

Abstract
Quinazolinediones (diones) are fluoroquinolone-like inhibitors of bacterial gyrase and DNA topoisomerase IV. To assess activity against mycobacteria, C-8-methoxy dione derivatives were compared with cognate fluoroquinolones by using cultured Mycobacterium smegmatis. Diones exhibited higher MIC values than fluoroquinolones; however, MICs for fluoroquinolone-resistant gyrA mutants, normalized to the MIC for wild-type cells, were lower. Addition of a 3-amino group to the 2,4-dione core increased relative activity against mutants, while alteration of the 8-methoxy group to a methyl or of the 2,4-dione core to a 1,3-dione core lowered activity against mutants. A GyrA G89C bacterial variant was strikingly susceptible to most of the diones tested; in contrast, low susceptibility to fluoroquinolones was observed. Many of the bacteriostatic differences between diones and fluoroquinolones were explained by interactions at the N terminus of GyrA helix IV revealed by recently published X-ray structures of drug-topoisomerase-DNA complexes. When lethal activity was normalized to the MIC in order to minimize the effects of drug uptake, efflux, and ternary complex formation, a 3-amino-2,4-dione exhibited killing activity comparable to that of a cognate fluoroquinolone. Surprisingly, the lethal activity of the dione was inhibited less by chloramphenicol than that of the cognate fluoroquinolone. This observation adds the 2,4-dione structural motif to the list of structural features known to impart lethality to fluoroquinolone-like compounds in the absence of protein synthesis, a phenomenon that is not explained by X-ray structures of drug-enzyme-DNA complexes.
INTRODUCTION
Fluoroquinolones are lethal antibacterial agents that are widely used to control many bacterial infections (23); for some diseases, such as multidrug-resistant tuberculosis, fluoroquinolones are key to successful treatment (4). As with other antimicrobials, fluoroquinolone use is threatened by an increasing prevalence of resistance (1). One way to address the resistance problem is to identify new derivatives that are particularly active with resistant mutants. For example, with some bacterial species, addition of an 8-methoxy substituent increases fluoroquinolone activity against mutants (8, 45, 46). Other examples include quinazolinediones, which have shown good activity with gyrase resistance mutants selected by fluoroquinolone treatment of Escherichia coli, Staphylococcus aureus, and Streptococcus pneumoniae (11, 16, 34). We found with E. coli that a quinazoline-2,4-dione was almost as active with gyrase mutants as with wild-type cells (12). How quinazolinediones behave with mycobacteria is unknown.
To compare quinolone-like compounds with regard to the likely effects of fluoroquinolone resistance mutations, sets of gyrase mutants are prepared in which the strains are otherwise isogenic. The MIC for the mutant is measured and related to the MIC for the wild type to correct for differences in uptake and efflux. The ratio of the MIC for the mutant to the MIC for the wild type defines a parameter termed antimutant activity. The goal has been to identify compounds for which this MIC ratio is near unity, since that should narrow the mutant selection window, the drug concentration range in which resistant mutant subpopulations are selectively enriched and amplified (44). Selected compounds are then compared for activity against resistant subpopulations by population analysis (12, 38) to confirm that amplification of mutant subpopulations is restricted. These MIC-based measurements reflect only bacteriostatic activity. Methods are also available to compare agents by two types of rapid killing, one that requires ongoing protein synthesis and one that does not (10); these lethal properties are likely to be important for limiting the induction of resistant mutants (27). Thus, MIC measured with wild-type cells, which is universally used for antimicrobial discovery, is but one of several ways to evaluate compounds early in drug development. Indeed, consideration of resistant mutant studies and killing tests reveals activity-specific differences not seen with the wild-type MIC. Some of those differences can now be combined with structural models of drug-DNA-topoisomerase interactions to provide a more complete understanding of drug action.
In the present study, we compared a set of quinazolinediones with cognate fluoroquinolones for activity against several fluoroquinolone-resistant gyrA mutants of Mycobacterium smegmatis. This mycobacterial species is well suited for cellular studies of gyrase mutants because it lacks topoisomerase IV (6), an additional quinolone target present in many bacteria that increases the complexity of resistance studies. Moreover, M. smegmatis serves as a fast-growing model for fluoroquinolone-M. tuberculosis interactions (26, 29), which are likely to become increasingly important as the growing prevalence of multidrug-resistant tuberculosis causes fluoroquinolones to assume a larger role in chemotherapy (36). While quinazoline-2,4-diones had higher MICs against wild-type M. smegmatis than those observed for cognate fluoroquinolones, dione activity was affected little by resistance mutations. Moreover, the relative susceptibilities of GyrA variants differed strikingly between diones and fluoroquinolones, providing an opportunity to test new structural models describing drug-topoisomerase-DNA complexes. We also examined M. smegmatis for dione-specific lethal effects: replacement of the quinolone carboxyl with a dione enhanced lethal action in the presence of chloramphenicol, thereby revealing a new property that considerations of lethality must explain.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Mycobactium smegmatis mc2155, isolate KD1163, has been described previously (47); quinolone-resistant mutants were obtained by selection on agar plates containing various concentrations of ciprofloxacin (47). The strains used in the study, listed in Table 1, were grown on 7H10 agar or in 7H9 liquid medium, each containing 10% ADC (albumin-dextrose complex), 0.05% Tween, and 0.2% glycerol (18).
Table 1.
M. smegmatis strains used in this studya
| Strain | Relevant genotype |
|---|---|
| KD1163 | Wild type (laboratory isolate of mc2155) |
| KD2008 | gyrA(G89C) |
| KD1987 | gyrA(A91V) |
| KD1991 | gyrA(D95Y) |
See reference 39.
Susceptibility determinations.
For determination of MICs, cells were grown to mid-exponential phase, diluted to 104 to 105 CFU/ml in tubes containing fluoroquinolone or quinazolinedione, and incubated at 37°C for 2 days with shaking. Growth was determined by visual inspection, and the lowest quinolone or dione concentration that blocked growth was taken as the MIC. The MIC for population analysis was determined by inhibition of colony formation on drug-containing agar; MIC99 was defined as the drug concentration that blocked the formation of 99% of the colonies.
Lethal activity was assessed by incubation of exponentially growing bacteria in liquid cultures with either dione or fluoroquinolone for the times and at the concentrations indicated in the figures. Incubation was followed by dilution and enumeration of CFU by plating on drug-free 7H10 agar. Bacterial survival was expressed as a fraction of the CFU present at the time of drug addition.
Population analysis profiles.
Population analysis was performed as described previously (47). Briefly, a series of agar plates was prepared in which the concentration of the dione UING5-207 or the cognate fluoroquinolone (UING5-249) varied over a broad range. Wild-type M. smegmatis (isolate KD1163), grown to stationary phase in liquid medium, was applied to each plate in amounts that allowed a small number of colonies to form. These putative mutants were counted to obtain a preliminary score; the colonies were transferred to drug-free agar for a second round of growth; and then the colonies were transferred to agar containing the drug at the same concentration used initially for selection. Clones that showed growth of separated, individual colonies on drug-containing plates after transfer were scored as resistant mutants and were used to correct the preliminary score.
Fluoroquinolones and diones.
Ciprofloxacin, moxifloxacin, and Bay y 3114 were obtained from Bayer AG (West Haven, CT). PD160793 was a generous gift from John Domagala, Parke-Davis Division of Pfizer Corp. (Ann Arbor MI). PD161148 and additional 8-methoxy quinolones, the 8-methyl- and 8-methoxy-quinazoline-2,4-diones, and the pyrido[1,2-c]pyrimidine-1,3-dione were prepared using methods described previously (3, 12, 20, 35, 41). All compounds were dissolved to 10 mg/ml in either dimethyl sulfoxide (DMSO) (for diones) or 0.1 N NaOH (for quinolones) prior to use.
Molecular modeling.
Structures of fluoroquinolone/dione-DNA-topoisomerase IV complexes were obtained from the Protein Data Bank (PDB) and were displayed using WebLab ViewerLite. Modeling was performed by manual docking. Protein Data Bank accession numbers for individual structures are noted in the figure legends.
RESULTS
Effect of dione structure on wild-type MIC.
Since a C-8-methoxy group improves fluoroquinolone activity with gyrase resistance mutants (8, 25), we prepared a series of diones in which all but one had a C-8-methoxy group (compound structures are shown in Fig. 1). With wild-type M. smegmatis, the MICs of the diones were generally higher than those of comparable fluoroquinolones (Table 2 [compounds are arranged according to their C-7 ring structures, diagramed in Fig. 1 and represented by the letters a to d]). The higher MIC for diones was expected, because these compounds are subject to efflux (12, 17, 40), and with purified E. coli gyrase they exhibit lower activity than fluoroquinolones (33). Addition of a 3-amine to a 2,4-dione lowered the MIC 4- to 10-fold, depending on the structure of the C-7 ring, which is located at the other end of the molecule (Table 2; compare MICs for the wild type in lines 1 and 2, lines 5 and 6, lines 9 and 10, and lines 12 and 13). A 1,3-dione (2-amino-pyrido[1,2-c]pyrimidine-1,3-dione; UIJR1-100) had about the same bacteriostatic activity as its cognate 3-amino-2,4-dione (UING5-207) (Table 2); changing the 8-methoxy group to an 8-methyl group lowered the dione MIC about 8-fold (Table 2, UING5-207 versus UIJR-048). Thus, ways exist to alter the bacteriostatic activity of diones against wild-type cells.
Fig. 1.

Structures of diones and fluoroquinolones used in this study. The numbering system for core atoms of dione and quinolone structures is shown in the fluoroquinolone structure on the lower left.
Table 2.
MICs of quinolones and diones against wild-type M. smegmatis
| Line no. | Compound | Compound typea | C-7 groupb | MIC (μg/ml)c |
|||
|---|---|---|---|---|---|---|---|
| wt | G89C mutant | A91V mutant | D95Y mutant | ||||
| 1 | UING5-47 | C-8-OMe 2,4-dione | R1 = a | 100 ± 11.8 | 50 ± 11.8 | 200 ± 0 | 400 ± 0 |
| 2 | UING5-157 | C-8-OMe 3-amino 2,4-dione | R1 = a | 12.5 ± 3.5 | 6.25 ± 2.7 | 50 ± 0 | 100 ± 0 |
| 3 | Bay y 3114 | C-8-H fluoroquinolone | R1 = a | 0.031 ± 0 | 2 ± 0.24 | 0.5 ± 0 | 0.5 ± 0.06 |
| 4 | Moxifloxacin | C-8-OMe fluoroquinolone | R1 = a | 0.062 ± 0 | 2 ± 0.12 | 0.25 ± 0.06 | 0.25 ± 0 |
| 5 | UING5-48 | C-8-OMe 2,4-dione | R1 = b | 100 ± 11.8 | 200 ± 0 | 200 ± 24 | 200 ± 24 |
| 6 | UING5-209 | C-8-OMe 3-amino 2,4-dione | R1 = b | 10 ± 1.2 | 10 ± 1.2 | 5 ± 0.65 | 10 ± 1.2 |
| 7 | PD160793 | C-8-H fluoroquinolone | R1 = b | 0.062 ± 0 | 4 ± 0.9 | 0.5 ± 0.06 | 2 ± 0.5 |
| 8 | PD161148 | C-8-OMe fluoroquinolone | R1 = b | 0.031 ± 0 | 0.5 ± 0.24 | 0.25 ± 0.12 | 0.5 ± 0.24 |
| 9 | UING5-63 | C-8-OMe 2,4-dione | R1 = c | 50 ± 6.5 | 200 ± 24 | 200 ± 24 | 200 ± 0 |
| 10 | UING5-159 | C-8-OMe 3-amino 2,4-dione | R1 = c | 12.5 ± 3.5 | 6.25 ± 1.2 | 25 ± 4.7 | 25 ± 4.7 |
| 11 | UING5-248 | C-8-OMe fluoroquinolone | R1 = c | 0.125 ± 0 | 4 ± 0.9 | 1 ± 0.12 | 2 ± 0 |
| 12 | UING5-200 | C-8-OMe 2,4-dione | R1 = d | 25 ± 6.5 | 25 ± 6.5 | 50 ± 0 | 100 ± 11.8 |
| 13 | UING5-207 | C-8-OMe 3-amino 2,4-dione | R1 = d | 5 ± 1.2 | 2.5 ± 0 | 5 ± 1.2 | 5 ± 1.2 |
| 14 | UIJR1-100 | C-8-OMe 1,3-dione | R1 = d | 5 ± 0 | 2.5 ± 0 | 10 ± 1.1 | 40 ± 9.4 |
| 15 | UIJR-048 | C-8-Me 3-amino-2,4-dione | R1 = d | 0.625 ± 0.12 | 0.156 ± 0.01 | 0.625 ± 0 | 2.5 ± 0.12 |
| 16 | UING5-249 | C-8-OMe fluoroquinolone | R1 = d | 0.062 ± 0 | 1 ± 0.23 | 0.125 ± 0.01 | 0.125 ± 0.01 |
OMe, methoxy; Me, methyl.
For the structures represented by the letters a, b, c, and d, see Fig. 1.
Values are averages ± standard deviations. Where the standard deviation is zero, at least three replicates had identical values. wt, wild type.
Effects of quinolone and quinazoline-2,4-dione structures on antimutant activity.
To relate drug structure to inhibition of mutant growth, several fluoroquinolones and diones were compared for their abilities to block the growth of a set of otherwise isogenic gyrA mutants. In this assay, activity data are expressed as the ratio of the MIC for the mutant to the MIC with the wild type (antimutant activity). The C-8-methoxy group of moxifloxacin lowered this ratio by a factor of 2, as seen in a comparison with the C-8-H compound Bay y 3114 (Fig. 2A). Two corresponding 2,4-diones (UING5-47 and UING5-157), each of which has a C-8-methoxy group and the same C-7 ring system as moxifloxacin, lowered the ratio even more (Fig. 2A). A similar phenomenon was observed when the compounds contained an ethyl-substituted piperazinyl ring attached to position C-7 (Fig. 2B). These data illustrate the general effect of the C-8-methoxy group and the 2,4-dione structure on antimutant activity.
Fig. 2.

Activities of fluoroquinolones and diones against M. smegmatis mutants. The MICs of each compound with wild-type M. smegmatis and three GyrA variants, KD2008 (G89C) (shaded bars), KD1987 (A91V) (filled bars), and KD1991 (D95Y) (open bars), were determined. Then the ratio of the MIC with the mutant to the MIC with the wild type was calculated. Compounds are grouped in panels according to the C-7 ring structure. (A) Compounds in the moxifloxacin series (R1 = a); (B) compounds in the PD161148 series (R1 = b); (C) compounds in the UING5-207 series (R1 = d, except for UING5-159). Similar results were obtained in replicate experiments.
Since pilot studies and work with E. coli (12) have indicated that the 2,4-dione UING5-207 had a particularly low ratio of the MIC for the mutant to the MIC with the wild type, we conducted several additional comparisons among derivatives of this compound. First, a 1,3-dione (UIJR1-100) was found to be less active than UING5-207 by the antimutant activity test (Fig. 2C). Reduced antimutant activity was also seen with an 8-methyl-2,4-dione derivative (UIJR-048) and with a 2,4-dione lacking a 3-amino group (UING5-200). A change in the C-7 ring system from an aminomethyl-substituted pyrrolidine group (UING5-207) to the amino pyrrolidine substituent (UING5-159) generated only slightly less antimutant activity. These data indicate that M. smegmatis UING5-207 has the best antimutant activity of the compounds tested.
The set of compounds related to UING5-207 was especially active with the GyrA G89C substitution (Fig. 2). Indeed, for eight diones, the MIC with this mutant was at or below the MIC with the wild type (Table 2; Fig. 2). In contrast, the cognate fluoroquinolones were more active with the A91V and D95Y mutants than with the G89C mutant relative to wild-type cells (Table 2; Fig. 2). The preference was readily seen when the ratios of the MIC for C89 to the MIC for Y95 were compared (Table 3). Three diones were outliers: UING5-48, UING5-63, and UING5-209 failed to show greater activity with the G89C variant (Table 3). UING5-48 and UING5-63 differed from the other two 2,4-diones (UING5-47 and UING5-200) only in the C-7 ring structure; UING5-209 also differed from its cognate 3-amino-2,4-diones only in the C-7 ring structure. These results indicate that C-7 substituents can affect binding interactions with GyrA in the 89-to-95 region of helix IV, which is far from the C-7 position in published X-ray structures of related compounds (2, 21, 22, 43).
Table 3.
Effect of C-7 ring structure on activity with G89C and D95Y GyrA variants of M. smegmatis
| Compound name | Compound typea | C-7 groupb | MIC ratio (89C/95Y)c |
|---|---|---|---|
| Moxifloxacin | Fluoroquinolone | a | 8 |
| PD161148 | Fluoroquinolone | b | 1 |
| UING5-248 | Fluoroquinolone | c | 2 |
| UING5-249 | Fluoroquinolone | d | 8 |
| UING5-47 | 2,4-Dione | a | 0.125 |
| UING5-48 | 2,4-Dione | b | 1 |
| UING5-63 | 2,4-Dione | c | 1 |
| UING5-200 | 2,4-Dione | d | 0.25 |
| UING5-157 | 3-NH2-2,4-dione | a | 0.062 |
| UING5-209 | 3-NH2-2,4-dione | b | 1 |
| UING5-159 | 3-NH2-2,4-dione | c | 0.2 |
| UING5-207 | 3-NH2-2,4-dione | d | 0.5 |
| UIJR1-100 | 1,3-Dione | d | 0.062 |
| UIJR-048 | 8-Me-2,4-dione | d | 0.062 |
8-Methoxy except for UIJR-048. Me, methyl.
The C-7 ring structures represented by the letters a through d are diagramed in Fig. 1.
Ratio of the MIC for the G89C mutant to that for the D95Y mutant.
Effect of 2,4-dione on selection of resistant mutants.
Population analysis was performed with M. smegmatis to assess the selection of resistant mutants by the 3-amino-2,4-dione UING5-207. Wild-type cells were grown to stationary phase and were applied to agar plates containing various concentrations of either UING5-207 or its cognate fluoroquinolone, UING5-249. After incubation of agar plates, colonies were counted and retested for susceptibility to the selecting compound. The fraction of cells recovered as mutants was then expressed as a function of the MIC99 (Fig. 3). The point at which extrapolation of each population analysis profile intersected the dashed line in Fig. 3 (unlabeled arrows) was taken to be the mutant prevention concentration (MPC, defined empirically as the drug concentration at which the fraction of colonies recovered is less than 1 in 1010 [9]). Population analysis profiles were similar at the low drug concentrations expected to select nongyrase mutants (47), while the 3-amino-2,4-dione was more restrictive at the higher drug concentrations where gyrase mutants are known to be selected by fluoroquinolones (47). Thus, the results of population analysis at high drug concentrations are consistent with the antimutant activity measured with known gyrase resistance mutants. This observation supports the idea that the ability of a compound to block the growth of known mutants parallels its ability to restrict the growth of undefined mutant subpopulations.
Fig. 3.
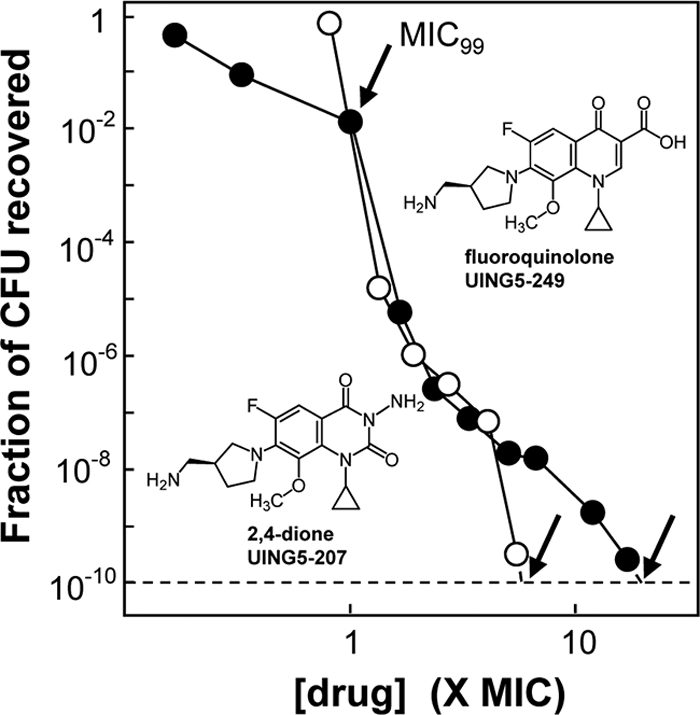
Effect of the dione versus quinolone core structure on mutant subpopulation enrichment. Wild-type M. smegmatis was grown to stationary phase and was then applied to agar plates containing the indicated concentrations of the 2,4-dione UING5-207 (open circles) or the cognate fluoroquinolone UING5-249 (filled circles) as a function of the drug concentration that blocks colony formation for 99% of the CFU in the culture (MIC99). The fraction of mutant cells recovered was determined as described in Materials and Methods. Unlabeled arrows indicate the MPC/MIC99 ratio. Similar results were obtained in replicate experiments; the average MPC/MIC99 ratio ± standard deviation was 20 ± 3.3 for UING5-249 and 6 ± 1.6 for UING5-207.
Effect of 2,4-dione structure on lethal activity.
In previous work with E. coli, we found that a 3-amino-2,4-dione (UING5-207) exhibits the same rapid killing as its cognate fluoroquinolone when drug concentrations are normalized to MIC (12). Killing was also observed when exponentially growing M. smegmatis was treated with various concentrations of UING5-207 for 18 h (Fig. 4A, open circles). Indeed, the compound killed cells with the same concentration dependence observed for the cognate fluoroquinolone (compare open circles in Fig. 4A with those in Fig. 4B); moxifloxacin was about 3-fold more lethal when the 99% lethal dose (LD99) was compared (Fig. 4C).
Fig. 4.
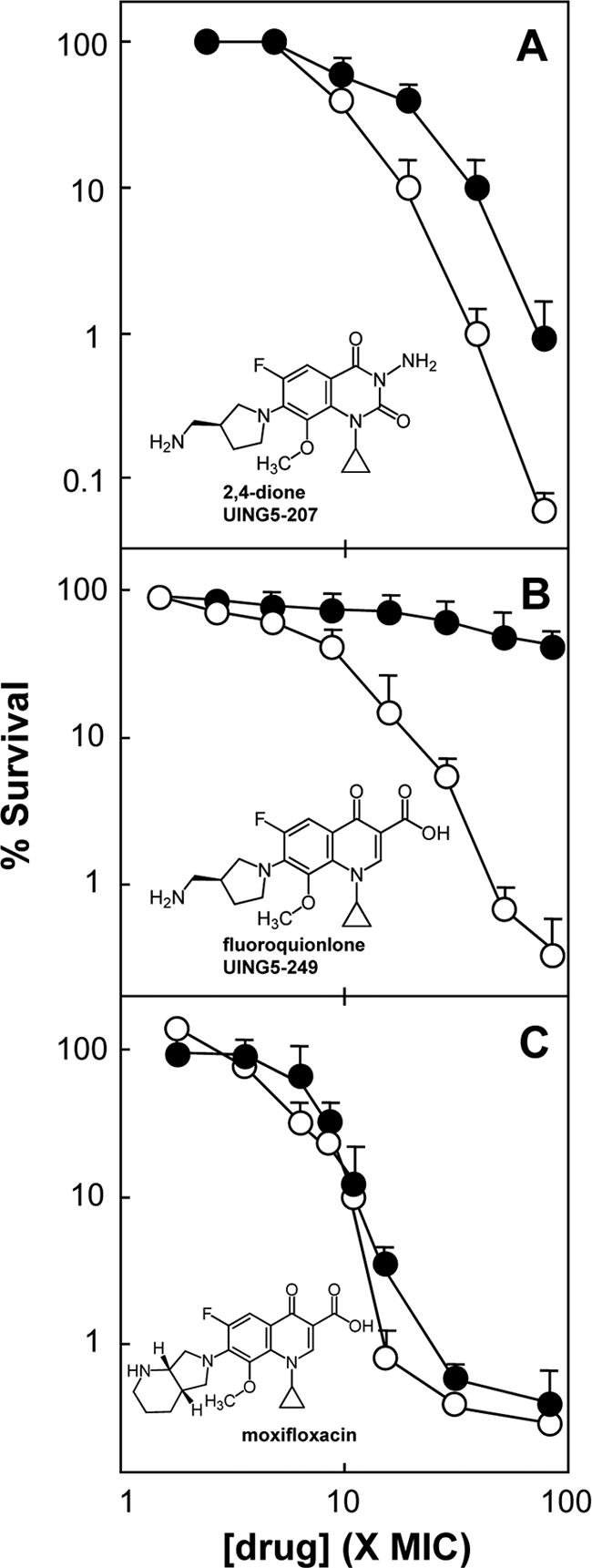
Effect of the dione versus quinolone core structure on lethal action against M. smegmatis. Growing cultures of wild-type M. smegmatis were either left untreated (open circles) or treated with chloramphenicol at 20 μg/ml (filled circles) for 1 h and were then exposed to the indicated concentrations of a 2,4-dione (UING5-207) (A), a fluoroquinolone (UING5-249) (B), or moxifloxacin (C). After 18 h, cultures were assayed for viable colony numbers for the determination of the percentage of survival. Similar results were obtained in replicate experiments.
Earlier work with killing by quinolones revealed that only a subset of quinolone-class agents can kill cells when protein synthesis is blocked (15, 26, 28). With E. coli and mycobacteria, the ability of fluoroquinolones to kill cells in the presence of chloramphenicol, an inhibitor of protein synthesis, depends on the structure of the C-7 substituent (26, 29, 31). This phenomenon was also observed in the present work: the lethal activity of the fluoroquinolone UING5-249 was blocked by chloramphenicol, while that of moxifloxacin was not (Fig. 4B and C). These compounds differ only in the structure of their C-7 ring systems. The lethal activity of UING5-207 was inhibited by chloramphenicol to a lesser extent than that observed with the fluoroquinolone (compare Fig. 4A and B). Thus, the core ring systems (2,4-dione versus 3-carboxylate-substituted quinolone) also affect lethal action in the absence of protein synthesis.
Modeling of 2,4-dione binding to gyrase and DNA.
Several X-ray crystal structures in which gyrase or topoisomerase IV is complexed to DNA and a fluoroquinolone or dione (2, 21, 22, 43) serve as the only available models for M. smegmatis gyrase-drug-DNA interactions (type II topoisomerase structures are very similar). In the X-ray structures, the fluoroquinolone and dione cores intercalate into DNA at the sites of nicks. The C-7 substituents extend into the GyrB/ParE region of gyrase/topoisomerase IV, and the 3-carboxylate/dione ends of the drugs point toward α-helix IV of GyrA/ParC (Fig. 5), where the major fluoroquinolone resistance substitutions are located. The 4-keto and 3-carboxylate groups of the fluoroquinolone coordinate Mg2+ and form a water-mediated binding contact with an aspartic acid of GyrA (amino acid 95 in M. smegmatis [Fig. 6A]). The carboxylate group also forms a hydrogen bond and a salt bridge with Arg-129. In many bacteria, the amino acid at the position equivalent to 91 in M. smegmatis is serine, which can form hydrogen bonds with the fluoroquinolone 3-carboxylate (43). In M. smegmatis, amino acid 91 is alanine, a hydrophobic amino acid that does not form hydrogen bonds. This alanine lowers the fluoroquinolone susceptibility of M. smegmatis and many other mycobacterial species (13, 14), presumably because of the loss of this drug-topoisomerase hydrogen bond.
Fig. 5.

Fluoroquinolone/dione-gyrase/topoisomerase IV-DNA complex. The structure of moxifloxacin bound to Acinetobacter baumannii topoisomerase IV was taken from Protein Data Bank access code 2XKK to show how the quinolones orient on type II topoisomerases while intercalating into DNA. Moxifloxacin is shown as a space-filling model. The C-7 ring system is at the upper end of each of the two moxifloxacin molecules; the carboxyl moiety is at the lower end, near GyrA helix IV.
Fig. 6.

Models of quinolone- and dione-topoisomerase-DNA complexes derived from published X-ray structures. In all cases amino acids are numbered according to the M. smegmatis gyrase numbering scheme for consistency with the text; data are from structures obtained with topoisomerase IV. (A) X-ray structure (access code 2XKK) of a fluoroquinolone (moxifloxacin) bound to a cleaved DNA type II topoisomerase complex. ParE/GyrB residue T500 and ParC/GyrA residues A91 and D95 have been modeled into the structure according to the amino acid sequence of M. smegmatis GyrA. The C-7 ring system interacts with ParE/GyrB as shown in the inset. The 3-carboxyl and 4-oxo groups interact with ParC/GyrA, particularly with D-95 through hydrogen bonding with Mg2+-coordinated water and with R-129 through hydrogen bonding and electrostatic attraction. Data are for Acinetobacter baumannii topoisomerase IV and are taken from reference 43. (Insets) Positioning of ParE/GyrB amino acids with respect to the C-7 group of moxifloxacin (top) (43) or its counterpart in the 2,4-dione PD0305970 (taken from reference 21 and modeled onto moxifloxacin). (B) X-ray structure of a 2,4-dione (PD0305970) interacting with type II topoisomerase and DNA. The C-7 ring system interacts with ParE/GyrB, while the N-3 amino and C-2 oxygen form hydrogen bonds with R-129. Data are for Streptococcus pneumoniae topoisomerase IV (PDB access code 3LTN) and are taken from reference 21. (Inset) Positioning of ParE/GyrB amino acids with the C-7 ring system. (C) Superposition of a fluoroquinolone (FQ) and a 3-amino-2,4-dione in drug-topoisomerase IV-DNA complexes. Positions are shown for moxifloxacin (PDB access code 2XKK) and PD0305970 (PDB access code 3LTN). (D) Proposed effects of three resistance substitutions on the fluoroquinolone-topoisomerase interaction. For illustrative purposes, three amino acid substitutions (G89C, A91V, and D95Y) are shown in the same figure as space-filling models along with moxifloxacin as shown in panel A. Y95 eliminates Mg2+ coordination with C-3 carboxylate; V91 increases the hydrophobicity around the fluoroquinolone carboxylate; and C89 causes R-129 to rotate, possibly disrupting the carboxyl-arginine hydrogen bond (shown as a red dotted line) and salt bridge. (E) Proposed effects of three resistance substitutions on the dione-topoisomerase interaction. For illustrative purposes, all three amino acid substitutions (G89C, A91V, and D95Y) are shown in the same figure as space-filling models along with PD0305970. The 89C, 91V, and 95Y substitutions do not interfere with interactions between the dione and the enzyme. (F) Comparison of interactions between ParC/GyrA helix IV and diones. 3-Amino-2,4-dione (left) and 2,4-dione (right) are shown as space-filling models interacting with helix IV of topoisomerase IV, also as a space-filling model (PDB access code 3LTN). The 3-amino dione has an extra hydrogen bond with R-129 and fits more closely with helix IV.
The X-ray structure of a 3-amino-2,4-dione complexed with DNA and S. pneumoniae topoisomerase IV shows the dione oriented in the same way as that seen for fluoroquinolones (20). Diones lack a 3-carboxyl group and do not coordinate Mg2+ (34) (Fig. 6B); consequently, there is no bridging contact with D95 and no need for Mg2+ to mitigate carboxylate-carboxylate repulsion. Thus, dione activity is not as sensitive as fluoroquinolone activity to substitutions for the negatively charged amino acid at position 95. The dione is also shifted away from GyrA helix IV by about 1.5 Å (Fig. 6C), which would make diones less sensitive to the steric effects of resistance substitutions in helix IV. Moreover, modeling suggests that the 3-amino diones can form a hydrogen bond network with Arg-129 of GyrA (Fig. 6B). Variants at position 129 are expected to be highly resistant to diones based on this model (Fig. 6B); however, such variants are unlikely to be viable, due to the involvement of Arg-129 in the gyrase catalytic reaction (24, 32, 37). Thus, the formation of a key contact between diones and Arg-129, an amino acid that cannot be altered without affecting cell viability, may be a significant factor in antimutant activity.
The substitution of cysteine at position 89, which strongly reduces susceptibility to fluoroquinolones (Fig. 2), is expected to contribute a hydrophobic environment near the 3-carboxylate of fluoroquinolones and a steric clash with Arg-129 (Fig. 6D) (the latter would disrupt interactions of Arg-129 with the fluoroquinolone carboxylate group). The X-ray structure of fluoroquinolone complexed with topoisomerase IV and DNA also shows a gap of about 1 Å between the fluoroquinolone and the helix IV surface. If the comparable region of M. smegmatis gyrase were filled by fluoroquinolone, a substitution of cysteine at position 89 would clash with the fluoroquinolone carboxylate (Fig. 6D).
Manual docking of the dione UING5-207 with topoisomerase IV modeled to contain resistance substitutions (Fig. 6E) shows the absence of the steric clash between Cys-89 and Arg-129 that is present in comparable fluoroquinolone-containing complexes (the orientation of Arg-129 is altered by an interaction with the dione 2-oxo group [compare Fig. 6E and D]). This factor could contribute to the absence of protection against the dione by the 89-Cys substitution. Manual docking also suggests that the 89-Cys substitution might favor the Arg-129 orientation seen in dione-containing complexes, which could contribute to greater activity of diones against this mutant than against wild-type cells. The dione 3-amino group would also improve the fit between the dione and helix IV, which would explain the lower MIC for 3-amino diones than for 3-H diones (Table 2). The hydrogen bond formed by the 3-amino group and the flanking DNA base of the bottom strand would explain differences in nucleotide sequence preference between diones and fluoroquinolones when cleavage is catalyzed by S. pneumoniae gyrase (34).
Manual docking also showed that fluoroquinolones and diones differ in the interaction of their C-7 rings with GyrB/ParE. The insets in Fig. 6 show that the dione interacts more closely than the fluoroquinolone with Arg-482 and Glu-501. This explains the greater effect of these amino acid substitutions on dione MICs than on fluoroquinolone MICs (34). Since the C-7 rings are far from GyrA helix IV in the X-ray structures, the current models do not easily explain the influence of the C-7 ring structure on the MIC of an 89C variant relative to a 95Y variant that was observed for some diones (Table 3). C-7 ring structure may have effects on binding that are not reflected by the static nature of the X-ray structures.
DISCUSSION
The work described above probed drug-gyrase interactions with M. smegmatis using structural variations in GyrA, fluoroquinolones, and quinazolinediones. To examine relationships between drugs and gyrase structures for possible differences in interactions, the MIC for each drug was measured with three gyrA mutants and was normalized to the wild-type MIC. The ratio of the MIC for the mutant to the MIC for the wild type was lower for all diones tested than for their cognate fluoroquinolones (Fig. 2). This general result is consistent with the observation that fluoroquinolones and diones select different resistance mutations (34) and is largely explained by models of fluoroquinolone- and dione-topoisomerase-DNA complexes derived from X-ray analyses (2, 21, 22, 43). Since the MICs of the diones for M. smegmatis (Table 2) and M. tuberculosis (data not shown) were high, considerable refinement will likely be required to make diones into clinically useful agents. However, comparisons between diones and fluoroquinolones provide support for the mutant selection window hypothesis (Fig. 2 and 3) and the biological relevance of the reported X-ray structures.
In the X-ray structures, the 3-carboxyl group of fluoroquinolones points toward GyrA/ParC helix IV, and the C-7 ring system of the drugs points into the GyrB/ParE region (Fig. 5). Resistance substitutions, such as A91V and D95Y, are expected to interfere with fluoroquinolone binding by loss of magnesium coordination and by steric hindrance. Diones lack a 3-carboxylate group and do not orient within the complex as closely to GyrA helix IV as cognate fluoroquinolones. These observations explain why 2,4-diones are not as active as cognate fluoroquinolones when inhibition of purified gyrase or topoisomerase IV activity is examined (2- to 20-fold difference with E. coli and S. aureus enzymes [33]). However, 2,4-dione activity with purified gyrase is not reduced as much as fluoroquinolone activity by fluoroquinolone-resistant substitutions in helix IV of GyrA (33). In the present work, the GyrA G89C substitution raised MICs for fluoroquinolones, while for many diones it lowered MICs, in some cases below the level with the wild type (Table 2; Fig. 2). The effects of the 89C substitution are explained by differences in fluoroquinolone and dione interactions near the N terminus of helix IV. The absence of this effect with E. coli (12) probably arises from differences in the structures of E. coli and mycobacterial GyrA in the region immediately around the position equivalent to 89 (42).
Among the diones tested, the 8-methyl-2,4-dione (UIJR-048) had the lowest MIC for wild-type cells, an observation consistent with previous work showing that an 8-methyl moiety typically imparts a lower MIC for the wild type than an 8-methoxy group with several Gram-positive and Gram-negative organisms (11, 40). One likely explanation for this phenomenon is that 8-methyl diones fit better in the nicked DNA site of ternary complexes, perhaps because the methyl group at position 8 orients in the plane of the core ring system. In contrast, an 8-methoxy group sticks out of the plane of the core ring system, where it might interfere with intercalation into the nicked site. We are working to make cognate 8-methyl fluoroquinolones in order to test this possible difference in complex binding with quinolones.
The activity of the 8-methoxy dione UING5-207 was less sensitive than that of the 8-methyl derivative (UIJR-048) to some resistance mutations when data were normalized to wild-type activity (Fig. 2C). This difference suggests that a more intimate fit with gyrase and/or DNA might lower the absolute MIC for the methyl derivative but raise its sensitivity to structural changes caused by resistance mutations. Thus, MICs for wild-type cells and for resistant mutants may reflect divergent structure-function activities. Indeed, in all cases examined, fluoroquinolones exhibited a lower MIC for wild-type cells (Table 2), but the cognate diones had a lower ratio of mutant MIC to wild-type MIC (Fig. 2). Consequently, simply making a more potent antimicrobial (lowering the MIC) will not necessarily produce agents that are more effective at restricting the emergence of resistance. In fact, a very potent drug with high-affinity binding to gyrase via a very intimate lock-and-key fit will likely be hypersusceptible to resistance mutations. To achieve good antimutant activity concurrent with a low MIC for the wild type, diones and quinolones may need to have increased binding with the nicked DNA moiety in the drug-enzyme-DNA complexes, and/or they may need to maintain bridging capabilities to GyrA helix IV residues in spite of resistance substitutions. Another possibility is to form binding contacts with functionally required amino acids that cannot be sites of resistance mutation (e.g., R-129). Testing of these ideas requires biochemical studies with purified wild-type and mutant gyrases to eliminate possible differences among compounds with respect to efflux, uptake, and nonspecific interactions with other cellular proteins.
The effects of the C-7 ring structure on the activities of diones and fluoroquinolones against bacteria with resistance substitutions in GyrA helix IV are not readily explained by structural models of drug-topoisomerase-DNA complexes, because the C-7 rings point toward GyrB/ParE, not GyrA/ParC (Fig. 5). In the present work, the ratio of the MIC for the 89C mutant to the MIC for the 85Y mutant differed among both fluoroquinolones and 2,4-diones in which the only structural differences were in the C-7 ring structure (Table 3). C-7 ring effects were also observed in earlier work with M. smegmatis: an 89C substitution raised the MIC more for a fluoroquinolone that had an N-ethyl on its C-7 piperazinyl ring than for a C-ethyl derivative (39). Explaining C-7 ring effects may require postulating an additional step in drug binding that places the C-7 ring close to amino acid 89, perhaps as part of drug binding that occurs prior to DNA cleavage (7, 19). C-7 structure may also affect drug intercalation into the nicked site of DNA, possibly altering the distance between the drug and helix IV. Regardless of the explanation, it is clear that currently available X-ray structures fail to explain C-7 ring effects or the 2-step nature of complex formation (7, 19).
The different lethal effects of various fluoroquinolones and diones also remain unexplained by models describing ternary complexes. For killing, the structure of the C-7 ring is particularly important, especially when protein synthesis is blocked by chloramphenicol (26, 30, 31). In the present work, this phenomenon was seen when the lethal effects of UING5-249 and moxifloxacin were compared (Fig. 4). Surprisingly, a dione eliminated most of the inhibitory effect of chloramphenicol (Fig. 4). Thus, the dione/carboxyl end of the compounds may also be important for killing nongrowing cells. Rapid killing, which requires higher drug concentrations than ternary complex formation, correlates with chromosome fragmentation (5, 31). A current problem is to understand how the drug binding seen in X-ray structures leads to chromosome fragmentation.
ACKNOWLEDGMENTS
We thank Marila Gennaro, Hiroshi Hiasa, and Richard Pine for critical comments on the manuscript.
This work was supported by NIH grants R01-AI73491, R01-GM3071721, and R01-AI87671l.
Footnotes
Published ahead of print on 7 March 2011.
REFERENCES
- 1. Agrawal D., Udwadia Z., Rodriguez C., Mehta A. 2009. Increasing incidence of fluoroquinolone-resistant Mycobacterium tuberculosis in Mumbai, India. Int. J. Tuberc. Lung Dis. 13:79–83 [PubMed] [Google Scholar]
- 2. Bax B., et al. 2010. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 19:935–940 [DOI] [PubMed] [Google Scholar]
- 3. Beylin V., et al. 2007. The preparation of two, preclinical amino-quinazolinediones as antibacterial agents. Org. Process Res. Dev. 11:441–449 [Google Scholar]
- 4. Caminero J., Sotgiu G., Zumla A., Migliori G. 2010. Best drug treatment for multidrug-resistant and extensively drug-resistant tuberculosis. Lancet Infect. Dis. 10:621–629 [DOI] [PubMed] [Google Scholar]
- 5. Chen C.-R., Malik M., Snyder M., Drlica K. 1996. DNA gyrase and topoisomerase IV on the bacterial chromosome: quinolone-induced DNA cleavage. J. Mol. Biol. 258:627–637 [DOI] [PubMed] [Google Scholar]
- 6. Cole S. T., et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544 [DOI] [PubMed] [Google Scholar]
- 7. Critchlow S. E., Maxwell A. 1996. DNA cleavage is not required for the binding of quinolone drugs to the DNA gyrase-DNA complex. Biochemistry 35:7387–7393 [DOI] [PubMed] [Google Scholar]
- 8. Dong Y., Xu C., Zhao X., Domagala J., Drlica K. 1998. Fluoroquinolone action against mycobacteria: effects of C-8 substituents on growth, survival, and resistance. Antimicrob. Agents Chemother. 42:2978–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong Y., Zhao X., Domagala J., Drlica K. 1999. Effect of fluoroquinolone concentration on selection of resistant mutants of Mycobacterium bovis BCG and Staphylococcus aureus. Antimicrob. Agents Chemother. 43:1756–1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Drlica K., Malik M., Kerns R. J., Zhao X. 2008. Quinolone-mediated bacterial death. Antimicrob. Agents Chemother. 52:385–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ellsworth E., et al. 2006. 3-Aminoquinazolinediones as a new class of antibacterial agents demonstrating excellent antibacterial activity against wild-type and multidrug resistant organisms. J. Med. Chem. 49:6435–6438 [DOI] [PubMed] [Google Scholar]
- 12. German N., Malik M., Rosen J., Drlica K., Kerns R. 2008. Use of gyrase resistance mutants to guide selection of 8-methoxy-quinazoline-2,4-diones. Antimicrob. Agents Chemother. 52:3915–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guillemin I., Jarlier V., Cambau E. 1998. Correlation between quinolone sensitivity patterns and sequences in the A and B subunits of DNA gyrase in mycobacteria. Antimicrob. Agents Chemother. 42:2084–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guillemin I., et al. 1999. Purification and inhibition by quinolones of DNA gyrases from Mycobacterium avium, Mycobacterium smegmatis and Mycobacterium fortuitum bv. peregrinum. Microbiology 145:2527–2532 [DOI] [PubMed] [Google Scholar]
- 15. Howard B. M., Pinney R. J., Smith J. T. 1993. 4-Quinolone bactericidal mechanisms. Arzneimittelforschung 43:1125–1129 [PubMed] [Google Scholar]
- 16. Huband M. D., et al. 2007. In vitro and in vivo activities of PD 0305970 and PD 0326448, new bacterial gyrase/topoisomerase inhibitors with potent antibacterial activities versus multidrug-resistant gram-positive and fastidious organism groups. Antimicrob. Agents Chemother. 51:1191–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hutchings K., et al. 2008. Synthesis and antibacterial activity of the C-7 side chain of 3-aminoquinazolinediones. Bioorg. Med. Chem. Lett. 18:5087–5090 [DOI] [PubMed] [Google Scholar]
- 18. Jacobs W. R., et al. 1991. Genetic systems in mycobacteria. Methods Enzymol. 204:537–555 [DOI] [PubMed] [Google Scholar]
- 19. Kampranis S., Maxwell A. 1998. The DNA gyrase-quinolone complex, ATP hydrolysis and the mechanism of DNA cleavage. J. Biol. Chem. 173:22615–22626 [DOI] [PubMed] [Google Scholar]
- 20. Kimura Y., Atarashi S., Takahashi M., Hayakawa I. 1994. Synthesis and structure-activity-relationships of 7-[3-(1-aminoalkyl)pyrrolidinyl]quinolone and 7-[3-(1-aminocycloalkyl)pyrrolidinyl]quinolone antibacterials. Chem. Pharm. Bull. 42:1442–1454 [DOI] [PubMed] [Google Scholar]
- 21. Laponogov I., et al. 2010. Structural basis of gate-DNA breakage and resealing by type II topoisomerases. PLoS One 5:e11338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laponogov I., et al. 2009. Structural insight into the quinolone-DNA cleavage complex of type IIA topoisomerases. Nat. Struct. Mol. Biol. 16:667–669 [DOI] [PubMed] [Google Scholar]
- 23. Linder J., Huang E., Steinman M., Gonzales R., Stafford R. 2005. Fluoroquinolone prescribing in the United States: 1995 to 2002. Am. J. Med. 118:259–268 [DOI] [PubMed] [Google Scholar]
- 24. Liu Q., Wang J. 1998. Identification of active-site residues in the “GyrA” half of yeast DNA topoisomerase II. J. Biol. Chem. 273:20252–20260 [DOI] [PubMed] [Google Scholar]
- 25. Lu T., Zhao X., Drlica K. 1999. Gatifloxacin activity against quinolone-resistant gyrase: allele-specific enhancement of bacteriostatic and bactericidal activity by the C-8-methoxy group. Antimicrob. Agents Chemother. 43:2969–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Malik M., Drlica K. 2006. Moxifloxacin lethality with Mycobacterium tuberculosis in the presence and absence of chloramphenicol. Antimicrob. Agents Chemother. 50:2842–2844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malik M., Hoatam G., Chavda K., Kerns R., Drlica K. 2010. Novel approach for comparing the abilities of quinolones to restrict the emergence of resistant mutants during quinolone exposure. Antimicrob. Agents Chemother. 54:149–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malik M., Hussain S., Drlica K. 2007. Effect of anaerobic growth on quinolone lethality with Escherichia coli. Antimicrob. Agents Chemother. 51:28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Malik M., et al. 2005. Lethality of quinolones against Mycobacterium smegmatis in the presence or absence of chloramphenicol. Antimicrob. Agents Chemother. 49:2008–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malik M., et al. 2010. Effect of N-1/c-8 ring fusion and C-7 ring structure on fluoroquinolone lethality. Antimicrob. Agents Chemother. 54:5214–5221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malik M., Zhao X., Drlica K. 2006. Lethal fragmentation of bacterial chromosomes mediated by DNA gyrase and quinolones. Mol. Microbiol. 61:810–825 [DOI] [PubMed] [Google Scholar]
- 32. Okada Y., et al. 2000. Assignment of functional amino acids around the active site of human DNA topoisomerase II alpha. J. Biol. Chem. 275:24630–24638 [DOI] [PubMed] [Google Scholar]
- 33. Oppegard L., et al. 2010. Comparison of in vitro activities of fluoroquinolone-like 2,4- and 1,3-diones. Antimicrob. Agents Chemother. 54:3011–3014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pan X., Gould K., Fisher L. 2009. Probing the differential interactions of quinazolinedione PD 0305970 and quinolones with gyrase and topoisomerase IV. Antimicrob. Agents Chemother. 53:3822–3831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rosen J. D., German N., Kerns R. J. 2009. Efficient synthesis of the 2-amino-6-chloro-4-cyclopropyl-7-fluoro-5-methoxy-pyrido[1,2-c]pyrimidine-1,3-dione core ring system. Tetrahedron Lett. 50:785–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rustomjee R., et al. 2008. A phase II study of the sterilising activities of ofloxacin, gatifloxacin and moxifloxacin in pulmonary tuberculosis. Int. J. Tuberc. Lung Dis. 12:128–138 [PubMed] [Google Scholar]
- 37. Schmidt B., Burgin A., Deweese J., Osheroff N., Berger J. 2010. A novel and unified two-metal mechanism for DNA cleavage by type II and IA topoisomerases. Nature 465:641–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sieradzki K., Roberts R., Haber S., Tomasz A. 1999. The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N. Engl. J. Med. 340:517–523 [DOI] [PubMed] [Google Scholar]
- 39. Sindelar G., et al. 2000. Mutant prevention concentration as a measure of fluoroquinolone potency against mycobacteria. Antimicrob. Agents Chemother. 44:3337–3343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tran T., et al. 2007. Structure-activity relationships of 3-aminoquinazolinediones, a new class of bacterial type-2 topoisomerase (DNA gyrase and topo IV) inhibitors. Bioorg. Med. Chem. Lett. 17:1312–1320 [DOI] [PubMed] [Google Scholar]
- 41. Tran T., et al. 2005. Facile synthesis of substituted 3-amino-1H-quinazoline-2,4-diones. J. Heterocyclic Chem. 42:669–674 [Google Scholar]
- 42. Tretter E., Schoeffler A., Weisfield S., Berger J. 2010. Crystal structure of the DNA gyrase GyrA N-terminal domain from Mycobacterium tuberculosis. Proteins 78:492–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wohlkonig A., et al. 2010. Structural basis of quinolone inhibition of type IIA topoisomerases and target-mediated resistance. Nat. Struct. Mol. Biol. 17:1152–1153 [DOI] [PubMed] [Google Scholar]
- 44. Zhao X., Drlica K. 2002. Restricting the selection of antibiotic-resistant mutants: measurement and potential uses of the mutant selection window. J. Infect. Dis. 185:561–565 [DOI] [PubMed] [Google Scholar]
- 45. Zhao X., et al. 1998. Killing of Staphylococcus aureus by C-8-methoxy fluoroquinolones. Antimicrob. Agents Chemother. 42:956–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhao X., Xu C., Domagala J., Drlica K. 1997. DNA topoisomerase targets of the fluoroquinolones: a strategy for avoiding bacterial resistance. Proc. Natl. Acad. Sci. U. S. A. 94:13991–13996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou J., et al. 2000. Selection of antibiotic resistant bacterial mutants: allelic diversity among fluoroquinolone-resistant mutations. J. Infect. Dis. 182:517–525 [DOI] [PubMed] [Google Scholar]