

Abstract
Novel classes of antimicrobials are needed to address the challenge of multidrug-resistant bacteria such as methicillin-resistant Staphylococcus aureus (MRSA). Using the architecture of the MRSA interactome, we identified pyruvate kinase (PK) as a potential novel drug target based upon it being a highly connected, essential hub in the MRSA interactome. Structural modeling, including X-ray crystallography, revealed discrete features of PK in MRSA, which appeared suitable for the selective targeting of the bacterial enzyme. In silico library screening combined with functional enzymatic assays identified an acyl hydrazone-based compound (IS-130) as a potent MRSA PK inhibitor (50% inhibitory concentration [IC50] of 0.1 μM) with >1,000-fold selectivity over human PK isoforms. Medicinal chemistry around the IS-130 scaffold identified analogs that more potently and selectively inhibited MRSA PK enzymatic activity and S. aureus growth in vitro (MIC of 1 to 5 μg/ml). These novel anti-PK compounds were found to possess antistaphylococcal activity, including both MRSA and multidrug-resistant S. aureus (MDRSA) strains. These compounds also exhibited exceptional antibacterial activities against other Gram-positive genera, including enterococci and streptococci. PK lead compounds were found to be noncompetitive inhibitors and were bactericidal. In addition, mutants with significant increases in MICs were not isolated after 25 bacterial passages in culture, indicating that resistance may be slow to emerge. These findings validate the principles of network science as a powerful approach to identify novel antibacterial drug targets. They also provide a proof of principle, based upon PK in MRSA, for a research platform aimed at discovering and optimizing selective inhibitors of novel bacterial targets where human orthologs exist, as leads for anti-infective drug development.
INTRODUCTION
Recent increases in antibiotic resistance among bacterial pathogens such as methicillin-resistant Staphylococcus aureus (MRSA), coupled with a dearth of new antibiotic development over the past 3 decades, have created major problems in the clinic. As such, there is an urgent need to identify novel, high-quality drug targets that can be used to develop new classes of highly effective antimicrobials. While antibiotics in current use have emerged almost exclusively from the whole-cell screening of natural products and small-molecule libraries, recent advances in genomic sciences, target identification, and assay development have enabled target-driven drug discovery approaches. The majority of these efforts, however, focused exclusively on unique bacterial targets of toxicity. Linked to this is the concern that new antibiotics targeting pathogen-specific proteins will likely exert the same level of selective pressures on the pathogen as did their predecessors, leading inevitably to the development of antibiotic resistance (30, 32, 41, 42). To avoid or minimize this problem, new antibiotic development strategies based on modern integrative knowledge of bacterial cellular processes and mechanisms of bacterial pathogenesis are critically needed.
One such strategy is the use of large-scale, genome-wide protein interaction networks in bacteria for initial target selection. Bacterial interactomes have the potential to provide invaluable insights into systems biology by allowing the analysis of biomolecular networks supported by specific protein-protein interactions. Thus, bacterial interactomes have great potential to expand our understanding of pathways and subnetworks and to identify highly connected essential hubs as potential novel antibacterial drug targets. Moreover, given that hubs are generally essential for network integrity, they are expected to be less prone to genetic mutations and subsequent resistance emergence due to the network centrality-lethality rule (12). To this end, we recently mapped the architecture of a protein interaction network (PIN) between 608 proteins of S. aureus MRSA252 (7). As a result of this analysis, pyruvate kinase (PK), the product of a single-copy gene, was identified as a highly connected hub protein in MRSA. Furthermore, we also found that PK is absolutely essential for S. aureus viability based upon PK antisense and gene disruption experiments (44). The essential requirement for PK for bacterial growth was also reflected by its high enzymatic activity during the exponential phase of the S. aureus life cycle. Taken together, these findings provide a clear rationale for selecting PK as a novel, candidate drug target (44).
PK (EC 2.71.40) catalyzes the final step in glycolysis with the irreversible conversion of phosphoenolpyruvate (PEP) to pyruvate with the concomitant phosphorylation of ADP to ATP (38). As PK plays a major role in the regulation of glycolysis, its inhibition leads to the interruption of carbohydrate metabolism and energy depletion. Moreover, both the substrate and the product of this reaction feed into a number of biosynthetic pathways, placing PK at a pivotal metabolic intersection. The X-ray crystal structures of several PKs from different species (e.g., Escherichia coli, Leishmania mexicana, Bacillus stearothermophilus, cat, rabbit muscle, human erythrocyte, and Saccharomyces cerevisiae) reveal a generally conserved architecture (16, 18, 29, 35, 36). PKs exist as homotetramers of identical subunits, each consisting of three to four domains: the N-terminal, A, B, and C domains (Fig. 1). The N-terminal helical domain is absent in prokaryotic bacterial PKs. The active site lies at the interface of the A and B domains in each of the four subunits, while the binding site for the allosteric effector appears to be located in the C domain (Fig. 2) (16, 18, 37). Allosteric regulation, in particular, provides mechanisms for the tight modulation of PK enzymes. Tetramerization occurs through interactions between the A and C domains of adjacent subunits so that bordering A domains constitute the A—A or “large” interface and adjacent C domains form the C—C or “small” interface between monomers (Fig. 1 and 2) (37).
Fig. 1.

Structure of the MRSA PK tetramer showing the domain boundaries and tetramer architecture. Domains were the A domain (yellow), B domain (blue), C domain (red), and extra-C-terminal domain (CT) (green). Each monomer has been colored to facilitate the identification of subunits in the tetramer. The large (A—A) and small (C—C) interfaces between monomers are shown as dashed lines.
Fig. 2.

Sequence alignment showing the C domains of MRSA PK and human PK isoforms. GenBank accession numbers are YP_041163.1 for MRSA252 (this study), NP_872270.1 for human M1 (hM1), NP_002645.3 for human muscle isozyme M2 (hM2), NP_000289.1 for human LR1 (hLR1), and NP_870986.1 for human LR2 (hLR2). Sequences were aligned by using ClustalW2 software (3). Amino acids in boldface type indicate effector binding sites: the binding sites of the sugar, 1-phosphate, and 6-phosphate moieties of FBP are indicated by closed circles, open squares, and closed squares, respectively. The poorly conserved residues are shaded in the MRSA PK sequence. Absolutely conserved, highly conserved, and poorly conserved residues are marked by asterisks, colons, and dots, respectively.
Due to the evolutionary distances between bacterial PKs (e.g., MRSA) and human isoenzymes, PK in MRSA possesses different kinetic properties and regulatory mechanisms than do human isozymes. These differences can be attributed to differences in protein sequences and structures (Fig. 1 and 2). MRSA PK is allosterically regulated by AMP and ribose 5-phosphate (R5P), which induces the conversion of the active R state of the enzyme from the inactive T state (44). In contrast, mammals possess four isoenzymes of PK, one (M1) of which is constitutively active and the other three (M2, L, and R) of which are allosterically regulated by fructose 1,6-bisphosphate (FBP) (23). Recently, it was shown that the allosteric transition between the inactive T state and the active R state involved a symmetrical 6° rigid-body rocking motion of the A- and C-domain cores in each of the four subunits. This also involved the formation of eight essential salt bridge locks across the C—C interface that provided tetramer rigidity and a 7-fold increase in the enzyme activity (22).
The selective targeting of human PK M2 containing a unique region for allosteric regulation in the C domain recently provided an opportunity to target cell metabolism for cancer therapy despite the high homology (95% identity) between M1 and M2 splice variants (39). Furthermore, despite the high degree of similarity between eukaryotic PKs, they have recently been drawing a lot of attention as novel targets for antitrypanosomal, antileishmanial, and antimalarial drugs (6, 8, 9, 26, 27). Therefore, sequence divergence between MRSA PK and human PKs is of particular interest for the design of selective inhibitors that specifically target the bacterial enzyme.
Sequence alignment between MRSA PK and human PKs revealed particular sequence divergence in the C domain (e.g., sites for effector binding) (Table 1 and Fig. 1 and 2) as well as 2 unique amino acid deletions in the A and C domains, which are involved in the formation of the A—A and C—C interfaces between the adjacent PK subunits. These regions provide tetramer rigidity and efficient catalytic activity of the enzyme (44) (Fig. 1). The distinctive MRSA PK C domain is of particular interest for the design of selective inhibitors that act specifically against bacteria. This was suggested by the results of several previously reported studies indicating that single mutations in the effector binding sites or sulfate removal led to an altered stability of C—C interfaces along with surprisingly profound effects on the rates of enzyme catalysis due to altered dynamics of the enzyme conformation (10, 13, 14, 20, 37).
Table 1.
Percent identities between domains of MRSA PK and the corresponding domains of human PK isoforms and PKs from E. faecium, S. pneumoniae, E. coli, and P. aeruginosaa
| PK | % identity (% similarity) |
||
|---|---|---|---|
| A domain | B domain | C domain | |
| Human | |||
| M1 | 57 (74) | 40 (57) | 30 (50) |
| M2 | 57 (74) | 40 (57) | 35 (55) |
| LR1 | 56 (70) | 36 (56) | 33 (55) |
| LR2 | 56 (70) | 36 (56) | 33 (55) |
| Bacterial | |||
| E. faecium PK | 78 (85) | 60 (77) | 57 (68.0) |
| S. pneumoniae PK | 48 (63) | 37 (66) | 40 (57) |
| E. coli PK1 | 63 (77) | 41 (62) | 37 (56) |
| E. coli PK2 | 48 (67) | 35 (55) | 33 (48) |
| P. aeruginosa PK1 | 47 (67) | 28 (55) | 32 (55) |
| P. aeruginosa PK2 | 47 (66) | 32 (52) | 31 (47) |
GenBank accession numbers are YP_041163.1 for MRSA PK, NP_872270 for human PK isoform M1, AAA36449.1 for human PK isoform M2, NM_000298 for human PK isoform LR1, NM_181871.3 for human PK isoform LR2, NP_814779.1 for E. faecium PK, YP_816275.1 for S. pneumoniae PK, NP_310410 for E. coli isoform PK1, NP_310591 for E. coli isoform PK2, NP_250189.1 for P. aeruginosa isoform PK1, and NP_253019.1 for P. aeruginosa isoform PK2.
The recent determination of the crystal structure of S. aureus PK (R. Zoraghi et al., unpublished data; P. Axerio-Cilies et al., unpublished data) and comparisons with human PKs have highlighted significant structural differences that motivated us to utilize a rational, structure-based approach to identify MRSA-specific PK inhibitors that preferentially bind to MRSA PK. In the present study, the atomic resolution structure of MRSA PK, virtual database screening, and enzyme activity assays were used in combination to identify low-molecular-weight, selective inhibitors of MRSA PK. Medicinal chemistry was used to improve the potency and structural features of the in silico hits. The discovery of these compounds indicates for the first time that evolutionarily conserved, essential, highly connected bacterial proteins such as MRSA PK with structural differences from their mammalian orthologs have clear potential as novel, high-quality antibacterial drug targets.
MATERIALS AND METHODS
Bacterial strains and compounds.
An epidemic methicillin-resistant S. aureus (MRSA) strain sequenced at the Sanger Centre (MRSA252; NRS71), S. aureus RN4220 (NCTC8325; NRS144), an MRSA strain sequenced at The Institute for Genomic Research (TIGR) (COL; NRS100), and a community-acquired MRSA strain sequenced at the National Institute of Technology and Evaluation, Tokyo, Japan (USA400; MW2; NRS123) were obtained from the NARSA (Network on Antimicrobial Resistance in S. aureus). Methicillin-sensitive S. aureus (MSSA) (ATCC 25923), Streptococcus pneumoniae (ATCC 49619), Streptococcus pyogenes (ATCC 700294), Listeria monocytogenes (ATCC 19115), Enterococcus faecium (ATCC 35667), Enterococcus faecalis (ATCC 29212), and Enterococcus faecium ATCC 700221 (vancomycin-resistant enterococcus [VRE]) were obtained from the Global Bioresource Center, ATCC. Multidrug-resistant (MDR) MRSA, Staphylococcus saprophyticus, Staphylococcus haemolyticus, Staphylococcus epidermidis, Acinetobacter baumannii, and extended-spectrum β-lactamase (ESBL)-producing Klebsiella pneumoniae strains were clinical isolates (Vancouver General Hospital, Vancouver, British Columbia, Canada). Escherichia coli DYE330 and Pseudomonas aeruginosa PAO1 were from the laboratory of B. B. Finlay. Escherichia coli IMP was obtained from Julian Davies (University of British Columbia, Vancouver, British Columbia, Canada). We obtained Pseudomonas aeruginosa PAO200, which is a PAO1 mexAB-oprM deletion strain, and Pseudomonas aeruginosa PAO75, which is PAO1 carrying mutations in mexAB-oprM, mexAB-oprM, mexCD-oprJ, mexEF-oprN, mexJK, mexXY, and opmH pscC, from Herbert Schweizer (Colorado State University).
Compounds identified by in silico screening were purchased from Chembridge, Chemdiv, IBScreen, Enamine, Asinex, Maybridge, Nanosyn, InterBioScreen, Timtec, and Aurora. Analogs of potent in silico hits were synthesized in the laboratory of R. N. Young. Compound stocks (10 mM) were prepared in dimethyl sulfoxide (DMSO) and stored at −20°C.
Generation of PK constructs.
Genomic DNA of MRSA Sanger strain MRSA252 extracted using DNeasy tissue kit (Qiagen) was used as a template to generate His-tagged MRSA PK. Human cDNA from the MCF-7 breast cancer cell line (courtesy of J. Wong, BC Cancer Research Center, Canada) was used as a template to generate the full-length human M2 PK enzyme. The following primer sets were used, creating appropriate restriction sites (NdeI and XhoI sites are underlined): M27F (5′-CTACATATGAGAAAAACTAAAATTGTATG-3′) and M27R (5′-GTTCTCGAGTTATAGTACGTTTGCATATCCTTC-3′) for the cloning of MRSA PK and hM2F (5′-GATCATATGATGTCGAAGCCCCATAGTGAAGCC-3′) and hM2R (5′-GTTCTCGAGTCACGGCACAGGAACAACACGCATG-3′) for the cloning of the human M2 PK isoform. The resulting PCR fragments for each construct were cloned into the unique NdeI and XhoI sites of the bacterial expression vector pET-28a(+) (Novagen). This step resulted in plasmids pET-28a-MRSA and pET-28M2, which generated N-terminal His-tagged recombinant MRSA PK and human M2 PK. The sequence and the correct reading frame of all constructs were verified by sequencing. Human M1, R, and L PK constructs in pET-28-a(+) vectors (courtesy of L. Cantley, Harvard Medical School, Boston, MA) were used to generate relevant recombinant His-tagged human PK isoforms (8, 9).
Expression and purification of recombinant His-tagged MRSA PK and human PKs.
MRSA and human constructs in pET-28a(+) were used to express relevant recombinant PK proteins in E. coli BL-21(DE3). The proteins were expressed and purified by using Ni-nitrilotriacetic acid (NTA) agarose (Qiagen) according to the manufacturer's protocol. Briefly, cells were grown to an absorbance of 0.4 to 0.5 at 600 nm in 2× YT medium and then induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) for 3 h at 20°C. Cells were lysed by sonication on ice (three 10-s bursts with a 30-s recovery between bursts) in lysis buffer (0.2 mg/ml lysozyme, 50 mM Tris [pH 7.5], 10 mM MgCl2, 200 mM NaCl, 100 mM KCl, 10% glycerol, 10 mM imidazole, 0.5% NP-40, and 1 mM dithiothreitol [DTT] containing Complete protease inhibitor). Cell lysates were cleared by centrifugation (18,000 × g in a Beckman JA-20 rotor) for 20 min at 4°C, and PK isoforms were purified by batch binding to Ni-NTA resin. The resins were then packed into columns (1 by 2 cm) and washed with 400 column volumes of lysis buffer containing 30 mM imidazole. His-tagged PK isoforms were eluted with the same buffer containing 300 mM imidazole. The proteins were dialyzed overnight at 4°C against 2,000 volumes of an ice-cold solution containing 30 mM Tris (pH 7.5), 25 mM KCl, 5 mM MgCl2, 10% glycerol, and 1 mM DTT to remove imidazole. All purification steps were done at 4°C; enzymes were flash-frozen and stored at −70°C. The enzymatic activity of frozen protein preparations was stable for at least 10 months and up to 5 freeze-thaw cycles. The purity and physical integrity of proteins were assessed by using SDS-polyacrylamide gel electrophoresis (PAGE) followed by Coomassie blue staining. The protein concentration was estimated by a Bradford assay (Bio-Rad protein assay) using bovine serum albumin as a standard.
In silico screening and CADD.
The crystal structure (Protein Data Bank [PDB] accession number 1T5A) for human PK was obtained from the PDB (30) and used for a subsequent structural superimposition with the MRSA PK structure (Axerio-Cilies et al., unpublished) to identify an unique suitable small-molecule binding pocket in MRSA PK. Details of the in silico screening and computer-aided drug discovery (CADD) will be described elsewhere. In summary, a total of 135 compounds were selected and purchased for biochemical and biological assays based upon a primary in silico screen of the ZINC chemical database, version 5.0 (containing 3.3 million compounds), to identify hit compounds predicted to specifically bind to a distinct cavity in MRSA PK. Subsequently, a second group of 73 compounds were selected in a secondary in silico similarity search for analogs of IS-63 (a positive hit identified in the primary screen) using version 8.0 of the ZINC compound library, containing ∼8.7 million compounds (15). These were prioritized for further characterization.
Measurement of pyruvate kinase activity.
Candidate MRSA PK inhibitors identified by CADD and medicinal chemistry programs were assayed for their abilities to inhibit enzymatic activities of MRSA PK and human PKs. The PK activity was determined by using a continuous assay coupled to lactate dehydrogenase (LDH), in which the change in absorbance at 340 nm owing to the oxidation of NADH was measured by using a Benchmark Plus microplate spectrophotometer (Bio-Rad) (8). The reaction mixture contained 60 mM Na+-HEPES (pH 7.5), 5% glycerol, 67 mM KCl, 6.7 mM MgCl2, 0.24 mM NADH, 5.5 units l-LDH from rabbit muscle (Sigma-Aldrich), 2 mM ADP, and 10 mM PEP (i.e., close to the Michaelis-Menten constant [Km] of MRSA PK so that the IC50s should approximate the inhibition constant [Ki]) in a total volume of 200 μl. Reactions were initiated by the addition of one of the PK enzymes (15 nM) to the mixture. PK activity proportional to the rate of the change at 340 nm was expressed as specific activity (μmol/min/mg), which is defined as the amount of PK that catalyzes the formation of 1 μmol of either product per minute. Inhibitors were dissolved in DMSO, with the final concentration of the solvent never exceeding 1% of the assay volume. IC50s were calculated by curve fitting on a four-parameter dose-response model with a variable slope using Graphpad Prism 5.0 (GraphPad Software, Inc., La Jolla, CA). In all studies, less than 10% of the total PEP was exhausted during the reaction. Reactions were performed at 30°C for 5 min. All values determined represent three measurements, each in triplicate, unless mentioned otherwise. Mode-of-inhibition and Ki values were determined by simultaneously changing the inhibitor concentration (0 to 400 nM) and substrate PEP concentration (2 to 20 mM) while keeping the level of the ADP substrate fixed at 2 mM. The resulting curve at each inhibitor concentration was fitted by nonlinear regression to the allosteric sigmoidal kinetic model using Graphpad Prism software. Ki values were obtained by nonlinear regression curve fitting using the following equation (30):
| (1) |
In vitro susceptibility testing.
The antimicrobial activities of PK inhibitor candidates were determined by using the 96-well microtiter standard 2-fold serial broth microdilution method, as described by the CLSI (formerly NCCLS) (24), with the various Gram-positive and Gram-negative bacterial species mentioned above. Briefly, bacteria (from a single colony) were grown overnight in brain heart infusion (BHI) broth (Difco), harvested by centrifugation, and then washed twice with sterile distilled water. Each stock solution of compounds in DMSO was diluted with BHI broth to prepare serial 2-fold dilutions in the range of 500 to 0.06 μM. One hundred microliters of the broth containing about 5 × 105 CFU/ml of bacteria was used to inoculate each well of a 96-well plate containing 100 μl of the same medium with the indicated concentrations of candidate inhibitors. Culture plates were incubated for 24 h at 37°C, and the optical density at 600 nm (OD600) was measured by using a Benchmark Plus microplate spectrophotometer (Bio-Rad). To control for intrinsic absorbance by the inhibitors, control series containing inhibitor dilutions but no cells were run for every experiment, and the resulting absorbance values were subtracted as background from the experimental readings. We defined the MIC as the lowest concentration of test compound leading to a complete inhibition of cell growth in relation to compound-free control wells, as determined by optical density measurements. We read MICs by the OD; however, we also read MICs by eye for all species, including S. pneumoniae, to confirm the MIC indicated by the absorbance. These values were always concordant for all species, including S. pneumoniae. Erythromycin, vancomycin, and ciprofloxacin were used as reference compounds. All assays were run in triplicate. Experiments were replicated at least three times to verify reproducibility under the above-described conditions. To investigate the influence of serum on the in vitro antibacterial activities of PK lead compounds, the MICs of compounds NSK4-77 and NSK5-15 along with vancomycin (control) were determined for S. aureus ATCC 25923 and a community-acquired MRSA (CA-MRSA) strain (USA400) in the presence of 10% heat-inactivated fetal calf serum (FCS). Calf serum inactivated at 56°C for 30 min was added to BHI broth to a final concentration of 10% (vol/vol), with the pH adjusted to 7.2.
Determination of mammalian cytotoxicity.
The cytotoxic activities of compounds were determined for HeLa 229 cells (ATCC CCL-2-0.1) in microtiter cultures by measuring the dehydrogenase activity using a CellTiter 96 AQueous One solution cell proliferation assay (Promega, Madison, WI) according to the manufacturer's protocol. Briefly, freshly passaged cells were seeded into microtiter wells (2 × 104 cells/well) in medium containing 10% heat-inactivated fetal calf serum and grown for 24 h. The original medium was then removed and replaced with medium containing the desired concentration of the compound or solvent control (i.e., DMSO). Plates were incubated for 24 h at 37°C in a humidified incubator with a 5% CO2 atmosphere. At the end of the growth period, cells were lysed by the addition of 20 μl of CellTiter 96 AQueous One solution, and the incubation was continued for another 3 h at 37°C. The production of formazan was determined at 490 nm with the Benchmark Plus microplate spectrophotometer (Bio-Rad). To control for intrinsic absorbance, control series containing inhibitor dilutions but no cells were run for every experiment, and the resulting absorbance values were subtracted as background from the experimental readings. Growth in compound-free control wells was considered 100%, and the percentage of growth inhibition was calculated for each compound concentration. Cytotoxicity was quantified as the CC50, the concentration of compound that inhibited 50% of the conversion of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) to formazan (19). The “selectivity index” is defined as the ratio of the mammalian cell cytotoxicity to the MIC for S. aureus (i.e., CC50/MIC). Positive-control measurements were performed with xanthohumol (HeLa cell CC50 = ∼9 μg/ml). All assays were performed three times in triplicate.
Determination of pyruvate levels.
Cultures of S. aureus RN4220 with initial suspensions of approximately 1 × 106 CFU/ml in BHI broth supplemented with either 1% DMSO (control) or the highest sublethal concentration (i.e., 1.25 μg/ml presented 80% growth inhibition) of compound AM-165 (test) were incubated at 37°C for 3 h. Independent cultures of S. aureus RN4220 with either 1% water (control) or the highest sublethal concentrations of ciprofloxacin (0.25 μg/ml presented ∼70% growth inhibition) or vancomycin (0.5 μg/ml presented 75% growth inhibition) were incubated as negative controls. Before cell harvesting, the turbidity of the cultures was measured as the OD600, and samples were taken to determine the number of cells (CFU/ml). Cell pellets were washed twice with phosphate-buffered saline (PBS) and lysed for 30 min at 37°C in lysis buffer (25 mM Tris-HCl [pH 7.5], 120 μg/ml lysozyme, 120 μg/ml lysostaphin, 0.05% Triton X-100, 20 μg/ml DNase I, 2 mM MgCl2), followed by vortexing using a FastPrep FP120 cell disrupter (Qbiogene, Inc., Carlsbad, CA). Insoluble cell debris was removed by centrifugation at 25,000 × g for 10 min. Soluble cell extracts were used to determine pyruvate levels by the colorimetric measurement of pyruvate oxidation using a Pyruvate Determination assay kit (Bio Vision, Mountain View, CA), with reference to a pyruvate standard curve prepared according to the manufacturer's instructions.
Time-kill studies.
Time-kill analyses were performed according to CLSI method M26-A.20 (25). Staphylococcus aureus ATCC 25923 was cultured overnight at 37°C in BHI broth. Cells were diluted in medium to an initial OD600 of 0.1 (equal to 107 CFU/ml) and incubated with shaking for 2 h at 37°C to achieve logarithmic growth. The culture was then diluted in medium to adjust the cell density to approximately 107 CFU/ml. Either compound NSK5-15, adjusted to final concentrations of 1, 2, 4, and 8 times the MIC, or vancomycin, adjusted to final concentrations of 2 and 4 times the MIC, was then added. Aliquots (0.1 ml) of the cultures were removed at 0 h, 2 h, 4 h, 6 h, and 24 h of incubation, and serial 10-fold dilutions were prepared in saline as needed. The number of viable cells on drug-free BHI plates was determined after 24 h of incubation. Rates of killing were determined by measuring the reduction in numbers of viable bacteria (log10 CFU/ml) at 0 h, 1 h, 2 h, 4 h, 6 h, and 24 h with fixed concentrations of the compound. Experiments were performed in duplicate. If plates contained fewer than 10 CFU/ml, the number of colonies was considered to be below the limit of quantitation. Samples of cultures containing the compound were diluted at least 10-fold to minimize drug carryover to the BHI plates. Bactericidal activity is defined as a ≥3-log reduction in the initial CFU count within 24 h. The compound was considered to be bacteriostatic at the concentration that reduced the original inoculum by 0 to 3 log10 CFU/ml within 24 h.
Selection for resistant mutants.
Changes in the susceptibilities of bacteria to NSK5-15 were monitored during serial passages of S. aureus RN4220 in BHI broth containing the highest sublethal compound, and results are displayed as the highest sublethal compound concentration for each culture for each day. Two independent cultures of S. aureus RN4220 were grown in 96-well assay plates in the presence of several concentrations of compound (0.125× to 32× the MIC). Cultures were recovered from the well with highest compound concentration that exhibited growth (e.g., >15% of untreated controls). For the subsequent passages, the cells were grown in the liquid culture with the highest sublethal inhibitor concentration of the last passage with a starting inoculum of 5 × 105 CFU/ml of bacteria. This process was repeated for 25 passages, and results are displayed as the highest sublethal compound concentration for each culture for each passage. After passages 5, 10, 15, 20, and 25, colonies were isolated from cultures before the determination of the MIC. Changes in the susceptibilities of RN4220 to fusidic acid were monitored for 10 passages as a positive control.
RESULTS
Expression and purification of PK proteins.
Recombinant His-tagged PK proteins, including MRSA PK and human PK isoforms (M1, M2, L, and R), were expressed, purified to near homogeneity (>98%), and detected by SDS-PAGE (12% [wt/vol] gel), as described in Materials and Methods. The estimated molecular mass of each of the respective bands on SDS-PAGE gels correlated well with that predicted based on the amino acid composition of each PK protein (Fig. 3), indicating that all PK constructs were expressed as full-length proteins. The structural integrity of each protein was verified by the pattern of migration on SDS-PAGE gels and the allosteric properties of each construct (data not shown). All PKs demonstrated enzyme activity comparable to those reported previously (data not shown) (4, 17, 40, 44). MRSA and human PK proteins were used to characterize the biochemical properties of candidate MRSA PK inhibitors.
Fig. 3.
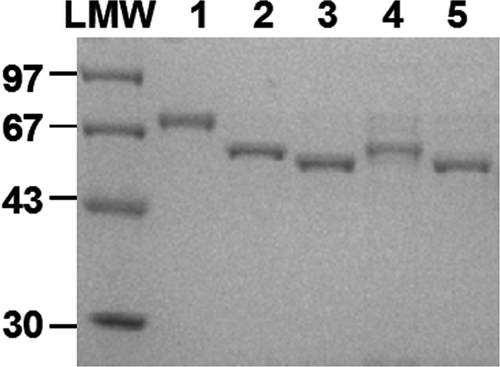
SDS-PAGE of purified MRSA PK and human PK isoforms. Five-microgram aliquots of MRSA PK and human M1, M2, R, and L PK proteins purified to homogeneity through the Ni-NTA chromatography step, as described in Materials and Methods, were applied onto lanes 1 to 5, respectively. Low-molecular-weight (LMW) standards (Pharmacia Biotech) were applied as size markers. Proteins were stained with Coomassie brilliant blue dye.
In silico screening for putative MRSA PK inhibitors.
The structural differences between MRSA and human PKs in the C domain were exploited to identify a unique suitable small-molecule binding pocket in the MRSA tetramer as the target for computer-aided drug discovery (CADD) by superimposing MRSA and human protein structures. The pocket is accessible to small molecules in MRSA PK, whereas it is partially blocked in human PKs by substituted amino acid residues. Details of in silico screening, CADD, and the nature of the distinct binding cavity distinct binding cavity of PK in MRSA will be described elsewhere (Axerio-Cilies et al., unpublished).
In vitro screening of putative in silico MRSA PK inhibitors.
Recombinant MRSA PK was used for biochemical inhibitor studies to examine a total of 135 compounds that were identified from an in silico screening of the ZINC compound library, version 5.0, against the unique MRSA binding pocket. For the initial screening, all compounds were used at a concentration of 50 μM, with a substrate concentration of 10 mM PEP, which is close to the Km for the PK in MRSA (e.g., 6.6 mM) so that the IC50s should approximate the Ki. Thirty-one (22.5%) of the compounds tested exhibited a significant effect (≥50% inhibition) on MRSA PK enzyme activity at 50 μM. Subsequently, these compounds were screened against human PK isoforms (M1, M2, R, and L) in search of a compound(s) that selectively inhibits bacterial PK. This screening resulted in the identification of an acyl hydrazone-based compound (IS-63) (molecular weight = 359.4) as a potent (IC50 of 0.85 μM) and selective MRSA PK inhibitor (with ≥45-fold selectivity over human PKs) (Table 2). However, IS-63 demonstrated poor antibacterial activity, most likely due to a lack of cellular penetration. Thus, a second round of in silico similarity searches was performed for compound IS-63 against the ZINC compound library, version 8.0 (15). Seventy-three analogs were selected and tested in a biochemical assay screen against MRSA PK and human PKs to identify compound IS-130 (molecular weight = 373.2) with markedly improved potency (IC50 of 0.091 μM), selectivity (with ≥1,370-fold selectivity over human PKs) (Table 2), and antibacterial activity (35% growth inhibition at 25 μM). No further inhibition of growth was achieved beyond 25 μM, probably due to compound instability over the time course of the experiment (e.g., 24 h) or limited membrane permeability. Subsequently, the toxic effects of IS-130 on human HeLa 229 cells were evaluated. The results indicated that IS-130 had no significant growth-inhibitory effects on HeLa cells (Table 2) at a concentration of up to 400 μM. Despite the fact that IS-130 exhibited only modest antibacterial potency, it was a small and efficient ligand with selective on-target activity with an apparently limited ability to penetrate the bacterial cell. It therefore provided a good starting point for future lead optimization through structure-activity relationship (SAR) programs.
Table 2.
Two acyl hydrazone-based in silico hits with potent selective inhibitory activity against MRSA PKa

All values represent the means of data from three independent experiments, each performed in triplicate. Antibacterial and human cell toxicity activities of compounds were evaluated as described in Materials and Methods, using S. aureus strain RN4220 and HeLa 293 cells, respectively, at a compound concentration of 100 μM.
Lead optimization: fragment-based approach.
A medicinal chemistry program was undertaken to synthesize IS-130 analogs, first to determine the structure dependencies of inhibition and second to introduce structural modifications that increase both the PK-inhibitory activity and antibacterial potency of the inhibitors. Details of these studies will be reported elsewhere (R. N. Young et al., unpublished data). The potency and selectivity of each derivative toward MRSA PK (IC50) as well as antibacterial activities (MIC) were measured to direct the iterative rounds of synthetic chemistry. Data obtained for key compounds derived from these studies are summarized in Table 3. Notably, the replacement of the indole sp2-hybridized nitrogen of IS-130 (R1) with a carbon atom (analog NSK4-66) resulted compounds with comparable inhibitory activities, but no significant antibacterial activity against MRSA was observed. An ethyl analog (AM-168), however, did show antibacterial activity, suggesting that increased lipophilicity might improve cell penetration and thus lead to enhanced antibacterial activity.
Table 3.
Inhibition of MRSA PK and antibacterial activities of synthesized derivatives of IS-130 with notable structure-activity relationshipsa

The MIC was determined for S. aureus RN4220, as determined by the broth microdilution method.
IS-130 is the hit compound.
The substitution of the indole NH with a methyl substituent conferred a robust increase in antibacterial activity, presumably via improved cell membrane penetration, leading to compounds with MIC values at the low-micromolar range. Compounds NSK4-77, NSK5-15, and AM-165, which exhibited the best combination of PK-inhibitory and antibacterial activities, were selected for further characterization. The relationship between the MIC and IC50 will be discussed elsewhere (Young et al., unpublished); however, the data presented here indicated that the greasiness and size of the R2 substituent increased while the MIC decreased, which may well be due to better permeability or better detergent action, to better enzyme inhibition, or to a combination of these.
PK lead compounds selectively inhibit MRSA PK.
To determine if the compounds were capable of acting selectively against MRSA PK, the inhibitory effects of the three most potent compounds with antibacterial activity (e.g., NSK4-77, NSK5-15, and AM-165) on the M1, M2, R, and L PK isoforms were tested in single-enzyme catalytic assays (Table 4). PK compounds with potent antibacterial activity displayed submicromolar (0.16 to 0.38 μM) IC50s toward bacterial PK, with a marked 180- to 940-fold selectivity over the human isoenzymes (Table 4). Therefore, PK lead compounds appeared to be a suitable starting point to develop highly specific antimicrobial agents.
Table 4.
Inhibition of enzymatic activities of MRSA PK and human PK isoforms by selected compoundsa
| PK isoform | NSK4-66 |
NSK4-77b |
NSK5-15b |
AM-165b |
||||
|---|---|---|---|---|---|---|---|---|
| Avg IC50 (μM) ± SD | Fold selectivity | Avg IC50 (μM) ± SD | Fold selectivity | Avg IC50 (μM) ± SD | Fold selectivity | Avg IC50 (μM) ± SD | Fold selectivity | |
| MRSA PK | 0.08 ± 0.01 | 1 | 0.38 ± 0.08 | 1 | 0.19 ± 0.01 | 1 | 0.16 ± 0.01 | 1 |
| Human M1 | 376 ± 6 | 4.4 × 103 | 178 ± 8 | 4.7 × 102 | 40 ± 3 | 2.2 × 102 | 99 ± 5 | 6.0 × 102 |
| Human M2 | 269 ± 17 | 3.2 × 103 | 251 ± 12 | 6.6 × 102 | 35 ± 2 | 1.9 × 102 | 187 ± 11 | 1.1 × 103 |
| Human R | 408 ± 27 | 4.8 × 103 | ND | ND | 35 ± 3 | 1.9 × 102 | 146 ± 12 | 8.9 × 102 |
| Human L | 369 ± 17 | 4.3 × 103 | ND | ND | 34 ± 2 | 1.8 × 102 | 155 ± 15 | 9.4 × 102 |
Values are averages ± deviations of values determined from two independent experiments performed in triplicates. ND, not determined.
Compound with potent antibacterial activities.
PK lead compounds are noncompetitive inhibitors.
To determine the nature of the inhibition by PK lead compounds, kinetic parameters of MRSA PK inhibition were determined with assay mixtures containing various concentrations of substrate (PEP) and different fixed concentrations of each inhibitor (e.g., NSK4-77 and NSK5-15) (Fig. 4). The maximal velocity (Vmax) and Michaelis-Menten constant (Km) were determined for each assay as described in Materials and Methods. The data suggested that inhibition by PK compounds could not be overcome by increasing the concentration of the substrate, indicating that the small-molecule inhibitor and the substrate (PEP) bind to distinct sites on the enzyme. Furthermore, the results presented in Fig. 4 show that the Km values remained unchanged (i.e., 6.5 mM) in the absence and presence of several fixed concentrations of both NSK4-77 (Fig. 4A) and NSK5-15 (Fig. 4B), whereas the Vmax was significantly decreased (e.g., up to 80%). Thus, PK lead compounds are noncompetitive inhibitors with respect to PEP, with inhibition constant (Ki) values of 269 ± 63 nM and 276 ± 82 nM for NSK4-77 and NSK5-15, respectively.
Fig. 4.

Inhibition studies with NSK4-77 and NSK5-15. (A) NSK4-77 was used at 0 nM (○), 100 nM (□), 200 nM (▿), and 400 nM (♢). (B) NSK5-15 was used at 0 nM (○), 50 nM (□), 100 nM (▿), and 200 nM (♢). The enzymatic activity of PK was assayed as described in Materials and Methods. The data shown are derived from one of two experiments performed in triplicate. Error bars indicate ranges of values within the single experiment shown.
PK lead compounds exhibit staphylococcus-specific antibacterial activity.
To investigate the antibacterial properties of PK inhibitors, the three most potent compounds from the focused SRA study were tested in vitro for their antibacterial activities against a diverse panel of bacterial species and strains. The results in Table 5 indicate that PK lead compounds showed effective in vitro antibacterial activities against all strains and species of staphylococci that were tested (MIC, 1.4 to 19.2 μg/ml), including methicillin-susceptible S. aureus (MSSA) (e.g., RN4220 and ATCC 25923), methicillin-resistant S. aureus (e.g., MRSA252, COL, and MW2), and a multidrug-resistant S. aureus (MDRSA) isolate that was resistant to many of the major classes of antibiotics. Compounds NSK5-15 and AM-165 also displayed MICs ranging from 1.4 to 5.1 μg/ml for other staphylococcal strains such as Staphylococcus epidermidis, Staphylococcus haemolyticus, and Staphylococcus saprophyticus (Table 5). PK compounds were inactive in vitro (MIC range, >73 to >93 μg/ml) against a range of Gram-negative human pathogens tested, such as Acinetobacter and Pseudomonas bacteria (Table 5). However, as shown in Table 5, the PK compounds were found to be active against a range of Gram-positive bacterial species, including several antibiotic-resistant strains. These compounds demonstrated superior antibacterial activities against Enterococcus faecalis, Enterococcus faecium, and vancomycin-resistant E. faecium (MICs, 0.25 to 2 μg/ml) as well as against Streptococcus pneumoniae and Streptococcus pyogenes (MICs, 0.5 to 8 μg/ml). Therefore, PK compounds demonstrated potent antibacterial activities against staphylococci, enterococci, and streptococci. Interestingly, the fact that PK hits are active against Streptococcus and Enterococcus species but not against E. coli and P. aeruginosa might be explained by the significantly higher domain identities between MRSA PK and Enterococcus and/or Streptococcus PKs than those between MRSA PK and E. coli and/or P. aeruginosa (Table 1).
Table 5.
Antibacterial activities of three potent PK inhibitors against selected staphylococci, nonstaphylococcal Gram-positive pathogens, and Gram-negative pathogens compared to standard antibioticsa
| Strain | MIC (μg/ml) |
|||
|---|---|---|---|---|
| NSK4-77 | NSK5-15 | AM-165 | Control | |
| Gram-positive bacteria | ||||
| Staphylococcus aureus RN4220 | 4.8 | 1.4 | 2.5 | 0.5,c 1,d 0.5e |
| Staphylococcus aureus ATCC 25923 | 9.6 | 2.9 | 5.0 | 0.5,c 1d |
| Staphylococcus aureus CA-MRSA (USA400) | 9.6 | 2.9 | 5.0 | 0.75,c 1d |
| Staphylococcus aureus HA-MRSA (COL) | 9.6 | 5.8 | ND | 0.30c |
| Staphylococcus aureus HA-MRSA252 | 9.6 | 2.9 | 5.0 | >10,c 0.5d |
| Staphylococcus aureus MDRSAb | 9.6 | 1.4 | 2.5 | >16c |
| Staphylococcus epidermidis | 9.6 | 1.4 | 2.5 | 1,d >4e |
| Staphylococcus haemolyticus | 19.2 | 2.9 | 5.0 | 0.1e |
| Staphylococcus saprophyticus | 9.6 | 2.9 | 5.0 | 1,d 0.5e |
| Enterococcus faecalis ATCC 29212 | >64 | 2 | >64 | 2-4,d <0.03e |
| Enterococcus faecium ATCC 35667 | >64 | 1 | >64 | 2,d 0.125e |
| Enterococcus faecium ATCC 700221 (VRE) | 8 | 0.25 | ND | >64,d 64e |
| Listeria monocytogenes ATCC 19115 | >77 | >93 | ND | 25e |
| Streptococcus pneumoniae ATCC 49619 | 0.5 | 1 | 0.5 | <0.03,c 0.25d |
| Streptococcus pyogenes ATCC 700294 | 8 | 8 | 8 | <0.125,c 2d |
| Gram-negative bacteria | ||||
| Acinetobacter baumannii X270295 | >77 | >186 | >162 | 0.8e |
| Escherichia coli DY330 | >77 | >93 | ND | >100,d 0.2e |
| Escherichia coli IMP | >125 | >125 | ND | 0.5,d 0.005e |
| ESBL-producing Klebsiella pneumoniae | ND | >93 | >81 | >50e |
| Pseudomonas aeruginosa PAO1 | >193 | >233 | >162 | 0.25e |
| Pseudomonas aeruginosa PAO200 | ND | >233 | >162 | 0.05e |
| Pseudomonas aeruginosa PAO75 | ND | >233 | >162 | 0.025e |
| Salmonella enterica serovar Typhimurium SL1344 | >193 | >233 | ND | <0.1e |
MICs were determined in BHI broth for all bacterial strains as described in Materials and Methods, except for enterococcal species and streptococcal species, for which MICs were determined with CAMHB (cation-adjusted Mueller-Hinton broth) (containing 20 mg of Ca2+/liter and 10 mg of Mg2+/liter) and CAMHB containing 2 to 5% laked horse blood, respectively. ND, not determined; HA-MRSA, hospital-acquired MRSA.
Resistant to cefazolin, clindamycin, ciprofloxacin, erythromycin, penicillin, oxacillin, and methicillin.
Value for erythromycin.
Value for vancomycin.
Value for ciprofloxacin.
PK lead compounds exhibit favorable selectivity indices (CC50/MIC).
The cytotoxicities of the three lead compounds were determined by using HeLa cells (cultured in medium containing 10% heat-inactivated fetal calf serum) in a 1-day incubation assay as described in Materials and Methods. The results shown in Fig. 5A show that PK lead compounds exhibited little cytotoxicity against mammalian cells (i.e., less than 30% of cell death at 500 μM, equal to 193, 232, and 202 μg/ml of NSK4-77, NSK5-15, and AM-165, respectively), with CC50 values of >200 μg/ml for all three compounds tested. The selectivity indices (CC50/MIC) for NSK4-77, NSK5-15, and AM-165 evaluated in relation to their very potent antibacterial activity (Fig. 5B) against MDRSA were 20, 80, and 40, respectively, indicating remarkable selectivity for bacterial versus mammalian cells in the modes of action of these compounds.
Fig. 5.

Cytotoxicity assessment of PK lead compounds against HeLa cells (A) compared to that of the MDRSA strain (B). Compounds NSK4-77 (○), NSK5-15 (□), and AM-165(▿) were used at 0 to 500 μM to determine CC50 and MIC values as described in Materials and Methods. The data presented are representative of three experiments performed in triplicate.
In order to investigate the influence of serum on the antibacterial efficacies of PK lead compounds in vitro, MICs of NSK4-77, NSK5-15, and vancomycin (control) in the presence of 10% heat-inactivated fetal calf serum were determined to be 19.2, 5.8, and 2 μg/ml, respectively, for S. aureus ATCC 25923 and 9.6, 5.8, and 1 μg/ml, respectively, for an S. aureus CA-MRSA strain (USA400). Compared with the values shown in Table 5 (generated in the absence of serum), it is clear that the presence of as much as 10% fetal calf serum did not significantly affect the antibacterial activities of PK lead compounds.
Bactericidal activity of the most potent PK compounds.
To determine if bioactive PK compounds were bactericidal, the activity of NSK5-15 was assessed by using S. aureus as a model organism. Figure 6 shows representative time-kill curves for compound NSK5-15 against MSSA ATCC 25923. Vancomycin was also used as a comparator drug. As shown in Fig. 6, maximum killing for NSK5-15 was observed at concentrations of 4× the MIC, with a 3-log drop in the numbers of CFU/ml occurring by 24 h after the addition of the compound, consistent with a bactericidal mode of action. Just slightly less than a 3-log drop in CFU/ml was also observed for NSK5-15 at 1× and 2× the MIC, suggesting a dose-dependent effect on staphylococcal killing up to a concentration equal to 4× the MIC. Killing was less rapid with 4× the MICs of NSK5-15 compared with those of vancomycin for the same strain. Similar rates of killing by NSK5-15 at 4× the MICs for an MDRSA strain were observed (data not shown). These results indicate that PK lead compounds are bactericidal against both MSSA and MDRSA.
Fig. 6.
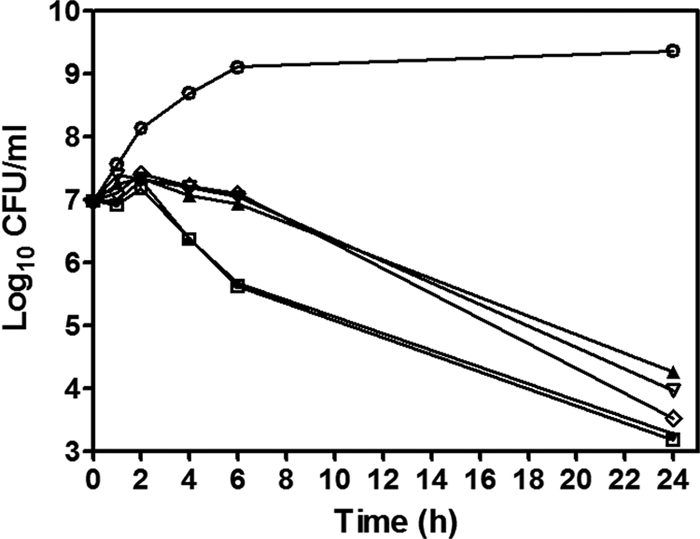
Time-kill curve of S. aureus incubated with AM-165. S. aureus ATCC 25923 was incubated with vehicle alone (e.g., DMSO) (○) as a growth control; AM-165 at 1× the MIC (▴), 2× the MIC (▿), 4× the MIC (♢), and 8× the MIC (▵); or vancomycin at 2× the MIC (□) and 4× the MIC (●) and sampled at the indicated time points. The log10 values of CFU/ml were plotted versus time as described in Materials and Methods. The data shown are from one of three independent experiments with similar results.
Resistance studies.
To assess the potential for cells to become resistant to the antibacterial effects of PK lead compounds, we tried to generate resistant mutants by using S. aureus RN4220. Cells were passaged for up to 25 consecutive generations in the presence of sublethal concentrations of NSK5-15 or for 10 generations in the presence of sublethal concentrations of fusidic acid. As shown in Fig. 7, after 25 subcultures of NSK5-15 in the presence of compound, the relative MIC of NSK5-15 for S. aureus remained stable, and mutants with significant increases in resistance to the compounds (>4× the MIC) were not detected. In contrast, mutants able to grow with concentrations up to 32× to 128× the (initial) MIC of fusidic acid appeared within 5 to 10 passages, indicating the emergence of resistant mutants.
Fig. 7.
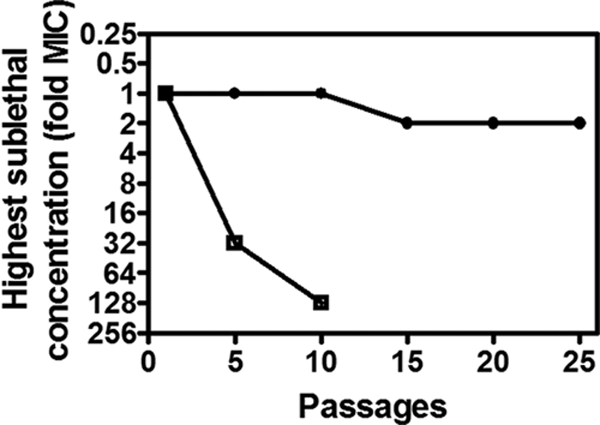
Serial passage of S. aureus RN4220 in either NSK5-15 (●) or fusidic acid (□). The highest sublethal concentration of compound (denoted as fold MIC) is plotted versus the number of days of serial passage for each compound, as described in Materials and Methods.
PK lead compounds reduced pyruvate production.
The target selectivity of the lead compound was determined by measuring pyruvate concentrations in S. aureus RN4220 strains incubated either with vehicle (i.e., DMSO) as a control or with sublethal concentrations of compound AM-165. As shown in Fig. 8, the pyruvate concentration was significantly reduced (by 4.3-fold) in AM-165-treated cells (0.19 ± 0.12 nmol per 5 × 107 cells) compared to the control cells (0.86 ± 0.31 nmol per 5 × 107 cells), indicating that essential PK (16) is the target of lead compounds for the inhibition of bacterial growth. Interestingly, as shown in Fig. 8, ciprofloxacin had no effect on the intracellular pyruvate level, which remained unchanged in ciprofloxacin-treated cells (0.82 ± 0.15 nmol per 5 × 107 cells) compared to the control cells (0.81 ± 0.19 nmol per 5 × 107 cells), while the intracellular pyruvate concentration increased slightly in vancomycin-treated cells (0.98 ± 0.21 nmol per 5 × 107 cells) compared to the control cells (0.79 ± 0.10 nmol per 5 × 107 cells). These results together with the demonstration that PK lead compounds inhibited PK enzymatic activity directly (Fig. 4 and Table 4) provide direct validation that the compounds are targeting PK. This mechanism of action was further confirmed with the recent availability of the X-ray structure of MRSA PK in complex with PK lead inhibitors (Axerio-Cilies et al., unpublished).
Fig. 8.

Pyruvate concentrations in S. aureus cells challenged with PK lead compound AM-165. Pyruvate concentrations were measured in S. aureus RN4220 cells incubated in the absence (control) or presence of the highest sublethal concentration of AM-165 (e.g., 1.25 μg/ml), ciprofloxacin (0.25 μg/ml), or vancomycin (0.5 μg/ml), as described in Materials and Methods. The data presented are means ± standard deviations (SD) of data from three independent experiments, each performed in duplicates.
DISCUSSION
The development of resistance to clinically important antibiotics by bacterial pathogens and potential biowarfare agents pose major threats to public health (2, 5, 21, 28). It is also clear that resistance is more likely to develop when newly introduced antibiotics target similar pathways or have structures that are chemically similar to agents already in use. Thus, new antibacterial compounds, possessing novel scaffolds and unique mechanisms of action, are urgently needed to combat the serious challenge of antimicrobial drug resistance in the clinic (28). In this study, we describe the strategy of using bacterial interactomes as a novel starting point for the identification of highly connected, essential hub proteins as potential drug targets. Since these hubs are critical to network integrity and stability, they should be ideal, novel targets for new antimicrobials. Using this strategy, MRSA PK was identified as a novel, potential target. Importantly, we found that the crystal structure of MRSA PK revealed structural differences between human and bacterial PKs (Zoraghi et al., unpublished; Axerio-Cilies et al., unpublished) (Fig. 1) that could potentially be exploited for drug development, thereby avoiding toxicity to host cells. The virtual screening of a commercially available database of low-molecular-weight compounds using computer-aided drug design (CADD) identified a novel class of potential inhibitors that appeared in silico to bind to MRSA PK selectively and not to human PK (Axerio-Cilies et al., unpublished). A subsequent experimental evaluation of candidate in silico binders led to the identification and characterization of a novel, potent, and selective PK inhibitor, IS-130 (Tables 2 and 3). Medicinal chemistry was then used to optimize the potency and selectivity of IS-130 (Young et al., unpublished), resulting in the identification of PK lead compounds (NSK4-77, NSK5-15, and AM-165) with submicromolar IC50 values (i.e., 0.16 to 0.38 μM) for MRSA PK and 180- to 1,100-fold selectivity toward the bacterial enzyme (Tables 3 and 4).
The anti-PK lead compounds that we identified had superior antistaphylococcal activity (Table 5) with favorable selectivity indices (CC50/MIC) over mammalian cells in vitro (Fig. 5). The latter finding suggests that these compounds were also selective for bacterial PK versus the mammalian isoenzymes in vivo. These candidate PK inhibitors, as structurally novel antibacterial agents, were found to have bactericidal activity, as a 3-log drop in the numbers of CFU/ml of both MSSA and MRSA strains was observed within 24 h after the addition of the compound (Fig. 6).
We believe that the basis for the selective targeting of staphylococcal PK versus human PKs is related to a structurally differentiated site located near the small interface formed by two PK protein monomers (Axerio-Cilies et al., unpublished). Compared to MRSA PK, the regions flanking the pocket in human PKs were poorly conserved in terms of amino acid sequence and were partially blocked by several amino acid residues that make the site inaccessible in human PKs.
The nature of this compound binding pocket also influenced the sensitivities of various other bacterial PKs to the novel candidate PK inhibitors. Some of the amino acids involved in forming the compound binding site in MRSA PK were not well conserved among other bacterial PKs (e.g., some Gram-negative bacteria), leading to the absence of favorable binding and a corresponding loss of antibacterial activity. Thus, the presence of favorable amino acid residues in the pocket located near the small interface formed by two protein monomers in bacterial PKs (e.g., MRSA) is crucial for high-affinity compound binding to the interface. This appears to explain the basis for the spectrum of Gram-positive antibacterial activity. In addition to higher levels of sequence (Table 1) and structural divergence in Gram-negative bacteria, the presence of two PK isoforms in a number of Gram-negative bacteria (e.g., E. coli, P. aeruginosa, Salmonella enterica serovar Typhi, Legionella pneumophila, and K. pneumoniae) may also contribute to poor antibacterial activities against Gram-negative organisms.
We used a medicinal chemistry approach to develop staphylococcus-specific drugs as noncompetitive inhibitors of PK. Further chemical optimization of these molecules could lead to the development of novel drugs with a reduced likelihood of being rendered ineffective due to the action of bacterial resistance mechanisms. This was suggested by the finding that we could not readily select for resistance to lead PK compounds during prolonged passage in culture in the presence of sublethal concentrations of the compound (Fig. 7). The finding that resistant PK isoforms did not emerge may be related to the essential requirement for PK for S. aureus growth and survival coupled with its critically connected network position with a severely limited tolerance for mutation. Thus, the new chemotypes that we identified appear to be attractive starting points for further development as antistaphylococcal agents. It is noteworthy, however, that the PK lead compounds exhibited strong antibacterial activity against a range of other Gram-positive bacteria, such as enterococcal and streptococcal species, with MICs in the low-micromolar range as well (Table 5). This is a very interesting feature of the PK lead compounds, as these organisms are frequently multidrug resistant, with relatively few good treatment options. Therefore, with a further optimization of the structures, there is the potential to develop an agent with activity to include these Gram-positive pathogens. However, PK compounds did not exhibit global activities against Gram-positive bacteria, as these compounds were inactive against Listeria monocytogenes (Table 5).
The inactivity of PK lead compounds against Gram-negative bacterial species was not due to their inability to penetrate the outer membrane (OM). Our results indicated that the antibacterial activities of PK lead compounds (e.g., NSK4-66, NSK4-77, and NSK5-15) against E. coli IMP, an extremely permeable strain of E. coli K-12 that is sensitive to detergents and antibiotics normally blocked by the OM, was not improved (Table 5) compared to those against E. coli DY330 (control), as PK lead compounds were not able to inhibit growth even at 125 μg/ml (exhibited only 30 to 50% growth inhibition at 125 μg/ml), while MICs of vancomycin and ciprofloxacin for E. coli IMP were significantly reduced (0.5 μg/ml and 0.005 μg/ml, respectively) compared to the control (>100 μg/ml and 0.2 μg/ml, respectively). Furthermore, we investigated the effects of a number of PK lead compounds (e.g., NSK4-77 and NSK5-15) on both recombinant E. coli PK1 and PK2 enzymes (44), and the results showed that PK lead compounds had no inhibitory effect on E. coli pyruvate kinase enzymes, as they exhibited only 1 to 4% inhibition of E. coli PK1 and PK2 at 10 μM.
The inactivity of PK lead compounds against Gram-negative organisms was not likely due to efflux pump effects. We investigated the antibacterial effects of PK lead compounds on two strains of Pseudomonas aeruginosa, PAO200 carrying a mexAB-oprM deletion and PAO75 carrying mexAB-oprM, mexAB-oprM, mexCD-oprJ, mexEF-oprN, mexJK, mexXY, and opmH pscC deletions. Our results indicated that the antibacterial activities of PK lead compounds remained unchanged (Table 5) compared to the Pseudomonas aeruginosa PAO1 control strain, as they were not able to inhibit growth even at concentrations as high as 233 μg/ml (they exhibited only up to 30 to 35% growth inhibition at 233 μg/ml), while ciprofloxacin exhibited significantly reduced MICs for PAO200 and PAO75 strains (0.05 μg/ml and 0.025 μg/ml, respectively, versus 0.25 μg/ml for the control PAO1 strain).
Recently, we provided direct evidence to show that S. aureus PK is absolutely essential for the growth and viability of this key Gram-positive pathogen (44). This was based upon the finding that the inactivation of chromosomal pyk by the insertion of a group II intron at various positions of sense and antisense strands of pyk created a lethal phenotype. Furthermore, our results clearly demonstrated that the PK enzymatic activity increased during the growth of S. aureus, and this correlated with increased PK protein expression levels in the early phase of growth (44). Consistent with these findings, the PK gene was recently identified as being an absolutely essential gene for the survival of other bacteria, such as Haemophilus influenzae, S. pneumoniae, and Mycobacterium tuberculosis (1, 31, 34, 43). Furthermore, PK activity was shown to be important for the growth of both Gram-negative (e.g., E. coli) and Gram-positive (e.g., Bacillus subtilis) bacteria (11, 33), as PK mutant strains demonstrated substantially decreased growth rates. PK might therefore be a potentially useful drug target in these pathogens as well. Evidence to support this comes from our results demonstrating that PK lead compounds have promising antibacterial activity against S. pneumoniae as well (Table 5). Modeling of these particular bacterial PKs versus human PKs will be required to assess this potential approach.
In relation to the findings reported above, two related and important questions to address are what is the evidence that the lead compounds are actually acting through the inhibition of PK and how this leads to bacterial killing. In regard to the latter question, PK is required for feeding pyruvate into the Krebs cycle, and an inhibition of PK would have been expected to compromise bacterial viability due to a deficiency of ATP. Regarding the first question, at least four findings support the conclusion that PK is the target of the lead compounds. These findings include (i) the essential nature of S. aureus PK (44); (ii) the correspondence between MICs (Table 5) and IC50s (Tables 3 and 4) of PK lead compounds for the growth of staphylococcal strains and the inhibition of MRSA PK enzymatic activity, respectively; and (iii) significant reductions in intracellular pyruvate concentrations in S. aureus cells treated with PK lead compounds (Fig. 8). Additional direct evidence to support the conclusion that lead compounds were actually acting through the inhibition of PK is the atomic resolution structure of MRSA PK showing it to be in complex with one of the leads (Axerio-Cilies et al., unpublished). Moreover, the results of the cocrystallization experiment provided significant insight into the binding determinants for selectivity and potency. This information is important and may be capitalized upon for the design of novel antibacterial agents that are not compromised by existing resistance mechanisms. In addition, modeling based upon the crystal structure of PK will help to determine more comprehensively the actual potential for targeting bacterial PKs more widely.
The possibility that PK lead compounds might have another target(s) could not be completely ruled out at this point. However, the reduction in levels of intracellular pyruvate upon compound dosing correlates well and is good evidence of a specific mechanism of action. In fact, in reality, most of the compounds generally hit more than one target to some degree. Therefore, the possibility that the lack of resistance is due to an inhibition of another target(s) cannot be ruled out at this time.
In conclusion, our findings show that the use of sequence-based structural divergence in critical bacterial hub proteins that have human orthologs represents a promising means of identifying novel drug targets. The attractiveness of these targets derives from their essentiality, their critical PIN locations and importance to network integrity, and their lower tolerance for mutations, which may restrict the development of drug resistance. This strategy represents a unique approach, since historically, known antimicrobial targets used in the clinic have been unique to bacterial pathogens and absent from the host. Moreover, conventional antimicrobial drug targets have not been selected based upon their hierarchical positions and importance in bacterial protein interactomes, as was the case for MRSA PK in this study.
ACKNOWLEDGMENTS
This work was supported by funding from Genome Canada and Genome British Columbia, Vancouver General Hospital (VGH) and University of British Columbia Hospital Foundation, and the SARS Accelerated Vaccine Initiative through the Proteomics for Emerging Pathogen Response (PREPARE) Project. Computer equipment for the PREPARE Project's Computational Genomics research was also supported by an in-kind contribution from IBM Healthcare and Life Sciences.
We thank Roger Leger from Indel Therapeutics Ltd., who provided us with MIC values for enterococcal and streptococcal species. We also thank Diane Roscoe from VGH Microbiology for providing us with clinical isolates; Jason Wong from the BC Cancer Research Center for providing us with the MCF-7 breast cancer cell line; Lewis C. Cantley and Matthew G. Vander Heiden from the Department of Systems Biology, Harvard Medical School, for providing us with M2, R, and L human PK constructs; Julian Davies (University of British Columbia, Vancouver, British Columbia, Canada) for providing us with the Escherichia coli IMP strain; and Herbert Schweizer (Colorado State University) for providing us with Pseudomonas aeruginosa PAO200 and Pseudomonas aeruginosa PAO75.
Footnotes
Published ahead of print on 28 February 2011.
REFERENCES
- 1. Akerley B. J., et al. 2002. A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl. Acad. Sci. U. S. A. 99:966–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Alanis A. J. 2005. Resistance to antibiotics: are we in the post-antibiotic era? Arch. Med. Res. 36:697–705 [DOI] [PubMed] [Google Scholar]
- 3. Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 4. Baranowska B., Baranowski T. 1982. Kinetic properties of human muscle pyruvate kinase. Mol. Cell. Biochem. 45:117–125 [DOI] [PubMed] [Google Scholar]
- 5. Barrett C. T., Barrett J. F. 2003. Antibacterials: are the new entries enough to deal with the emerging resistance problems? Curr. Opin. Biotechnol. 14:621–626 [DOI] [PubMed] [Google Scholar]
- 6. Chan M., Tan D. S., Sim T. S. 2007. Plasmodium falciparum pyruvate kinase as a novel target for antimalarial drug-screening. Travel Med. Infect. Dis. 5:125–131 [DOI] [PubMed] [Google Scholar]
- 7. Cherkasov A., et al. 2011. Mapping the protein interaction network in methicillin-resistant Staphylococcus aureus. J. Proteome Res. 10:1139–1150 [Epub ahead of print.] [DOI] [PubMed] [Google Scholar]
- 8. Christofk H. R., et al. 2008. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230–233 [DOI] [PubMed] [Google Scholar]
- 9. Christofk H. R., Vander Heiden M. G., Wu N., Asara J. M., Cantley L. C. 2008. Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452:181–186 [DOI] [PubMed] [Google Scholar]
- 10. Collins R. A., Kelly S. M., Price N. C., Fothergill-Gilmore L. A., Muirhead H. 1996. Ligand-induced conformational changes in wild-type and mutant yeast pyruvate kinase. Protein Eng. 9:1203–1210 [DOI] [PubMed] [Google Scholar]
- 11. Cunningham D. S., et al. 2009. Pyruvate kinase-deficient Escherichia coli exhibits increased plasmid copy number and cyclic AMP levels. J. Bacteriol. 191:3041–3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fraser H. B., Hirsh A. E., Steinmetz L. M., Scharfe C., Feldman M. W. 2002. Evolutionary rate in the protein interaction network. Science 296:750–752 [DOI] [PubMed] [Google Scholar]
- 13. Hannaert V., et al. 2002. The putative effector-binding site of Leishmania mexicana pyruvate kinase studied by site-directed mutagenesis. FEBS Lett. 514:255–259 [DOI] [PubMed] [Google Scholar]
- 14. Ikeda Y., Tanaka T., Noguchi T. 1997. Conversion of non-allosteric pyruvate kinase isozyme into an allosteric enzyme by a single amino acid substitution. J. Biol. Chem. 272:20495–20501 [DOI] [PubMed] [Google Scholar]
- 15. Irwin J. J., Shoichet B. K. 2005. ZINC—a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 45:177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jurica M. S., et al. 1998. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 6:195–210 [DOI] [PubMed] [Google Scholar]
- 17. Kharalkar S. S., et al. 2007. Identification of novel allosteric regulators of human-erythrocyte pyruvate kinase. Chem. Biodivers. 4:2603–2617 [DOI] [PubMed] [Google Scholar]
- 18. Larsen T. M., Laughlin L. T., Holden H. M., Rayment I., Reed G. H. 1994. Structure of rabbit muscle pyruvate kinase complexed with Mn2+, K+, and pyruvate. Biochemistry 33:6301–6309 [DOI] [PubMed] [Google Scholar]
- 19. Marshall N. J., Goodwin C. J., Holt S. J. 1995. A critical assessment of the use of microculture tetrazolium assays to measure cell growth and function. Growth Regul. 5:69–84 [PubMed] [Google Scholar]
- 20. Mattevi A., et al. 1995. Crystal structure of Escherichia coli pyruvate kinase type I: molecular basis of the allosteric transition. Structure 3:729–741 [DOI] [PubMed] [Google Scholar]
- 21. Monroe S., Polk R. 2000. Antimicrobial use and bacterial resistance. Curr. Opin. Microbiol. 3:496–501 [DOI] [PubMed] [Google Scholar]
- 22. Morgan H. P., et al. 2010. The allosteric mechanism of pyruvate kinase from Leishmania mexicana: a rock and lock model. J. Biol. Chem. 285:12892–12898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Munoz M. E., Ponce E. 2003. Pyruvate kinase: current status of regulatory and functional properties. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 135:197–218 [DOI] [PubMed] [Google Scholar]
- 24. National Committee for Clinical Laboratory Standards. 2003. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. Approved standard, M7-A6. National Committee for Clinical Laboratory Standards, Wayne, PA [Google Scholar]
- 25. National Committee for Clinical Laboratory Standards. 1999. Methods for determining bactericidal activity of antimicrobial agents. Approved guideline M26-A. National Committee for Clinical Laboratory Standards, Wayne, PA [Google Scholar]
- 26. Nowicki M. W., et al. 2008. Design, synthesis and trypanocidal activity of lead compounds based on inhibitors of parasite glycolysis. Bioorg. Med. Chem. 16:5050–5061 [DOI] [PubMed] [Google Scholar]
- 27. Opperdoes F. R., Michels P. A. 2001. Enzymes of carbohydrate metabolism as potential drug targets. Int. J. Parasitol. 31:482–490 [DOI] [PubMed] [Google Scholar]
- 28. Pathania R., Brown E. D. 2008. Small and lethal: searching for new antibacterial compounds with novel modes of action. Biochem. Cell Biol. 86:111–115 [DOI] [PubMed] [Google Scholar]
- 29. Rigden D. J., Phillips S. E., Michels P. A., Fothergill-Gilmore L. A. 1999. The structure of pyruvate kinase from Leishmania mexicana reveals details of the allosteric transition and unusual effector specificity. J. Mol. Biol. 291:615–635 [DOI] [PubMed] [Google Scholar]
- 30. Rybak M. J. 2004. Resistance to antimicrobial agents: an update. Pharmacotherapy 24:203S–215S [DOI] [PubMed] [Google Scholar]
- 31. Sassetti C. M., Boyd D. H., Rubin E. J. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 48:77–84 [DOI] [PubMed] [Google Scholar]
- 32. Silver L. L., Bostian K. A. 1993. Discovery and development of new antibiotics: the problem of antibiotic resistance. Antimicrob. Agents Chemother. 37:377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Somerville G. A., et al. 2002. Staphylococcus aureus aconitase inactivation unexpectedly inhibits post-exponential-phase growth and enhances stationary-phase survival. Infect. Immun. 70:6373–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Song J. H., et al. 2005. Identification of essential genes in Streptococcus pneumoniae by allelic replacement mutagenesis. Mol. Cells 19:365–374 [PubMed] [Google Scholar]
- 35. Speranza M. L., et al. 1989. Primary structure of three peptides at the catalytic and allosteric sites of the fructose-1,6-bisphosphate-activated pyruvate kinase from Escherichia coli. Biol. Chem. Hoppe Seyler 370:211–216 [DOI] [PubMed] [Google Scholar]
- 36. Stuart D. I., Levine M., Muirhead H., Stammers D. K. 1979. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 A. J. Mol. Biol. 134:109–142 [DOI] [PubMed] [Google Scholar]
- 37. Tulloch L. B., et al. 2008. Sulphate removal induces a major conformational change in Leishmania mexicana pyruvate kinase in the crystalline state. J. Mol. Biol. 383:615–626 [DOI] [PubMed] [Google Scholar]
- 38. Valentini G., et al. 2000. The allosteric regulation of pyruvate kinase. J. Biol. Chem. 275:18145–18152 [DOI] [PubMed] [Google Scholar]
- 39. Vander Heiden M. G., et al. 2010. Identification of small molecule inhibitors of pyruvate kinase M2. Biochem. Pharmacol. 79:1118–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wang C., et al. 2001. Human erythrocyte pyruvate kinase: characterization of the recombinant enzyme and a mutant form (R510Q) causing nonspherocytic hemolytic anemia. Blood 98:3113–3120 [DOI] [PubMed] [Google Scholar]
- 41. Wright G. D. 2003. Mechanisms of resistance to antibiotics. Curr. Opin. Chem. Biol. 7:563–569 [DOI] [PubMed] [Google Scholar]
- 42. Yoneyama H., Katsumata R. 2006. Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci. Biotechnol. Biochem. 70:1060–1075 [DOI] [PubMed] [Google Scholar]
- 43. Zhang R., Ou H. Y., Zhang C. T. 2004. DEG: a database of essential genes. Nucleic Acids Res. 32:D271–D272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zoraghi R., et al. 2010. Functional analysis, overexpression, and kinetic characterization of pyruvate kinase from methicillin-resistant Staphylococcus aureus. Biochemistry 49:7733–7747 [DOI] [PubMed] [Google Scholar]