

Abstract
Alternate therapies are needed for treatment of secondary bacterial pneumonia following influenza. The immunomodulatory peptide P4 has shown promise in mouse models of primary pneumococcal infection. Mice infected with influenza virus and then challenged with Streptococcus pneumoniae were treated with a combination of P4 peptide and intravenous immune globulin. Survival was improved from 20% to 80% in treated mice relative to controls. Clinical cure correlated with increased clearance of bacteria and decreased lung consolidation. Greater trafficking of professional phagocytic cells to the site of pneumococcal infection coupled with enhanced opsonophagocytosis as manifest by decreased surface display of Fcγ receptors (FcγR) on neutrophils and macrophages were associated with P4 peptide treatment. This suggests that the mechanism of action for improved clearance of bacteria engendered by P4 is through improved uptake by phagocytes mediated by IgG Fc-Fcγ receptor interactions following antibody-mediated opsonophagocytosis of bacteria. Antibody-based therapies, when coupled with immune modulators, such as P4 peptide, may be an effective tool together with antibiotics in our armamentarium against severe pneumonia.
INTRODUCTION
Secondary bacterial pneumonia is a common complicating factor that contributes to the morbidity and mortality associated with epidemic and pandemic influenza (15). During the 1918 Spanish flu pandemic, 95% of fatal cases characterized by autopsy were associated with a bacterial etiology (18). Furthermore, in the recent H1N1 pandemic, examination of fatal and severe pneumonias identified a secondary bacterial pathogen in 25 to 56% of cases, with Streptococcus pneumoniae, Staphylococcus aureus, and Streptococcus pyogenes (group A streptococcus) identified as prominent superinfecting agents (1, 5, 6, 13, 18, 20, 24).
Secondary bacterial pneumonia as a complication of influenza is difficult to treat. Despite the use of appropriate antibiotic regimens in 95 to 99% of severe cases of bacterial superinfection during pandemic H1N1 influenza virus in 2009, a high mortality rate (14 to 46%) was observed. This suggests an inefficiency of antibiotic therapies for bacterial superinfections (4, 5, 7, 10, 20). Recent data from a murine model suggest that treatment of postinfluenza bacterial pneumonia with cell wall-active, β-lactam antibiotics exaggerates the inflammatory response to bacteria in the lung and therapies that do not cause lysis of Gram-positive pathogens may be a better alternative (8, 9, 12). However, as these pathogens continue to acquire resistance, alternatives to antibiotics or adjunctive therapies may be required for effective cure.
As early as 1891, patients with life-threatening bacterial pneumonia were treated with pathogen-specific immune sera derived from rabbits or horses, with drastic reductions in morbidity and mortality (3). In the preantibiotic era, passive immunotherapy was the primary mode of treatment for many infectious diseases, including diphtheria, tetanus, and scarlet fever (2). However, several factors impeded its continued use in the modern era, including toxicity and serum sickness, which occurred in 10 to 50% of patients. In 1937, the introduction of sulfonamides active against common agents of community-acquired pneumonia halted the widespread use of passive immunotherapy. Currently, antibody therapy is indicated as a treatment in only a few, select situations, such as for toxin neutralization (diphtheria, tetanus, and botulism) or for postexposure prophylaxis of viral infections (rabies, measles, hepatitis A and B, and Ebola viruses). However, several recent advances in antibody harvesting and monoclonal antibody production are prompting reconsideration of passive immunotherapy.
Several recent studies have sought to improve upon passive immunotherapy regimens for primary pneumococcal pneumonia through the use of an immunomodulatory peptide as adjunctive therapy for intravenous (i.v.) immune globulin (IVIG) (17, 23). P4 is a 28-amino-acid peptide derived from pneumococcal surface adhesin A (22). It has been used to successfully treat mice with otherwise fatal S. pneumoniae infections (23). This peptide has been found to increase the adherence and internalization of pneumococci and to activate host immune cells in vitro (22, 23). In this study, we sought to determine whether combined therapy with P4 and IVIG could be used to treat mice with severe secondary bacterial pneumonia following influenza. We hypothesized that the combined therapy of IVIG and P4 peptide would facilitate innate immune responses and reduce the pneumococcal burden following influenza infection, which encourages bacterial overgrowth. We report that this regimen rescues mice from severe influenza-pneumococcal coinfections, suggesting that it may serve as a viable alternative or adjunct to antibiotic therapy.
MATERIALS AND METHODS
Mice.
Six- to eight-week-old female BALB/c mice (Jackson Laboratory, Bar Harbor, ME) were maintained in a biosafety level 2 facility in the Animal Resource Center at St. Jude Children's Research Hospital (SJCRH). All experimental procedures were approved by the Animal Care and Use Committee at SJCRH and were done under general anesthesia with inhaled isoflurane (2.5%) (Baxter Healthcare Corporation, Deerfield, IL).
Viral and bacterial strains.
The St. Jude strain of mouse-adapted influenza virus A/Puerto Rico/8/34 (H1N1; PR8) was generated by reverse genetics and grown in Madin-Darby canine kidney (MDCK) cells followed by 2 passages through eggs for stock. The resulting allantoic fluid was stored at −80°C. The hemagglutinin and neuraminidase genes were sequenced to ensure that no inadvertent mutations were present, and the viral stock was further characterized in MDCK cells and eggs to determine the dose infectious for 50% of tissue culture wells (TCID50) or eggs (EID50). S. pneumoniae strain A66.1 (type 3) was obtained from Elaine Tuomanen at SJCRH and engineered to express luciferase (Kevin Francis and Jun Yu, Xenogen Corporation, Alameda, CA). S. pneumoniae was grown in Todd Hewitt broth (Difco Laboratories, Detroit, MI) to an optical density at 620 nm (OD620) of approximately 0.4, then frozen at −80°C, and mixed 2:1 with 5% sterile glycerol, and the titer was subsequently determined on tryptic soy agar (TSA; Difco Laboratories, Detroit, MI) supplemented with 3%, vol/vol, sheep erythrocytes.
Infectious model and treatment.
Groups of mice (group size is indicated in figure legends) were dosed intranasally with 30 TCID50s (equivalent to 0.25 doses lethal for 50% of mice [MLD50]) of PR8 in a volume of 100 μl phosphate-buffered serum (PBS). After 7 days, mice were dosed in a similar manner with 75 CFU of S. pneumoniae (0.1 MLD50). Forty-eight and seventy-two hours following bacterial challenge, mice were passively immunized with IVIG (100 μl/mouse i.v.), followed 20 min later with P4 peptide (100 μg/mouse in 100 μl of PBS i.v.). Control mice were administered 100 μl of PBS or P4 peptide. Experiments were conducted in 3 independent trials. IVIG and P4 peptide were obtained from the CDC (Atlanta, GA) and previously described elsewhere (23). The P4 peptide was dissolved in water at 10 mg/ml and was completely soluble at this concentration. The peptide was stored at −80°C in single-use aliquots. Prior to use, the peptide was sonicated for 3 min and then diluted to 1 mg/ml in PBS.
Titers and histopathological examination.
Immediately after euthanasia by CO2 inhalation, lungs from 4 mice per group were sufflated with PBS and removed. Each mouse's single left lobe was fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned at 4 μM, stained with hematoxylin and eosin, and examined microscopically for histopathological alterations. Each mouse's right lung lobes were placed into 0.5 ml of PBS and homogenized using a tissue homogenizer (Omni TH, Kennesaw, GA). Bacterial and viral titers were then determined on TSA or MDCK cells, respectively, as described previously (21). Microscopic evaluation of lungs was performed by an experienced veterinary pathologist (K.L.B.) blinded to the study purpose, design, and group composition. A semiquantitative grading scheme was utilized to score the overall character of the pneumonic process and the pathology specifically observed in the interstitium and terminal airways as described elsewhere (14), in which a score of 1 indicates mild perivascular inflammation and few leukocytes in alveolar spaces, a 2 indicates moderate perivascular inflammation and numerous leukocytes in alveolar spaces, with mild alveolar wall thickening, a 3 indicates marked perivascular inflammation, marked leukocyte infiltrates in alveolar spaces, moderate thickening of alveolar walls, focal alveolar wall necrosis, and airway epithelial necrosis with fibrin and edema, and a 4 indicates extensive coalescing inflammation with interstitial and airway necrosis, fibrin, edema, hemorrhage, and consolidation.
Cell populations in BALFs.
Following euthanasia by CO2 inhalation, the trachea was exposed and cannulated with a 24-gauge plastic catheter (BD Instye; Becton Dickinson, Sandy, UT). Lungs were lavaged twice with 1.5 ml Hanks balanced salt solution without divalent cations but with phenol red (Mediatech, Manassas, VA) supplemented with 1% fetal bovine serum (FBS) and 0.05 mM EDTA. Flow cytometry was performed on the bronchoalveolar lavage fluid (BALF) suspension via staining (1 μl/106cells). All cells were gated for granularity (FSChighxSSChigh), viability (DAPI−), single cells (FSCWlowxFSCA), and Ter119− (BD-Pharmaceuticals, clone TER-119), and specific markers were used to determine cell populations. For the macrophage population, cells were further gated as CD11b+ (BD Pharmaceuticals; clone M1/70), CD11c+ (BD Pharmaceuticals; clone HL3), Gr-1+ (BD Pharmaceuticals; clone RB6-8C5), and CD45R/B220− (eBiosciences; clone RA3-6B2). The gating strategy for neutrophils included GR1+, CD11b−, CD3e− (BD Pharmaceuticals; clone 145-2C11), and CD45R/B220−.
Statistical analyses.
A comparison of survival rates between groups of mice was done with the log rank chi-square test on the Kaplan-Meier survival data. Comparisons of bacterial titers, cell counts, weight loss, and histopathology scoring between groups were done using analysis of variance (ANOVA). A P value of <0.05 was considered significant for these comparisons. SigmaStat for Windows (SysStat Software, Inc., version 3.11) was utilized for all statistical analyses.
RESULTS
Treatment with IVIG and P4 peptide improves survival of coinfected mice.
Mice coinfected with influenza A virus followed 7 days later by S. pneumoniae had a higher rate of mortality (4 out of 5 mice died) than mice infected with a single pathogen (0 out of 5 mice died) (Fig. 1A). To potentially improve the survival in this murine coinfection model, dually infected mice were treated intravenously with IVIG followed 20 min later with peptide P4. Treatment with a combination of IVIG and P4 increased the survival rate from ≤30% (PBS-, IVIG-, or P4-only groups) to 80% (Fig. 1B). To document that all animals developed illness from the infections and assess potential toxic side effects from the drug therapy, weight loss was followed. All groups of mice exhibited similar degrees of morbidity as assessed by weight loss (Fig. 1C). To quantitate the location and extent of infection, luciferase-expressing pneumococci were utilized, and mice were imaged daily. All mice developed pneumonia as evidenced by bioluminescent imaging; the P4-and-IVIG-treated group successfully cleared the infection by the second day of treatment (Fig. 2). These findings indicate that the dual treatment rescues mice from secondary bacterial pneumonia following influenza.
Fig. 1.

P4 peptide combination therapy rescues mice from bacterial pneumonia following influenza. (A) Mice were mock infected with PBS (A66.1 group; n = 4) or infected with a sublethal dose of influenza A virus strain PR8 (PR8 group [n = 4] and PR8 + A66.1 group [n = 10]) and then challenged 7 days later with S. pneumoniae strain A66.1 (A66.1 and PR8 + A66.1 groups) or PBS (PR8 group). (B) Forty-eight and seventy-two hours after bacterial challenge, groups of virus-and-bacterium-coinfected mice (n = 10) were mock treated with PBS or treated with IVIG alone, P4 peptide alone, or IVIG followed by P4 peptide 20 min later. Morbidity represented by weight loss (C) and survival (B) were followed for 21 days. An asterisk indicates a significant (P < 0.05) difference in survival compared to that of the other groups by the log-rank test on the Kaplan-Meier data. In panel C, values indicate the mean and error bars the standard deviation of the measurements.
Fig. 2.

In vivo bioluminescent imaging of secondary bacterial pneumonia. Representative mice coinfected with influenza virus and pneumococci and either mock treated with PBS or treated with IVIG and P4 48 h and 72 h after bacterial challenge were imaged daily for localization and quantitation of bioluminescent bacteria.
Inflammation is reduced in the lungs of mice treated with IVIG and P4.
Lungs from coinfected mice that were mock treated with PBS or treated with IVIG and P4 together were compared by histopathological examination. On day 5 post-bacterial challenge, the interstitia of the control, mock-treated mice were found to be infiltrated by neutrophils and foamy macrophages, which created a massive consolidation throughout the lung (Fig. 3B and D). In the P4-treated mice, few macrophages, neutrophils, or lymphocytes were present in the interstitium, the hyperplastic changes were mild, and consolidation was limited to small areas of the lung (Fig. 3A and C). Using a semiquantitative scoring system, we found that mice that received the P4 therapy had generally lower histopathology scores than control mice, although variation between animals was present (Fig. 2E). These findings suggest that P4 treatment leads to a reduction in the extent and quality of inflammation in the lung, which contributes to improved outcomes.
Fig. 3.

Histopathology of lungs from mice treated with IVIG and P4 peptide. Lungs were removed from mice coinfected with influenza virus and pneumococci (n = 4 per group per time point). Group treated with IVIG and P4 peptide (A and C); PBS control group (B and D); single-blind, histopathology scores by a veterinary pathologist using a semiquantitative grading scheme (E). Representative photomicrographs taken 5 days after bacterial challenge at a magnification of ×4 (A and B) and ×40 (C and D) are presented. The extensive neutrophilic infiltrate observed with mock-treated mice (D) caused near-complete consolidation of the lungs (B), while the relatively mild inflammatory infiltrate found in IVIG-and-P4-treated mice (C) was limited to specific foci (A). In panel E, horizontal lines indicate the mean pathology score.
Dual treatment accelerates clearance of bacteria from lung.
Bioluminescent imaging of bacterial colonization in the lung suggested that combined P4 and IVIG therapy for treatment of bacterial pneumonia following influenza virus infection leads to a substantive reduction in the bacterial load. Therefore, bacterial burdens from the lungs of IVIG-and-P4-peptide-treated mice were compared to those of mock-treated mice which had received only PBS. After the initial treatment 48 h post-bacterial challenge, the bacterial burdens in both treated and mock-treated mice were similar (1.6 × 108 CFU/ml versus 3.6 × 108 CFU/ml, respectively) (Fig. 4). However, post-bacterial challenge, the dually treated mice were found to have a more than 300-fold difference in the bacterial load within the lungs by day 5 (Fig. 4). This suggests that the P4 treatment accelerated the clearance of pneumococci from the lung. In addition, the kinetics of viral clearance were examined post-bacterial challenge. Viral titers were detectable in 2 of 4 mice mock treated with PBS following the first treatment dose on postinfection day 2 (5.3 and 4.5 TCID50/ml lung tissue) but were not detected following the second treatment dose on day 3. Virus could not be detected at any time point following treatment with IVIG and P4.
Fig. 4.
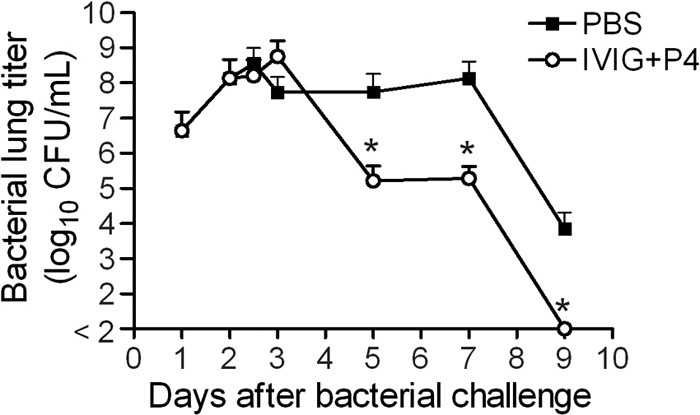
Bacterial lung load from mice treated with PBS or IVIG and P4. Lung titers were determined for groups of influenza-virus-and-pneumococcus-coinfected mice (n = 5) either mock treated with PBS or treated with IVIG and P4 peptide 48 and 72 h after bacterial challenge. Values indicate the mean, and error bars the standard deviation of the measurements. An asterisk indicates a significant (P < 0.05) difference in bacterial titer compared to mock-treated animals at that time point.
IVIG-and-P4 treatment increases professional phagocyte populations in the lung, with reduced surface expression of Fcγ receptors I/II (FcγRI/II).
With the accelerated clearance of pneumococci observed with mice that received combined therapy and in vitro studies indicating that P4 therapy increases adherence and phagocytosis of pneumococci (22, 23), enumeration of cell populations in the BALF was undertaken. Total cell counts, particularly the absolute numbers of white cells, increased in the airways over the first 48 h post-bacterial challenge, in concert with the increased bacterial load, and then decreased in the next 12 h (Fig. 5A). During the 2 days of treatment, the percents (data not shown) and numbers of both macrophages (Fig. 5B) and neutrophils (Fig. 5C) again increased in mice receiving IVIG and P4 but decreased in mice receiving PBS. By the time point immediately after the second treatment, the difference between groups was statistically significant (P < 0.05). These data suggest that combined therapy provides an accelerated innate immune response, leading to quicker resolution of the infection and a reduction in disease pathology. Given the degree of consolidation and infiltration of the airways visible in the histopathology on day 5 (Fig. 3), it was surprising that few cells were recovered from the BALF (Fig. 5); we hypothesize that recovery was impeded by the consolidative process such that most cells were in the parenchyma or marginated and inaccessible to washing at this stage.
Fig. 5.

Airway inflammatory cells in mice treated with PBS or IVIG and P4. BALF was obtained from groups of mice (n = 8 to 10) coinfected with influenza virus and pneumococci and mock-treated with PBS or treated with IVIG and P4 peptide 48 and 72 h after bacterial challenge. The relative numbers of total white blood cells (A), macrophages (B), and neutrophils (C) were determined by flow cytometry. The proportion of either macrophages (D) or neutrophils (E) displaying CD16/32 was determined by flow cytometry. Arrows indicate the timing of treatment relative to BALF collection. An asterisk indicates a significant (P < 0.05) difference in cell number or proportion compared to mock-treated animals at that time point.
In previous studies, P4 peptide was shown to increase the opsonophagocytosis activity of a broad range of phagocytic cells in vitro (22, 23). Our data showing decreased bacterial loads, associated with increased numbers of macrophages and neutrophils, suggested that enhanced opsonophagocytosis through these cells might explain the increased survival in our study. To probe the specific mechanism involved, we examined the expression of FcγR on the surface of airway phagocytes. We reasoned that this might provide indirect evidence of enhanced phagocytic capability since FcγR are responsible for the uptake of opsonized bacterium by binding the Fc portion of antibodies (19). BALFs were collected and stained for FcγR (CD16/32) expression on the cell surface of macrophages and neutrophils. As early as day 3 pretreatment (for macrophages) and day 3 posttreatment (for neutrophils), a significant (P < 0.05) reduction in FcγR cell surface display could be demonstrated (Fig. 5D and E). This persisted through day 5 post-bacterial superinfection. This decrease in surface FcγR expression in response to P4 therapy suggests that these receptors are internalized during phagocytosis of IgG-opsonized bacteria. This explains the requirement for type-specific antipneumococcal antibody (provided in this study as IVIG) to induce a clinical cure and implicates modulation of FcγR in the mechanism of action of P4 peptide.
DISCUSSION
Despite the use of antibiotics active against secondary bacterial pathogens, pneumonia following influenza has a high mortality rate. Due to a steady increase in the number of antibiotic-resistant strains, the effectiveness of strategies using antimicrobials alone is likely to decrease over time. Alternative or adjunctive therapies are therefore necessary for routine treatment of complications of seasonal influenza and in preparation for the next influenza pandemic. In this paper, we report that combination therapy with IVIG and P4 peptide effectively rescues mice from a fatal secondary bacterial pneumonia without the use of antibiotics. An increase in phagocytic cells in the airways and a decrease in surface display of FcγR correlated with decreased inflammation in the lungs and clinical cure. The requirement of both P4 and IVIG for reductions in mortality suggests a model in which type-specific antibody binds pneumococci, P4 enhances transit of macrophages and neutrophils into the airways and activates FcγRs on these professional phagocytes, and a robust opsonophagocytosis response results, clearing the infection. The decrease in bacterial load, in turn, leads to a decrease in the requirement for new inflammatory cells within the lung parenchyma, reducing pneumonic consolidation and acute lung injury.
P4 is a novel immunomodulatory peptide derived from the pneumococcal surface adhesin A protein. It has previously been shown to activate various cell lines and increase the opsonophagocytosis capabilities of host cells in vitro (23). This resulted in improved outcomes of primary pneumococcal pneumonia in mice (23). In the setting of prior influenza virus infection, however, the inflammatory milieu is quite different during bacterial lung infections, with enhanced infiltration of inflammatory cells into both the lungs and the airways, increased bacterial loads, and hypercytokinemia (14, 25). Thus, we sought in this study to determine whether therapy with IVIG and P4 would remain effective during fulminant coinfections. Remarkably, two doses of IVIG followed by P4, administered 24 and 48 h following the onset of severe bacterial pneumonia, substantially improved mouse survival. By both bioluminescent imaging and culture, this correlated with a significant reduction in bacterial lung load. P4 or IVIG alone each had no effect, confirming that enhanced utilization of natural defenses, rather than direct killing by the peptide, was responsible for bacterial clearance. Since bacterial lysis by antibiotics is associated with worse outcomes in secondary bacterial pneumonia following influenza (8), this alternate strategy may be superior to current practice either alone or as adjunctive therapy with antibiotics (16). Although 80% survival represents a significant beneficial outcome in this model, we think it likely that earlier treatment, sustained treatment, or combination treatment with antibiotics would likely improve the survival advantage further.
The mechanism by which P4 exerts its salutary effects is of obvious interest. The finding of increased transit of phagocytes into the airways, where the pneumococcus exerts its pathogenic effects, suggests modulation of chemotaxis or trafficking. Once present at this site, the rapid clearance of bacteria associated with decreased surface display of FcγRI and FcγRII on these phagocytes implicates P4 in enhancing opsonophagocytosis. The pathogen-specific, antibody-dependent P4 effect at this stage implies that FcγR recognition of the Fc portion of antibodies bound to pneumococci assists in the process. The engagement of FcγRs has been shown to activate a self-limiting inflammatory response in macrophages (19). Additionally, it has been shown that internalized antigen/IgG complexes are much more efficiently presented on major histocompatibility complex (MHC) class II molecules than internalized soluble antigens, most likely due to the specific binding to the surface FcγRs (11). The limited proinflammatory response elicited by binding of the FcγRs and the independent cell-activating properties of P4 peptide may work collectively to restore the proper balance of pro- and anti-inflammatory cytokines and improve the function of professional phagocytes, leading to a more favorable outcome and effective treatment of secondary bacterial pneumonia following influenza.
In summary, the combined therapy of IVIG and P4 improves the outcome of influenza and secondary bacterial pneumonia caused by pneumococci. The accelerated clearance of pneumococci and reduced infiltration of inflammatory cells into the lung parenchyma likely contribute to this favorable outcome, through mechanisms involving both the altered trafficking and enhanced capacity of professional phagocytes. Historically, passive immunotherapy was clinically utilized in the treatment of pneumonia until the introduction of antibiotics. With the increasing failure of strategies involving direct killing of pneumonic pathogens to effect cure, particularly in the subset of patients with severe pneumonia, such as that engendered by prior influenza virus infection, a return to modified versions of passive immunotherapy may be indicated. We demonstrate here that an immunomodulatory strategy coupled with antibody therapy can be an effective tool in our armamentarium against severe pneumonia. Further exploration in preclinical models and clinical studies of humans is indicated.
ACKNOWLEDGMENTS
This work was supported by public health service grant AI-76816, ALSAC, and the Centers for Disease Control and Prevention.
The findings and conclusions in this presentation are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention or the Agency for Toxic Substances and Disease Registry.
Footnotes
Published ahead of print on 7 March 2011.
REFERENCES
- 1. Centers for Disease Control and Prevention. 2009. Bacterial coinfections in lung tissue specimens from fatal cases of 2009 pandemic influenza A (H1N1)—United States, May-August 2009. MMWR Morb. Mortal. Wkly. Rep. 58:1071–1074 [PubMed] [Google Scholar]
- 2. Christian H. A. 1944. The principles and practice of medicine. D. Appleton-Century Company, Inc., New York, NY [Google Scholar]
- 3. Chudwin D. S. 1989. Prophylaxis and treatment of pneumococcal bacteremia by immune globulin intravenous in a mouse model. Clin. Immunol. Immunopathol. 50:62–71 [DOI] [PubMed] [Google Scholar]
- 4. Domínguez-Cherit G., et al. 2009. Critically ill patients with 2009 influenza A(H1N1) in Mexico. JAMA 302:1880–1887 [DOI] [PubMed] [Google Scholar]
- 5. Estenssoro E., et al. 2010. Pandemic 2009 influenza A in Argentina: a study of 337 patients on mechanical ventilation. Am. J. Respir. Crit. Care Med. 182:41–48 [DOI] [PubMed] [Google Scholar]
- 6. Gill J. R., et al. 2010. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Arch. Pathol. Lab. Med. 134:235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jain S., et al. 2009. Hospitalized patients with 2009 H1N1 influenza in the United States, April-June 2009. N. Engl. J. Med. 361:1935–1944 [DOI] [PubMed] [Google Scholar]
- 8. Karlström Å., et al. 2009. Treatment with protein synthesis inhibitors improves outcomes of secondary bacterial pneumonia after influenza. J. Infect. Dis. 199:311–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karlström Å., et al. Toll-like receptor 2 mediates fatal immunopathology in mice during treatment of secondary bacterial pneumonia following influenza. J. Infect. Dis., in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar A., et al. 2009. Critically ill patients with 2009 influenza A(H1N1) infection in Canada. JAMA 302:1872–1879 [DOI] [PubMed] [Google Scholar]
- 11. Manca F., et al. 1991. Effect of antigen/antibody ratio on macrophage uptake, processing, and presentation to T cells of antigen complexed with polyclonal antibodies. J. Exp. Med. 173:37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mandell L. A., et al. 2007. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin. Infect. Dis. 44(Suppl. 2):S27–S72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mauad T., et al. 2010. Lung pathology in fatal novel human influenza A (H1N1) infection. Am. J. Respir. Crit. Care Med. 181:72–79 [DOI] [PubMed] [Google Scholar]
- 14. McAuley J. L., et al. 2007. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe 2:240–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCullers J. A. 2006. Insights into the interaction between influenza virus and pneumococcus. Clin. Microbiol. Rev. 19:571–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. McCullers J. A., English B. K. 2008. Improving therapeutic strategies for secondary bacterial pneumonia following influenza. Future Microbiol. 3:397–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melnick N., et al. 2009. Evaluation of a novel therapeutic approach to treating severe pneumococcal infection using a mouse model. Clin. Vaccine Immunol. 16:806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Morens D. M., Taubenberger J. K., Fauci A. S. 2008. Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness. J. Infect. Dis. 198:962–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nimmerjahn F., Ravetch J. V. 2007. Fc-receptors as regulators of immunity. Adv. Immunol. 96:179–204 [DOI] [PubMed] [Google Scholar]
- 20. Palacios G., et al. 2009. Streptococcus pneumoniae coinfection is correlated with the severity of H1N1 pandemic influenza. PLoS One 4:e8540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peltola V. T., Murti K. G., McCullers J. A. 2005. Influenza virus neuraminidase contributes to secondary bacterial pneumonia. J. Infect. Dis. 192:249–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rajam G., et al. 2008. A functional epitope of the pneumococcal surface adhesin A activates nasopharyngeal cells and increases bacterial internalization. Microb. Pathog. 44:186–196 [DOI] [PubMed] [Google Scholar]
- 23. Rajam G., et al. 2009. A 28-aa pneumococcal surface adhesin A-derived peptide, P4, augments passive immunotherapy and rescues mice from fatal pneumococcal infection. J. Infect. Dis. 199:1233–1238 [DOI] [PubMed] [Google Scholar]
- 24. Shieh W. J., et al. 2010. 2009 pandemic influenza A (H1N1): pathology and pathogenesis of 100 fatal cases in the United States. Am. J. Pathol. 177:166–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Smith M. W., et al. 2007. Induction of pro- and anti-inflammatory molecules in a mouse model of pneumococcal pneumonia after influenza. Comp. Med. 57:82–89 [PMC free article] [PubMed] [Google Scholar]