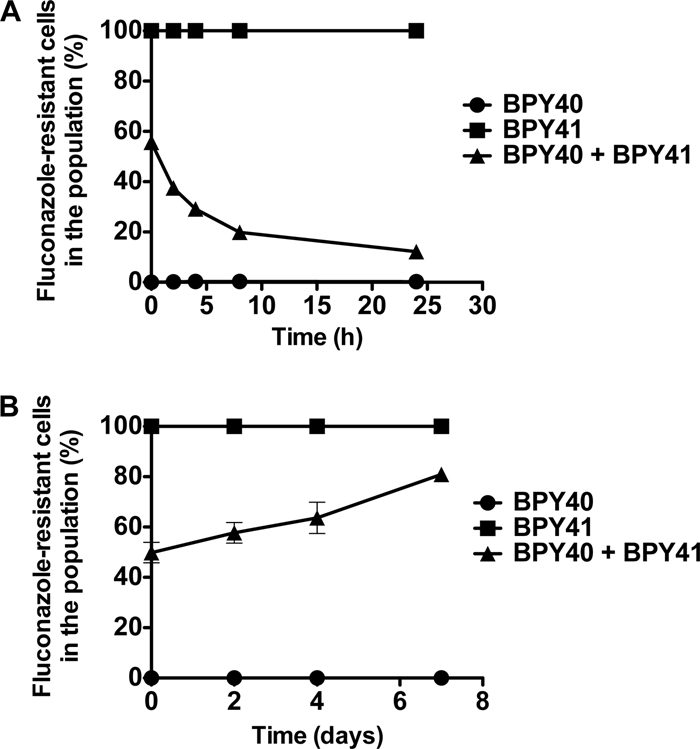
Fitness assays of azole-resistant and azole-susceptible isolates. (A) In vitro fitness assays. Strains BPY40 and BPY41 were inoculated in YEPD in single or mixed (1:1) cultures. Aliquots were taken from each culture in triplicate, and population ratios were calculated from the proportions of azole-susceptible and azole-resistant colonies plated onto YEPD agar as described in Materials and Methods. (B) In vivo fitness assays. Strains BPY40 and BPY41 were inoculated intravenously as single or mixed (1:1) cultures to three groups of immunocompetent mice as described in Materials and Methods. Kidneys were taken at given time points from sacrificed mice, and population ratios were measured as described above.
Expression profiles of BPY40 and BPY41.
To evaluate the impact of the petite mutation on the C. glabrata transcriptome, genome-wide transcriptional analysis was performed. For this purpose, C. glabrata custom arrays were designed from the Agilent eArray platform to determine changes in the expression profile of BPY41 relative to that of BPY40. These arrays contained two distinct 60-mer probe sets covering more than 98% of the C. glabrata nuclear genome as well as six of the eight mitochondrial protein-encoding genes. We chose here a one-color (Cy3) labeling since we were interested in comparing not only signals obtained from BPY41 and BPY40 but also those from other C. glabrata strains, which is reported in a separate study (15). Normalized expression profiles from BYP40 and BPY41 were compared to each other, and only genes with a P value of <0.05 were selected for further analysis.
Microarray experiments revealed that 379 genes were differentially regulated at least 2-fold between BPY40 and BPY41. Among them, 285 genes were upregulated and 94 downregulated. These genes are listed in Tables S1 and S2 in the supplemental material, respectively, and are grouped by probable function and ordered by expression level. Taking a regulation threshold at a value of ≥2, the microarray data were in agreement with separate verifications by qRT-PCR of CgCDR1 (CAGL0M01760g), CgCDR2 (CAGL0F02717g), CgSNQ2 (CAGL0I04862g), and CgPDR1 (CAGL0A00451g) (Fig. 5A). These genes were chosen since they are critical for the development of azole resistance in this yeast species (23, 34, 38). We also validated the microarray data by verifying the expression of genes encoding glycosylphosphatidylinositol (GPI)-anchored proteases (yapsins), some of which were regulated in BPY41 compared to BPY40 (Fig. 5B). The yapsin gene family (CgYPS1 to -11) is required for cell wall integrity, adherence to mammalian cells, survival in macrophages, and virulence (20). Except for YPS8, where changes in relative gene expression were 3-fold higher for microarray data than for qRT-PCR, the qRT-PCR data for these genes showed good agreement with relative expression data obtained with microarrays (Fig. 5B).
Fig. 5.

Relative gene expression levels of selected genes in C. glabrata isolates. (A) Expression of genes involved in azole resistance in BPY41 and DSY4339 as measured by qRT-PCR. Values are averages from three independent experiments and represent increase in gene expression relative to that in the azole-susceptible strain BPY40 (set at 1.00). qRT-PCR was performed as described by Sanguinetti et al. (35) for CgCDR1, CgCDR2, CgSNQ2 and as described by Ferrari et al. (14) for CgPDR1. Relative expression of genes in BPY41 obtained by microarray analysis is plotted for comparison. (B) CgYPS gene expression in BPY41 and DSY4339 as measured by qRT-PCR. Values are averages from three independent experiments and represent increase in gene expression relative to that in the azole-susceptible strain BPY40 (set at 1.00). No qRT-PCR data were available for CgYPS1 and CgYPS7. Relative expression of CgYPS genes in BPY41 obtained by microarray analysis is plotted for comparison.
Since C. glabrata and S. cerevisiae are genetically closely related, S. cerevisiae gene names and annotations were used to categorize the biological functions and processes of the corresponding C. glabrata genes, with the exception of known C. glabrata genes that are named with the Cg prefix. Several processes were enriched in genes upregulated in BPY41 compared to BPY40, among which are those related to the mitochondrial deficiency of BPY41 (carbohydrate metabolism, respiratory chain, and oxidation-reduction), those involved in the stress response, and also those involved in the sporulation process, even though this last process has not been yet identified as an active operating process in C. glabrata (29). As expected, genes associated with multidrug resistance and transport of antifungal agents, such as CgCDR1, CgCDR2, and CgSNQ2, were among the most upregulated genes in BPY41. Other ABC transporter genes, including YOR1 (CAGL0G00242g), YBT1 (CAGL0C03289g), and YCF1 (CAGL0L06402g), whose functions are not directly associated with antifungal resistance are among the upregulated genes belonging to the small-molecule transport process. A substantial number of genes involved in cell wall biogenesis/cell wall maintenance were also upregulated in BPY41 compared to BPY40. Of special interest are GPI-anchored aspartic proteases (CgYps3, CgYps8, CgYps9, and CgYps10) belonging to a family of proteins involved in the pathogenesis of C. glabrata, several Mnt3 (mannosyl transferase) homologues, and other cell surface proteins (Aga1 and Muc1). The upregulation of the genes encoding these proteins could have an impact on the interactions with the host. It is interesting to observe that several genes involved in DNA damage (YIM1, MEC3, and MAG1) and repair (MND1, RAD7, and HSM3) are upregulated in BPY41. Because mitochondrial dysfunction can alter oxidative metabolism and generate toxic metabolites, it is possible that DNA stability could be affected and thus the upregulation of these genes could reflect a protective effect. Interestingly, a recent study established whole-genome connections with mitochondria and identified the nuclear DNA repair machinery as being dependent on mitochondrial functions (27). Lastly, it is interesting to observe that besides the upregulation of CgPDR1, several other transcription factors are also upregulated in BPY41. Among them, both ROX1 and HAP1 control genes in a heme-dependent manner, and their upregulation could be a consequence of compensatory mechanisms due to mitochondrial dysfunction. Others, such as HAC1, encoding a basic leucine zipper (bZIP) transcription factor which participates to the unfolded protein response (UPR) pathway (44), and RPN4, which stimulates proteasome genes (17), reflect that BPY41 is under stress conditions. RLM1, which is involved in cell wall integrity (18), is upregulated, which is consistent with the upregulation of cell wall genes in BPY41.
Consistent with the petite phenotype in BPY41, the genes mostly downregulated in BPY41 compared to BPY40 were five mitochondrial protein-encoding genes (ATP6, OLI1 [or ATP9], COX1, COX2, and COX6). This result could be expected because of absence of mitochondrial DNA in BPY41. In addition to these genes, nuclear genes encoding cytochrome c oxidase (COX5B and COX13) and ATP synthase (ATP1 and ATP6) subunits were also downregulated (see Table S2 in the supplemental material). These genes fall into processes related to the respiratory electron transport chain and are specifically enriched in BPY41, which again could be expected from this respiratory-deficient isolate. Interestingly, several downregulated genes are enriched in the process of de novo purine nucleotide biosynthesis (ADE1, ADE5,7, ADE6, ADE12, ADE13, MTD1, and HPT1). These genes, as well as other genes downregulated in BPY41, including HIS4, SHM2, GCV1, and GCV2, are known to be regulated by the transcription factors Bas1 and Pho2 in S. cerevisiae (32). The downregulation of this pathway has implications in the formation of AMP and GMP, which themselves provide purine bases for DNA and RNA, as well as for a number of essential coenzymes (NAD, NADP, flavin adenine dinucleotide [FAD], and coenzyme A) and signaling molecules. Interestingly, FCY2, which encodes a purine permease, and HPT1, catalyzing the formation of IMP and GMP (belonging to the purine salvage pathway), are also downregulated in BPY41 compared to BPY40. Generally, these genes are feedback regulated by ATP/ADP levels (32). Probably the loss of mitochondrial function affects the ATP/ADP ratios, with associated regulatory effects.
Since coregulated genes may be controlled by the occurrence of similar cis-acting motifs which serve as recognition sites for specific transcription factors, we tested whether common regulatory motifs could be found in the genes commonly up- and downregulated in our strains. We used the Regulatory Sequence Analysis Tools (RSAT) (http://rsat.ulb.ac.be/rsat/index.html) and implemented a pattern discovery tool (oligo-analysis) for the occurrence of putative regulatory elements in all C. glabrata promoters of genes up- and downregulated in BPY41 (see details in Materials and Methods). When all promoters of upregulated genes were inspected, three major consensus patterns were observed (see Fig. S3 in the supplemental material), among which one ([T/G]CCACGG[A/G] resembled the PDRE sequence of PDR1 in S. cerevisiae (TCC[A/G][C/T]G[G/C][A/G]) (12). This consensus occurred in 8.8% of the upregulated genes and in 50% of the 20 most upregulated genes. The same consensus was absent, except for a single gene (CAGL0A01199g, DIP5 [dicarboxylic amino acid permease]) in the downregulated genes. These data therefore strongly suggest that CgPDR1 in C. glabrata has an important role in the upregulation of genes in a situation of mitochondrial deficiency. These data also support the results of other studies highlighting the role of this transcription factor under drug-induced conditions or in the presence of GOF mutations (14, 40, 42).
The second and third enriched consensus sequences, [A/T][A/G]GGGG, are similar to the “canonical” stress response elements (SRE), CCCCT and AGGGG, that function in both orientations and are bound by the transcription factors Msn2 and Msn4 in S. cerevisiae (see Fig. S3 in the supplemental material). Interestingly, a total of 20 genes that are upregulated in BPY41 are known to be regulated by the general environmental stress response in both C. glabrata and S. cerevisiae (24, 31). These genes include, for example, HSP12 and HSP42 (heat shock genes), TRX2 (encoding a thioredoxin isoenzyme protecting cells against oxidative and reductive stress), NHT1 (encoding a trehalase required for thermotolerance and for resistance to other cellular stresses), and several genes (GSY2, SGA1, and GDB1) involved in glycogen metabolism, which itself is involved in the stress response (43). The presence of the SRE consensus in genes upregulated in BPY41 is consistent with their enrichment in the stress response process, which was also revealed in transcriptional analysis of a petite mutant of S. cerevisiae (39).
Finally, our transcript profile analysis shows that a group of genes encoding GPI-anchored proteases (yapsins) were upregulated in BPY41 (Fig. 5B). The yapsin gene family (CgYPS1 to -11) is required for cell wall integrity, adherence to mammalian cells, survival in macrophages, and virulence (20).
DISCUSSION
Here we analyzed a pair of C. glabrata isolates, one of which is azole resistant with mitochondrial defects. We showed previously that azole resistance in C. glabrata obtained by a GOF mutation in CgPDR1 was associated with enhanced virulence in animal models (14). We demonstrate here that azole resistance obtained by mitochondrial dysfunction (absence of mitochondrial DNA in BPY41) without the need for CgPDR1 mutations is also coupled with increased virulence in the same animal model. The mitochondrial mutant BPY41, although not able to assimilate nonfermentable carbon sources formed from glucose in vitro, is still more virulent in animal models of infection (intravenous infection in mice and vaginal infection in rats) than its respiration-competent parent, BPY40. These results were confirmed by tissue burden analysis, where the higher CFU counts were associated with enhanced virulence. Moreover, the mitochondrial defect did not decrease but even increased the fitness of the azole-resistant isolate in vivo. These results were surprising, since it has been reported that mitochondrial dysfunction was associated with a strong loss of virulence in the systemic infection model in mice (7). The reasons for this discrepancy could be that different experimental conditions (for example, inoculum size and fungal burden sampling timing), different strains, and different mice were used in the two studies. We believe that the conditions that determined the occurrence of mitochondrial dysfunction had a major impact on the animal experimentation. In the study published by Brun et al. (7), mitochondrial mutants were obtained by ethidium bromide mutagenesis, which is a classical method to obtain such mutants at a high rate. In our study, we have obtained two types of mitochondrial mutants from the same parent (BPY40); one originated from a patient sample (BPY41) and the other from ethidium bromide mutagenesis (DSY4339). Both mitochondrial mutants lacked mitochondrial DNA and were azole resistant. They exhibited high expression of CgPDR1 target genes, although the relative expression levels were lower in the in vitro-generated petite mutant DSY4339. The two isolates showed similar growth kinetics in vitro (see Fig. S4 in the supplemental material). However, the in vitro-generated mutant was less virulent than both BPY40 and BPY41, which could not be explained simply by differences in growth kinetics. This result therefore recapitulates data obtained by Brun et al. (7) with the in vitro-generated mutant and therefore may provide an explanation for the above-mentioned discrepancies. Clearly there are some differences between the two mitochondrial mutants. It is possible that the in vivo-generated mutant underwent secondary compensatory mutations not necessary for the in vitro-generated mutant. Such compensatory events are known in other bacterial pathogens (1, 22). Until now few clinical mitochondrial mutants were reported among azole-resistant C. glabrata isolates (3, 14), which therefore challenges the clinical significance of mitochondrial dysfunction in this yeast species. While it is likely that mitochondrial mutants will arise during azole therapy, the sampling procedure for further analysis in clinical laboratories may be not sensitive enough to detect such mitochondrial mutants. In systemic infections, blood cultures are often the only source for further laboratory analysis, and this medium might not be optimal for the growth of mitochondrial mutants compared to respiratory-competent isolates and especially when mixed infections occur. In support of this hypothesis, we have shown here that the in vitro fitness of BPY41 is decreased compared to that of BYP40 and that during prolonged in vitro growth, the population of mitochondrial mutants tends to disappear (Fig. 4).
Transcript profile analysis of the clinical isolates by use of microarrays confirmed the mitochondrial deficiency of BPY41, given the high number of genes regulated in BPY41 that are related to energy metabolism. Compared to microarray data for “petite” mitochondrial mutants in S. cerevisiae (39), a striking degree of overlap was observed for genes upregulated in both species compared to those in a respiration-competent parent. These genes fall mainly into the categories of “mitochondrion organization and cellular respiration” and “amino acid and carbohydrate metabolism” (see Table S1 in the supplemental material).
The microarray data obtained here could also be compared with a separate study where an azole-resistant isolate (F15) obtained by direct selection on a fluconazole-containing medium was analyzed with an azole-susceptible parent (42). In their analysis, Vermitsky et al. (42) identified 78 and 31 genes that were up- and downregulated by more than 2-fold, respectively, compared to the wild type. These numbers are different from those obtained with BPY40 and BPY41 in the present study (285 and 94 genes upregulated and downregulated in BPY41 compared to BPY40, respectively). As summarized in Table S1 in the supplemental material, 48 genes out of the 78 upregulated genes listed by Vermitsky et al. (42) were found in both F15 and BPY41. Genes overlapping in the two studies belonged mainly to the categories of “small-molecule transport,” “stress response,” “DNA replication and damage response,” and “lipid, fatty acid, and sterol metabolism.” Strikingly, no gene in the category of “mitochondrion organization and cellular respiration” was found in common between F15 and BPY41, which is consistent with the major difference in mitochondrial function between the two isolates. Moreover, our study identified the entire set of genes within the category of “cell surface and cell wall” which were upregulated in BPY41 and not in F15. On the other hand, only six genes were commonly downregulated in both isolates (see Table S1 in the supplemental material), which may also reflect the impact of the mitochondrial defect in BPY41 on gene regulation.
Interestingly, we have observed that several CgYPS genes were upregulated in the mitochondrial mutant. The yapsin gene family (CgYPS1 to -11) is required for cell wall integrity, adherence to mammalian cells, survival in macrophages, and virulence (20). Within the yapsin family, CgYps1 and CgYps7 were shown to play major roles in these phenotypes. Interestingly, yapsin proteases are able to process a cell wall adhesin important for C. glabrata virulence, Epa1, which can be released from the cell wall and may therefore have an indirect effect on this important mediator of virulence (20). In our work, only CgYPS3, CgYPS8, CgYPS9, and CgYPS10 were upregulated, while CgYPS1 and CgYPS7 did not reach the upregulation threshold. The indirect consequence of the CgYPS gene expression profile will be that the contribution of these genes in virulence could be rather modest. Moreover, we tested the expression of the CgYPS genes in DSY4339. We hypothesized that CgYPS gene expression could be different in this strain, thus contributing to decreased virulence compared to BPY40 and BPY41. Figure 5B shows that CgYPS gene expression is similar in BPY41 and DSY4339, and thus yapsin genes (except CgYPS1 and CgYPS7) could not be responsible for the virulence difference. However, because yapsins were shown to process cell wall proteins (16, 20) and some of them are upregulated in BPY41, the expression profile of this gene family could have still a positive influence on the virulence of C. glabrata. In addition, other genes involved in cell wall biogenesis/cell wall maintenance were upregulated in BPY41, among them MNT3 (encoding mannosyl transferase) homologues and other cell surface genes (AGA1 and MUC1). The upregulation of these genes could have an impact on the interactions with the host. However, other genes might be critical in the association between azole resistance and enhanced virulence. For example, a parallel study undertaken in our laboratory has revealed that two highly upregulated genes in BPY41 (the ABC transporter gene CgCDR1 and CAGL0M12947g, a mitochondrial gene with still-unknown function) play an important role in the enhanced virulence phenotype of respiration-competent azole-resistant isolates (15). Increased virulence compared to that of the wild type has also been reported for a C. glabrata mutant lacking Ace2, a transcription factor which is a homologue of the Saccharomyces cerevisiae cell cycle-regulated transcription factor Ace2. In S. cerevisiae, Ace2 localizes specifically to the daughter nucleus during mitotic exit and regulates the expression of genes whose products are required for cell separation. Strikingly, the C. glabrata ace2 mutant exhibits cell separation defects and form clumps in liquid cultures (19). The basis for enhanced virulence of the ace2 mutant is still largely unknown; however, it is worth mentioning that the ace2 mutant possesses defects in cell wall biogenesis, which may influence the host response (19, 36). While formation of clumps was not observed in BPY41 here, we observed that the expression of several cell wall biogenesis genes was altered in this mutant. Cell wall alterations as a cause of increased virulence in both cases may be an attractive possibility to support increased virulence of these C. glabrata mutants.
It has been established that the transcription factor CgPDR1 plays a critical role in the expression of genes involved in azole resistance in both respiration-competent and -deficient C. glabrata isolates (14, 40). With the microarray data obtained here, we attempted to identify consensus sequences in the promoters of coregulated genes. We identified a consensus sequence with high similarity to the PDRE sequence established in S. cerevisiae. Vermitsky et al. (42) have proposed the consensus sequence TCC(AG)(TC)G(GC)(AG), and more recently Tsai et al. (41) have proposed a similar consensus sequence (Table 3 shows consensus sequence comparisons). The occurrence of these consensus sequences in the promoters of upregulated genes in BPY41 is similar; however, the consensus sequence proposed by Vermitsky et al. (42) occurs in a higher proportion of genes (13% versus 8.8%) and with a higher copy number among the selected genes (84 sites versus 37 sites) (Table 3). At present, these consensus sequences have been obtained only by in silico analysis but using different set of microarray data and different stringencies. It is still unknown whether CgPdr1 will bind to the promoters containing this consensus sequence, and therefore it will be necessary in the future to address CgPdr1 occupancy by chromatin immunoprecipitation (ChIP) on a genome-wide scale.
Table 3.
Consensus promoter elements of PDR1 genes from C. glabrata and S. cerevisiae
| Sequence | Matches in upregulated genes in this study, % (copy no.a) | Reference(s) |
|---|---|---|
| (T/G)CCACGG(A/G) | 8.8 (37) | This study |
| TCC(A/G)(C/T)G(G/C)(A/G) | 13 (84) | 11, 12 |
| TCC(A/G)(C/T)G(G/C)(A/G) | 13 (84) | 42 |
| TCC(A/G)(C/T)GGA | 7.7 (31) | 41 |
Number of binding sites of the given consensus sequence in the matching genes.
In conclusion, this work showed for the first time that mitochondrial dysfunction can positively affect C. glabrata virulence. The molecular basis for this property is still unclear, and the virulence factors responsible for enhanced virulence need to be identified. The virulence divergence between in vivo- and in vitro-generated mitochondrial mutants constitutes an interesting possibility for the further identification of virulence factors in C. glabrata, and these investigations are under way in our laboratory.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant 31003A_127378 from the Swiss Research National Foundation to D.S. M.S. was supported by a grant from the Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) “Lazzaro Spallanzani” (Strategic Research Program 2006, Italy) and from Università Cattolica del S. Cuore (Linea D1, 2010).
We thank Jacques Bille and Philippe Hauser for critical reading of the paper, Françoise Ischer for excellent technical assistance, and Marilena La Sorda for excellent technical assistance in performing animal experiments.
Footnotes
Supplemental material for this article may be found at http://aac.asm.org/.
Published ahead of print on 14 February 2011.
REFERENCES
- 1. Andersson D. I. 2006. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr. Opin. Microbiol. 9:461–465 [DOI] [PubMed] [Google Scholar]
- 2. Bethea E. K., Carver B. J., Montedonico A. E., Reynolds T. B. 2010. The inositol regulon controls viability in Candida glabrata. Microbiology 156:452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bouchara J. P., et al. 2000. In-vivo selection of an azole-resistant petite mutant of Candida glabrata. J. Med. Microbiol. 49:977–984 [DOI] [PubMed] [Google Scholar]
- 4. Brisse S., et al. 2009. Uneven distribution of mating types among genotypes of Candida glabrata isolates from clinical samples. Eukaryot. Cell 8:287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brun S., et al. 2003. Relationships between respiration and susceptibility to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 47:847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brun S., et al. 2004. Mechanisms of azole resistance in petite mutants of Candida glabrata. Antimicrob. Agents Chemother. 48:1788–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brun S., et al. 2005. Biological consequences of petite mutations in Candida glabrata. J. Antimicrob. Chemother. 56:307–314 [DOI] [PubMed] [Google Scholar]
- 8. Clinical and Laboratory Standards Institute. 2008. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 3rd ed. CLSI document M27-A3 Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 9. De Bernardis F., Lorenzini R., Cassone A. 1999. Rat model of Candida vaginal infection, p. 735–740 In Zak O., Sande M. (ed.), Handbook of animals models of infection. Academic Press, New York, NY [Google Scholar]
- 10. De Bernardis F., et al. 2000. Local anticandidal immune responses in a rat model of vaginal infection by and protection against Candida albicans. Infect. Immun. 68:3297–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeRisi J., et al. 2000. Genome microarray analysis of transcriptional activation in multidrug resistance yeast mutants. FEBS Lett. 470:156–160 [DOI] [PubMed] [Google Scholar]
- 12. Devaux F., et al. 2001. An artificial transcription activator mimics the genome-wide properties of the yeast Pdr1 transcription factor. EMBO Rep. 2:493–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dujon B., et al. 2004. Genome evolution in yeasts. Nature 430:35–44 [DOI] [PubMed] [Google Scholar]
- 14. Ferrari S., et al. 2009. Gain of function mutations in CgPDR1 of Candida glabrata not only mediate antifungal resistance but also enhance virulence. PLoS Pathog. 5:e1000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ferrari S., Sanguinetti M., Torelli R., Posteraro B., Sanglard D. Contribution of CgPDR1-regulated genes in enhanced virulence of azole-resistant Candida glabrata. PLoS One 6:e17589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gagnon-Arsenault I., Parise L., Tremblay J., Bourbonnais Y. 2008. Activation mechanism, functional role and shedding of glycosylphosphatidylinositol-anchored Yps1p at the Saccharomyces cerevisiae cell surface. Mol. Microbiol. 69:982–993 [DOI] [PubMed] [Google Scholar]
- 17. Ju D., Wang X., Ha S.-W., Fu J., Xie Y. 2010. Inhibition of proteasomal degradation of Rpn4 impairs nonhomologous end-joining repair of DNA double-strand breaks. PLoS One 5:e9877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jung U. S., Sobering A. K., Romeo M. J., Levin D. E. 2002. Regulation of the yeast Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase. Mol. Microbiol. 46:781–789 [DOI] [PubMed] [Google Scholar]
- 19. Kamran M., et al. 2004. Inactivation of transcription factor gene ACE2 in the fungal pathogen Candida glabrata results in hypervirulence. Eukaryot. Cell 3:546–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaur R., Ma B., Cormack B. P. 2007. A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc. Natl. Acad. Sci. U. S. A. 104:7628–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knudsen L. F., Curtis J. M. 1947. The use of the angular transformation in biological assays. J. Am. Stat. Assoc. 42:282–296 [DOI] [PubMed] [Google Scholar]
- 22. Maisnier-Patin S., Berg O. G., Liljas L., Andersson D. I. 2002. Compensatory adaptation to the deleterious effect of antibiotic resistance in Salmonella typhimurium. Mol. Microbiol. 46:355–366 [DOI] [PubMed] [Google Scholar]
- 23. Miyazaki H., et al. 1998. Fluconazole resistance associated with drug efflux and increased transcription of a drug transporter gene, PDH1, in Candida glabrata. Antimicrob. Agents Chemother. 42:1695–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moskvina E., Schuller C., Maurer C. T., Mager W. H., Ruis H. 1998. A search in the genome of Saccharomyces cerevisiae for genes regulated via stress response elements. Yeast 14:1041–1050 [DOI] [PubMed] [Google Scholar]
- 25. Muller H., Hennequin C., Gallaud J., Dujon B., Fairhead C. 2008. The asexual yeast Candida glabrata maintains distinct a and alpha haploid mating types. Eukaryot. Cell 7:848–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Orkwis B. R., Davies D. L., Kerwin C. L., Sanglard D., Wykoff D. D. 2010. Novel acid phosphatase in Candida glabrata suggests selective pressure and niche specialization in the phosphate signal transduction pathway. Genetics 186:885–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perocchi F., et al. 2006. Assessing systems properties of yeast mitochondria through an interaction map of the organelle. PLoS Genet. 2:e170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Posteraro B., et al. 2006. Azole resistance of Candida glabrata in a case of recurrent fungemia. J. Clin. Microbiol. 44:3046–3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richard G. F., Kerrest A., Lafontaine I., Dujon B. 2005. Comparative genomics of hemiascomycete yeasts: genes involved in DNA replication, repair, and recombination. Mol. Biol. Evol. 22:1011–1023 [DOI] [PubMed] [Google Scholar]
- 30. Richardson M., Lass-Florl C. 2008. Changing epidemiology of systemic fungal infections. Clin. Microbiol. Infect. 14(Suppl. 4):5–24 [DOI] [PubMed] [Google Scholar]
- 31. Roetzer A., et al. 2008. Candida glabrata environmental stress response involves Saccharomyces cerevisiae Msn2/4 orthologous transcription factors. Mol. Microbiol. 69:603–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rolfes R. J. 2006. Regulation of purine nucleotide biosynthesis: in yeast and beyond. Biochem. Soc. Trans. 34:786–790 [DOI] [PubMed] [Google Scholar]
- 33. Sanglard D., Ischer F., Bille J. 2001. Role of ATP-binding-cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob. Agents Chemother. 45:1174–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sanglard D., Ischer F., Calabrese D., Majcherczyk P. A., Bille J. 1999. The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob. Agents Chemother. 43:2753–2765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanguinetti M., et al. 2005. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob. Agents Chemother. 49:668–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stead D., et al. 2005. Proteomic changes associated with inactivation of the Candida glabrata ACE2 virulence-moderating gene. Proteomics 5:1838–1848 [DOI] [PubMed] [Google Scholar]
- 37. Thomas-Chollier, et al. 2008. RSAT: regulatory sequence analysis tools. Nucleic Acids Res. 36:W119–W127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Torelli R., et al. 2008. The ATP-binding cassette transporter-encoding gene CgSNQ2 is contributor to the CgPdr1-dependent azole resistance in Candida glabrata. Mol. Microbiol. 68:186–201 [DOI] [PubMed] [Google Scholar]
- 39. Traven A., Wong J. M., Xu D., Sopta M., Ingles C. J. 2001. Interorganellar communication. Altered nuclear gene expression profiles in a yeast mitochondrial DNA mutant. J. Biol. Chem. 276:4020–4027 [DOI] [PubMed] [Google Scholar]
- 40. Tsai H. F., Krol A. A., Sarti K. E., Bennett J. E. 2006. Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob. Agents Chemother. 50:1384–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsai H. F., et al. 2010. Microarray and molecular analyses of the azole resistance mechanism in Candida glabrata oropharyngeal isolates. Antimicrob. Agents Chemother. 54:3308–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vermitsky J. P., et al. 2006. Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol. Microbiol. 61:704–722 [DOI] [PubMed] [Google Scholar]
- 43. Wilson W. A., Wang Z., Roach P. J. 2002. Systematic identification of the genes affecting glycogen storage in the yeast Saccharomyces cerevisiae. Mol. Cell Prot. 1:232–242 [DOI] [PubMed] [Google Scholar]
- 44. Wimalasena T. T., et al. 2008. Impact of the unfolded protein response upon genome-wide expression patterns, and the role of Hac1 in the polarized growth, of Candida albicans. Fungal Genet. Biol. 45:1235–1247 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.