

Abstract
Proposed molecular classifiers may be overfit to idiosyncrasies of noisy genomic and proteomic data. Cross-validation methods are often used to obtain estimates of classification accuracy, but both simulations and case studies suggest that, when inappropriate methods are used, bias may ensue. Bias can be bypassed and generalizability can be tested by external (independent) validation. We evaluated 35 studies that have reported on external validation of a molecular classifier. We extracted information on study design and methodological features, and compared the performance of molecular classifiers in internal cross-validation versus external validation for 28 studies where both had been performed. We demonstrate that the majority of studies pursued cross-validation practices that are likely to overestimate classifier performance. Most studies were markedly underpowered to detect a 20% decrease in sensitivity or specificity between internal cross-validation and external validation [median power was 36% (IQR, 21–61%) and 29% (IQR, 15–65%), respectively]. The median reported classification performance for sensitivity and specificity was 94% and 98%, respectively, in cross-validation and 88% and 81% for independent validation. The relative diagnostic odds ratio was 3.26 (95% CI 2.04–5.21) for cross-validation versus independent validation. Finally, we reviewed all studies (n = 758) which cited those in our study sample, and identified only one instance of additional subsequent independent validation of these classifiers. In conclusion, these results document that many cross-validation practices employed in the literature are potentially biased and genuine progress in this field will require adoption of routine external validation of molecular classifiers, preferably in much larger studies than in current practice.
Keywords: predictive medicine, genes, gene expression, proteomics
INTRODUCTION
The advent of high-throughput molecular technologies has led some to hypothesize that comprehensive assessment of the genome, the transcriptome and the proteome may lead to the discovery of new molecular classifiers capable of classifying patients more accurately than existing traditional prognostic factors and biomarkers [1, 2]. This has led to a corresponding bounty of novel molecular classifiers for which high levels of predictive performance have been claimed, some of which have even received approval for clinical use [3]. However, the development of classifiers from high-dimensional data is a complex process involving multiple analytic decisions [4], each of which is subject to a number of potential methodological errors which may result in spuriously high levels of reported performance [5, 6]. Amidst the current proliferation of molecular classifiers, it is difficult to know whether novel classifiers with impressive performance represent true discoveries or spurious patterns of ‘noise discovery’ resulting from improper analysis of high-dimensional data [7–9].
Proper internal and external validation is essential for assessing the performance and generalizability of a classifier [10, 11]. However, the validation process itself can be susceptible to inappropriate application, and multiple authors have pointed to common misapplication of validation practices in the literature [10, 12–16]. Simple (re-substitution) analysis of a training set is well known to give biased (inflated) estimates of classification accuracy and over-fitting can occasionally be severe [17]. To avoid over-fitting, several internal validation methods have been widely used in the literature, including leave-one out and k-fold cross-validation, as well as the related family of bootstrap-based methods. However, several authors have demonstrated that inappropriate application of cross-validation leads to inflated estimates of classification accuracy [10, 14–20]. Various specific sources of bias have been described, with an accumulating literature using diverse terminology on ‘population selection bias’, ‘incomplete cross-validation’, ‘optimization bias’, ‘reporting bias’ and ‘selection of parameters bias’, among others [15–19]. There is increasing appreciation that proper and transparent design and reporting of these studies is important [8, 10, 21]. Moreover, it is also increasingly appreciated that it is essential to perform external validation of any classifier in external, independent data from those where it was developed [10, 20]. External validation may be performed by the team that developed the classifier or independent teams. Many of the biases that affect internal validation are addressed with stringent external validation, and the generalizability of the classifier becomes more robustly documented when the exercise is repeated successfully across multiple research teams and patient populations.
In this article, we assess the state of contemporary validation practices for novel molecular classifiers, with special focus on the use of external validation and its comparison against internal cross-validation. We have conducted an empirical assessment of a sample of studies which claim to develop and independently validate a novel molecular classifier in the same publication. We assessed the internal and external validation methods employed, and we obtained data relating to the classification accuracy of these classifiers as estimated from internal cross-validation, external validation from the same publication and any subsequent external validation efforts by the same, and different, research teams in other papers.
METHODS
Literature search and eligibility criteria
To obtain a sample of published studies reporting on the external validation of molecular classifiers, we performed a search of the MEDLINE database (through PubMed) on 23 June 2010 using the following terms: (molecular OR gene OR expression OR profile OR proteomic* OR metabolomic* OR microarray) AND (independent validation[tw] OR external validation[tw]). A single reviewer (PC) screened all abstracts and potentially eligible studies were retrieved in full text. Two reviewers (PC and IJD) reviewed all potentially eligible studies independently and discrepancies were resolved by consensus including a third reviewer (JPAI).
We set the following inclusion criteria: the article used molecular information to make binary classifications of human subjects into categories of interest; the molecular classifiers were not already well-established for clinical use for the condition of interest; external validation was performed in the paper; and classification results for internal and external validation were available in the form of sensitivity and specificity, or we could calculate these metrics based on other presented information. We did not consider animal studies, reviews, editorials, letters or other studies not reporting primary research findings or studies not published in English.
From each eligible study, we extracted the following information: first author and year of publication, categories into which subjects were being classified, the number of subjects in training and validation samples, whether these samples were independent, whether there were any gross differences between training and validation samples, whether the samples originated from a single or multiple centers, the technology used to identify molecules of interest (i.e. genotyping arrays, gene expression arrays, proteomic technologies), methods used for cross-validation, whether cross-validation results were averaged across all validations or represented the best performing classifier identified at the cross-validation phase, whether feature/variable selection was done so as to preserve the integrity of cross-validation test sets, whether multiple models were tested in the external validation data and the reported sensitivity and specificity of the classifier in cross-validation and independent validation.
We use here the term cross-validation to describe all variants thereof (leave-one out, k-fold, etc.) as well as bootstrap-based methods that can serve a similar purpose of estimating classification accuracy while avoiding over-fitting without using additional external samples. Conversely, we use the term external validation to describe procedures that determine predictive performance of a model in data that were not used in any aspect of the development of the model.
Power calculations for validation samples
To evaluate whether the independent validation phase in each study was sufficiently designed to detect a meaningful difference in sensitivity or specificity compared to cross-validation estimates (whenever cross-validation had been performed), we estimated the power of the independent validation phase in each study to detect a 20% decrease in sensitivity or specificity compared to the cross-validation phase at a one-sided alpha-level = 0.05 using an exact test for two binomial proportions. Of note, any cross-validation process induces correlation in the predictions due to re-use of data for building the multiple predictors in the cross-validation iterations. The cross-validated sensitivity and specificity proportions will have a true variance larger than what is estimated for binomial proportions. However, most articles that use cross-validation do not report variances or confidence intervals (CIs) that account for this correlation pattern, and it was impossible for us to calculate these without access to the raw data. Usually the difference in the variance estimates is small [22]. If anything, this deficiency suggests that the power estimates that we derived would tend to be slightly inflated.
Because many of the sample sizes were small and asymptotic assumptions were possibly violated, we used a simulation approach to calculating power [23]. For these analyses, we assumed that sample sizes and disease/healthy ratios in the simulated studies were equal to the ones observed in the each published study of interest; i.e. we treated the sample size and the distribution of disease and healthy individuals at each phase as determined by each study’s experimental design.
Comparison of external validation versus cross-validation
We compared the classification accuracy of the classifiers in the external validation phase versus what had been estimated in the cross-validation phase, for studies where both types of validation had been performed and reported. Classification accuracy was expressed by the diagnostic odds ratio (DOR), estimated as 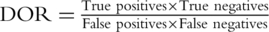 . We calculated the DOR for the cross-validation (DORcv) and independent validation (DORiv) and their variance, and then calculated the relative DOR (rDOR), i.e. the ratio of the two DORs for each study [24–26]. DORcv was set to be higher than one for all studies and rDOR values higher than one indicate that the classifier of interest performed worse in the external validation phase. The standard error of each ln(DOR) was calculated as
. We calculated the DOR for the cross-validation (DORcv) and independent validation (DORiv) and their variance, and then calculated the relative DOR (rDOR), i.e. the ratio of the two DORs for each study [24–26]. DORcv was set to be higher than one for all studies and rDOR values higher than one indicate that the classifier of interest performed worse in the external validation phase. The standard error of each ln(DOR) was calculated as 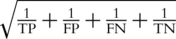 , with a continuity correction of k = 0.5 in studies where one of the denominators was zero. We used the sum of the variances of ln(DORcv) and ln(DORiv) to estimate the variance of the ln(rDOR). Then, we obtained the 95% CI for the ln(DOR) as
, with a continuity correction of k = 0.5 in studies where one of the denominators was zero. We used the sum of the variances of ln(DORcv) and ln(DORiv) to estimate the variance of the ln(rDOR). Then, we obtained the 95% CI for the ln(DOR) as 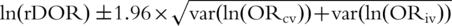 and reported the back-transformed (exponentiated) values. When the two DORs are calculated in samples from different individuals their covariance is zero, i.e. they are independent [25]. For reasons similar to those discussed in the power calculations section, the var(ln(DORcv)) estimates we obtained are lower than the ‘true’ variance values that account for the correlation pattern in cross-validation [27]. Therefore, to explore the effect of underestimating the var(ln(DORcv)), we performed a sensitivity analysis where we doubled the standard error of the ln(DORcv) and the summary results were similar (data not shown).
and reported the back-transformed (exponentiated) values. When the two DORs are calculated in samples from different individuals their covariance is zero, i.e. they are independent [25]. For reasons similar to those discussed in the power calculations section, the var(ln(DORcv)) estimates we obtained are lower than the ‘true’ variance values that account for the correlation pattern in cross-validation [27]. Therefore, to explore the effect of underestimating the var(ln(DORcv)), we performed a sensitivity analysis where we doubled the standard error of the ln(DORcv) and the summary results were similar (data not shown).
To summarize the results across all studies, we used the rDORs and their variance to perform meta-analysis using both fixed and random effect models [28]. Between-study heterogeneity was assessed using Cochran’s χ2-based Q-statistic and the I2 index [29, 30]. Heterogeneity was considered statistically significant at PQ < 0.10. I2 ranges between 0 and 100% and expresses the percentage of between study heterogeneity that is not explainable by chance. We also estimated the 95% CI of I2 estimates [29, 31].
In order for cross-validation to provide minimally biased estimates of prediction error, all aspects of the model-building process need to be cross-validated to maintain the integrity of the process, and the reported result should be the average of all cross-validations whenever multiple cross-validations have been performed. Feature selection bias occurs when feature or variable selection takes place outside the cross-validation loop, and optimization bias occurs when the best, rather than the average, cross-validation result is reported. Cross-validation performance may be inflated when either feature selection bias [16, 19] or optimization bias [10, 14, 19, 20] is present. We categorized studies based on whether either of these biases appeared to be present, and we performed subgroup analyses comparing the summary rDOR in such studies versus the summary rDOR in studies that do not suffer such biases.
We also explored random effects meta-regression of the rDOR over the following variables: year of publication, sample size of cross-validation sample (log-transformed), sample size of external validation sample (log-transformed), whether samples in the training phase originated from multiple centers, whether samples in the independent validation phase came from multiple centers and whether clear differences between training and validation samples were present [32, 33]. These analyses are entirely exploratory and should be interpreted with caution given the limited number of studies.
Statistical analyses were conducted using Stata version SE/11.1 (Stata Corp., College Station, TX, USA) and R version 2.11 (R Foundation for Statistical Computing, Vienna, Austria) [34, 35].
Subsequent validation efforts
In order to determine if subsequent validation efforts had been undertaken for any of the classifiers included in our study, we searched the ISI Web of Science database to identify all the research papers that cited the 35 included studies [36]. Timespan was ‘All Years’ and retrieved citations were limited to those classified as ‘Articles’ by ISI Web of Science. These papers were then reviewed at the abstract level in order to identify whether they reported on subsequent external validations of any of the classifiers we assessed. Articles that appeared to be validation efforts were examined in full text to identify whether the validation attempt used the exact classifier from the original publication, whether there was any overlap in authorship between the two papers (i.e. same or different research teams), and to extract the subsequent validation sample size and quantify the performance of the classifier in subsequent external validation.
RESULTS
Eligible studies analyzed
The PubMed search yielded 285 citations. After abstract level review, 92 articles were considered potentially eligible and were examined in full-text. Of these articles, 57 were excluded for the following reasons: 29 did not report classification results as sensitivity and specificity (e.g. they reported percentage overall accuracy or only presented ROC curves without selecting a specific operating point on them), 15 reported on molecular classifiers that were not binary, 6 did not pertain to molecular classifiers, 5 did not report primary data, 1 had no independent validation sample, and 1 did not pertain to humans. This left 35 articles that met the inclusion criteria and were included in the analyses [37–71].
Study characteristics and validation practices
Table 1 describes the characteristics of the included studies. The vast majority studies used gene expression or proteomic data, as would be expected from our search strategy. Most of the published classifiers pertained to cancer or cancer-related outcomes. The majority of studies were single center studies, and the median total sample size was 119 (IQR 62–192), with a median training/validation ratio of 1.3. The ratio of classification groups (i.e. ‘cases’ defined as the group with a diagnosis or outcome of interest where model sensitivity was determined, and ‘controls’ defined as the comparison group where model specificity was determined) was roughly 1:1. The median reported classification performance for sensitivity and specificity was 94 and 98%, respectively, in cross-validation and 88 and 81% in independent validation, already suggesting a decrease in performance from cross-validation to independent validation.
Table 1:
Characteristics of the 35 evaluated studies
| Characteristic, all included studies | Included studies (n = 35) | Characteristic, subset of studies using cross-validation | Included studies (n = 28) |
|---|---|---|---|
| Total sample size | 119 (62–192) | Cross-validation (CV) performed | |
| Training samples | 69 (29–103) | leave-one-out CV, n (percentage of CV subset) | 13 (46) |
| ‘Cases’ | 36 (15–55) | 4- to 10-fold CV, n (percentage of CV subset) | 13 (46) |
| ‘Controls’ | 24 (16–50) | Bootstrap, n (percentage of CV subset) | 2 (7) |
| Validation samples | 54 (33–81) | Feature selection preserves integrity of CV test sets | |
| ‘Cases’ | 23 (13–38) | Yes, n (percentage of CV subset) | 7 (25) |
| ‘Controls’ | 25 (11–40) | No, n (percentage of CV subset) | 18 (64) |
| Ratio of training to validation | 1.3 (0.8–2.1) | Unclear, n (percentage of CV subset) | 3 (11) |
| Training samples from multiple centers, n (%) | 9 (26) | Reported performance from CV | |
| Result is the average of all CV results, n (percentage of CV subset) | 3 (11) | ||
| Validation samples from multiple centers, n (%) | 13 (37) | ||
| Result is the best of many CV results, n (percentage of CV subset) | 22 (79) | ||
| Training and validation samples independent, n (%) | 35 (100) | ||
| Unclear, n (percentage of CV subset) | 3 (11) | ||
| Clear difference between training and validation samples, n (%) | 13 (37) | Sensitivity in training sample | 94 (84–100) |
| Specificity in training sample | 98 (85–100) | ||
| Sensitivity in independent validation | 88 (78–92) | ||
| Predicted outcome | Specificity in independent validation | 81 (68–95) | |
| Cancer related, n (%) | 27 (77) | ||
| Non-cancer related, n (%) | 8 (23) | ||
| Molecular feature used for classification, n (%) | |||
| DNA variation | 2 (6) | ||
| RNA (gene expression) | 19 (54) | ||
| Protein | 12 (34) | ||
| Other | 2 (6) | ||
| Classifier tested in validation is identical to that developed in training | |||
| Yes, n (%) | 34 (97) | ||
| Unclear, n (%) | 1 (3) | ||
| >1 model tested in validation data, n (%) | 10 (29) |
Values are median (IQR) unless otherwise specified.
Table 1 also presents data relating to other critical issues relating to study design and data analysis issues relating to classifier development. All of the included studies claimed independence of the training and external validation samples with no discernible overlap between the two. For the validation of a classifier, it is essential that the classifier tested in independent validation be identical to that developed in training (e.g. regression coefficients should be fixed and there should be no re-estimation of parameters or addition of features used for classification). In all but one study, it appeared clear that this principle had been maintained. It is common to develop many different classifiers on the training data; however, since there is often wide uncertainty around the performance estimates for classifiers, selecting the best classifier among many is likely to introduce a form of optimization bias. External validation can guard against this bias, but if many classifiers are moved forward to independent validation then independent validation may be subject to this bias as well. Ten (29%) of the included studies tested more than 1 classifier in the independent validation sample. Of these studies, six reported the results for all classifiers tested in independent validation.
Since the performance of classifiers may be dependent on the population to which the classifier is applied, training and validation samples should be similar, unless the authors intend to extend the applicability of a previously established classifier. Nearly 40% of the included studies had a clear difference between training and validation samples. Notable examples of such differences included discrepancies in the distribution of cancer stages between training and validation samples [45], differences in baseline characteristics (i.e. 68% males in healthy control group in training samples, 31% males in control group in validation) [51] or differences in sample specimen quality (in [71] 2% of gene expression arrays failed quality control in samples from one center, compared to 70% of samples from a second center; in classifier development, the training sample was comprised entirely of samples from the first center and roughly half of the validation sample was comprised of samples from the second center).
Feature selection bias and optimization bias were a potential concern in the majority of the included studies, with feature selection bias being likely in 64% and optimization bias in 79% of the 28 studies that performed internal cross-validation. Fifteen (54%) studies had evidence of both biases, 10 (36%) studies suffered from at least one of the two, two studies could not be classified for both types of bias and only one study was clearly exempt from either bias.
Power estimates for independent validation
In general, independent validation sample sizes were inadequate to detect even a large (20%) decrease in the sensitivity or specificity compared with the training set performance at the liberal false positive rate of 5%. Across studies, the median power of the independent validation phase was 36%, [IQR, = 21–61%] for sensitivity and 29%, [IQR, 15–65%] for specificity at a one-sided alpha = 0.05 for a decrease of 20% between phases. Three studies had power of 80% or higher in the independent validation cohort for sensitivity, and five studies had power of 80% or higher for specificity.
Comparison of classification accuracy in external versus cross-validation
As shown in Figure 1, the large majority of studies reported worse classification performance in external validation versus internal cross-validation, and the meta-analysis estimate of the rDOR was significantly different from one (summary random effects rDOR 3.26, 95% CI 2.04–5.21). There was no strong evidence for substantial heterogeneity of rDOR between these 28 studies (I2 = 0%, 95% CI 0–42%) and meta-analysis under a fixed-effects model produced identical results.
Figure 1:

Random effects meta-analysis of relative diagnostic odds ratios (rDORs) for 28 studies with cross-validation classification estimates and independent validation estimates for the same classifier. The majority of studies show worse classification performance in independent validation than in cross-validation, with a summary rDOR estimate of 3.26 (95% CI 2.04–5.21) (where rDOR=1 represents equal classification performance in cross-validation and independent validation).
Figures 2 and 3 show the rDORs forest plots for these 28 studies stratified by the likely presence of feature selection and optimization bias, respectively. In both of these analyses, the subgroups characterized by a high potential for bias show statistically significantly worse performance in validation than cross-validation (summary rDOR = 4.50, 95% CI 2.54–7.95 for selection bias and rDOR = 3.20, 95% CI 1.99–5.15 for optimization bias), whereas the subgroups without clear evidence of these biases do not show significantly worse performance (rDOR 1.75, 95% CI 0.66–4.60 and rDOR 5.62, 95% CI 0.40–78.80, respectively). However, the point estimate for the subgroup where optimization bias was unlikely is more extreme, and it should be acknowledged that the subgroup estimates have wide and overlapping CIs due to the small number of studies that did not suffer from these potential biases and the large variances of the rDORs.
Figure 2:

Random effects meta-analysis of relative diagnostic odd ratios (rDORs) for the 28 included studies, stratified by whether feature selection bias is likely to be present. The group with feature selection bias demonstrates significantly worse performance in independent validation than in cross-validation (rDOR 4.50, 95% CI 2.04–5.21), whereas this is not the case in the group that is unlikely to have this bias.
Figure 3:

Random effects meta-analysis of relative diagnostic odd ratios (rDORs) for the 28 included studies, stratified by whether optimization bias (i.e. reporting the best result from cross-validations versus the average result across all cross-validation) is likely to be present. The group with optimization bias demonstrates significantly worse performance in independent validation than in cross-validation (rDOR 3.20, 95% CI 1.99–5.15). The group that is unlikely to have bias had a more extreme but non-significant point estimate.
To explore other causes of heterogeneity in rDORs between studies, we performed meta-regressions with other variables, including trends over time, the ratio of training to validation samples, total sample size, multicenter versus single-center samples in training and validation and presence of a clear difference between training and validation samples, but none of these factors was significantly associated with magnitude of the rDOR (all P-values >0.1).
Subsequent external validations
We identified all research articles (n = 758) which cited any of the 35 studies included in our analyses and reviewed them at the abstract level in order to identify any subsequent efforts to validate these classifiers. Only three subsequent validation efforts were identified, two by the same research team that published the initial paper [72, 73], and one by an independent research team [74]. Only one study used the exact classifier (predicting local tumor response to pre-operative chemotherapy in breast cancer) described in the initial publication. In this article, the classifier was one of four tested classifiers, and the sample size was 100 (15 cases of residual breast cancer in breast tissue or lymph nodes, 85 with no residual disease). Using the same threshold determined in the primary publication, the performance of the classifier in subsequent validation for sensitivity and specificity was 60% and 74%, respectively, compared to 92% and 71% in the independent validation from the original publication, corresponding to a rDOR of 6.86 (95% CI, 0.60–78.71) for these two independent validations [42, 73]. Accounting for uncertainty in both studies, the power to detect a decrease of 20% in sensitivity or specificity was 16% and 37%, respectively. The second study, performed by the same research team as the original publication, examined the predictive ability of the same mitochondrial deletion mutation for prostate cancer, but in a different clinical scenario. The original classifier had been trained on biopsy samples from subjects with prostate cancer, though since multiple biopsies were available from the same patient, some samples contained only benign tissue. In the validation publication, the authors selected patients with initially benign prostate biopsies. From these initial biopsies, the authors developed a prediction tool to identify the ∼20% of subjects who developed biopsy-proven prostate cancer within 1 year. Citing the difference in patient populations and biopsy samples, the authors selected a new discriminatory cutoff in the validation paper [58, 72]. The third study, performed by a different research team, used gene expression data from the same three genes used in the original classifier, but the authors used different model-building methods (logistic regression in validation paper versus linear discriminant analysis in the original paper) to develop a classifier from the expression data of these genes [40, 74].
DISCUSSION
The development of molecular classifiers from high-dimensional data is subject to a number of pitfalls in study design, analytical methods and reporting of results. This review of internal and external validation practices in the recent literature provides an empirical view of the field, and highlights areas for future improvements. We identified examples of questionable analytic practices in the model-building phase, underpowered independent validation efforts and overly optimistic assessment of classification performance. Cross-validation practices yield optimistically biased estimates of performance compared to external validation. Even external validations may sometimes provide inflated accuracy results and there is a dearth of external replication efforts by totally independent groups.
Sample size
Many studies of molecular classifiers are fundamentally limited by sample size. In 11 of the 35 included studies, at least one of the classification groups in either training or validation included 10 or fewer subjects. Cross-validation error estimation methods may be inaccurate in small sample settings [75]. Even if validation efforts yield encouraging point estimates, the large uncertainty associated with these estimates would still preclude drawing strong conclusions about the classifier performance. Furthermore, small sample sizes are more prone to yield spuriously good classification accuracy due to chance. There is some empirical evidence that in the published literature of microarray cancer classifiers, early smaller studies tend to show more impressive classifier performance [76] than larger later studies [77]. Smaller studies may differ from larger studies due to true heterogeneity, chance or publication bias [78, 79]. Moreover, the majority of molecular classifier evaluations are single-center studies. This not only leads to restrictions in the number of participants enrolled, but it also limits the generalizability of the findings. Obtaining clinical samples can be challenging in certain research contexts, particularly for rare diseases. However, for most conditions, increased collaboration between groups or networks of investigators could readily address the issue of inadequate sample sizes. In setting such collaborations, uniformity or at least compatibility of sample collection and processing across collaborating centers is essential. Whenever large sample sizes cannot be attained, the use of appropriate cross-validation methods may be preferable to further reducing the sample size by sample-splitting.
Internal validation
The majority of studies in our sample used re-sampling methods (either bootstrap or k-fold cross-validation) to quantify classification performance in the training sample. There has been considerable debate regarding the appropriate application of cross-validation methods in the literature. In a critical review of cancer classifiers, Dupuy et al. [9] showed that most studies pursued inappropriate cross-validation practices. Both feature selection and classifier optimization bias can result in substantial inflation of classification performance estimates [5, 10, 14–16, 19]. Optimization has been termed ‘reporting bias’, ‘optimization of the data set bias’, ‘optimization of the method’s characteristics’ and ‘optimal selection of the classification method’ by previous investigators, depending on the exact mechanism that leads to the unwarranted optimized results [14, 15]. Among the studies we reviewed, 89% were potentially affected by at least one of these types of bias. The performance estimates from current cross-validation practices were optimistically biased compared to the results of external validation, and feature selection and optimization bias contribute to this over-optimism.
External validation
We also observed practices that may lead to biased estimates of performance even in external validation. Previous authors have pointed out the risks of occult sources of ‘data leak’ in which information from the external validation sample can leak into the development of a classifier and lead to overly optimistic external validation results [11]. Our study documents other ways in which the process of external validation may be subverted, the most common of which is the development and validation of multiple classifiers with incomplete reporting of all validation results. In this scenario, external validation may again yield overly optimistic estimates of performance when only the best replication results are reported. Often, it was difficult to deduce from a particular publication exactly how many models were tested.
Genuine differences versus bias
In ∼40% of studies that we examined, there was some identifiable difference between training and validation samples. Such differences may either reduce or increase the classifier performance in external validation versus cross-validation, depending on what choices are made in selecting samples for either stage. It is not possible to generalize and each study should be scrutinized on a case-by-case basis to decipher what direction bias may take. However, some biases are well-recognized in the literature, e.g. spectrum of disease bias [80–82] may inflate estimates of classification performance when extreme cases and/or hyper-normal controls are used. Early studies may use such designs and reach over-optimistic results that do not hold true when the classifier aims to discriminate between patient groups that are representative of those seen in typical clinical practice and healthy individuals [83]. Of course, it is desirable to extend the applicability of classifiers by testing them in novel target populations and different settings. However, such extensions are probably more appropriate for well-validated rather than novel classifiers that are proposed for the first time and have not been extensively validated.
Limitations
Our literature search was designed to yield a representative sample of publications on molecular classifiers with independent validation for binary outcomes. Thus, our findings did not include studies pertaining to continuous outcomes or time-to-event outcomes such as duration of survival or disease-free survival, and they are also not generalizable to studies that report a molecular classifier without making a claim for independent validation. Given our search strategy, some classification methods such as nuclear magnetic resonance or infrared spectroscopy were not well-represented in our sample. Because our study aims to compare cross-validation and external validation results from the same publication, it is unclear whether studies without external validation would have used cross-validation more rigorously. Some studies intended to use cross-validation for model selection; in these cases, one should not use the biased results from cross-validation to describe classification accuracy. In addition, while the number of included studies was sufficient to show a statistically significant difference in classification performance between cross-validation and independent validation, it was clearly insufficient for subgroup analyses. Thus, there may yet be unidentified predictors of heterogeneity in validation performance that our subgroup analyses were not adequately powered to detect.
Our power analyses and rDOR calculations may have been influenced by the fact that data pairs (predicted cross-validation class and the ‘true’ class) for each sample used in cross-validation are not independent across subjects; the dependency arises due to the repeated use of the ‘true’ classes in the cross-validation process [22, 27]. This would have tended to make our power calculations anti-conservative (i.e., power would have been even lower than we calculated). For the same reason, we may have underestimated the var(DORcv), leading to confidence interval coverage far from the intended coverage probability. Sensitivity analysis by artificially inflating the standard error of the DORcv estimate did not affect our conclusions. Simulation studies have demonstrated that when the ‘true’ DOR is large the confidence interval coverage of the DORcv estimate will be approximately correct. In our dataset DORcv estimates were extremely large (median 177.7, IQR: 26.31–680) providing some reassurance; however, because the DOR parameter value in each study is unknown, inferences drawn from resampling-based procedures should be interpreted with caution [27]. We note that such inferences are highly prevalent in the published literature.
We extracted data using strict definitions of practices likely to incur optimization or feature selection bias, but some aspects of our data extraction were limited by poor reporting and diversity of analytic techniques resulting in a certain opacity of methods [10, 14]. Proliferation of methods and increased availability of software for data analysis do not necessarily imply a problem, but when coupled with unclear reporting they limit the ability to reproduce research findings [84]. Attempts to reproduce the published results of analyses of high-dimensional data sets, both in proteomic [6] and gene expression [4] research, demonstrate the necessity of further, independent replication efforts. However, independent replication efforts for our study sample were almost non-existent. Across the 35 studies included in our analyses, we only identified one further attempt at strict replication (i.e. replication of the exact classifier reported in the original publication).
Our study also identified some areas in which proper validation practices were consistently applied. When independent validation was claimed, there were no instances in which overt overlap between training and validation samples was noted. Similarly, nearly all studies provided specific reassurance that the molecular classifier subjected to external validation was the exact same classifier developed in the training sample, though in most instances the details provided were insufficient to allow for close examination of this claim. Moreover, this does not guarantee that the same exact classifier is also used when external validation is attempted in other studies by the same or independent groups of researchers.
Concluding comments
As a synopsis, Table 2 catalogues some major biases that can affect the development of molecular classifiers at the stage of cross-validation or external validation. Some of these biases are uncommon, while others are quite prevalent. Opaque reporting may occasionally hinder an exact appreciation of these biases.
Table 2:
Sources of bias in internal and external validation
| Internal validation |
| Selection bias |
| Feature selection: discriminative ‘features’ (variables) are selected for model inclusion on the entire data sample, failing to respect the integrity of the cross-validation test sets |
| Optimization bias |
| Multiple models: multiple models are built in the training sample, each of these models is evaluated in cross-validation, and the best performing model is selected |
| Optimization of parameters: cross-validation is used for parameter tuning/coefficient estimation. The best performing model within the cross-validation loop is selected, and the cross-validation result pertaining to this model is reported, rather than the average of all cross-validations |
| External validation |
| Breaches of Independence |
| Sample overlap: training and validation samples are not truly independent because there is some overlap between the two samples |
| Data leak: independence between training and validation samples is not maintained, often through the process of feature selection on both samples or forms of data filtering that are related to outcome information |
| Selection bias |
| Selection of the data set: multiple validation efforts in multiple data sets are attempted, but only the best one is reported |
| Optimization bias |
| Multiple models: multiple models are moved forward to external validation, but only the results of the best performing model are reported |
| Parameter re-estimation: model parameters, coefficients or cut points are re-estimated in the validation data rather than keeping the values determined in the training sample |
| Both/either internal or external validation |
| Biases stemming for selection of population and study design |
| Spectrum of disease bias: classification performance may be inflated when extreme cases are compared against hyper-normal controls |
| Different design and population selection features between internal and external validation: direction of bias may vary on a case-by-case basis |
The advent of high-throughput genomic and proteomic data is likely to be transformative for biology and medicine. However, reaping the benefits of quantitative changes in the availability of data requires also qualitative changes in the way research is performed, reported and validated. Our empirical evaluation reinforces the importance of transparent reporting and rigorous independent external validation of classifiers developed from high-dimensional data.
Key Points.
The development of molecular classifiers is subject to a number of pitfalls which can lead to overly optimistic reports of classification performance.
Many studies employ cross-validation methods which result in overly optimistic performance estimates.
Validation in an independent data set is crucial in establishing the reliability and generalizability of a molecular classifier, though this process can itself be susceptible to bias when inappropriate methods are used.
There is a dearth of subsequent independent replication efforts for published molecular classifiers.
FUNDING
National Institutes of Health (K08HL102265 to P.J.C. and UL1 RR025752 to Tufts-Clinical Translational Science Institute); Research Scholarship from the ‘Maria P. Lemos’ Foundation (to I.J.D.). The content is solely the responsibility of the authors and does not necessarily represent the offcial views of the National Center for Research Resources or the National Institutes of Health.
Supplementary Material
Biographies
Peter Castaldi is a physician-researcher and Assistant Professor of Medicine at the Institute for Clinical Research and Health Policy Studies at Tufts Medical Center. His principal research interests relate to the application of genomic discoveries to clinical practice.
Issa Dahabreh is a Research Associate at the Institute for Clinical Research and Health Policy studies at Tufts Medical Center. His research interests are focused on methods for evidence synthesis and their application to molecular medicine.
John Ioannidis serves as professor on the faculty of the medical schools at Stanford University, University of Ioannina (currently on leave), and Tufts University (adjunct appointment). His interests include evidence-based medicine and genomics and the dissection of sources of bias with empirical methods.
References
- 1.Lander ES. Array of hope. Nat Genet. 1999;21:3–4. doi: 10.1038/4427. [DOI] [PubMed] [Google Scholar]
- 2.Ioannidis JPA. Expectations, validity, and reality in omics. J Clin Epidemiol. 2010;63:950–9. doi: 10.1016/j.jclinepi.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 3.van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 4.Ioannidis JP, Allison DB, Ball CA, et al. Repeatability of published microarray gene expression analyses. Nat Genet. 2009;41:149–55. doi: 10.1038/ng.295. [DOI] [PubMed] [Google Scholar]
- 5.Simon R, Radmacher MD, Dobbin K, et al. Pitfalls in the use of DNA microarray data for diagnostic and prognostic classification. J Natl Cancer Inst. 2003;95:14–8. doi: 10.1093/jnci/95.1.14. [DOI] [PubMed] [Google Scholar]
- 6.Baggerly KA, Morris JS, Coombes KR. Reproducibility of SELDI-TOF protein patterns in serum: comparing datasets from different experiments. Bioinformatics. 2004;20:777–85. doi: 10.1093/bioinformatics/btg484. [DOI] [PubMed] [Google Scholar]
- 7.Diamandis EP. Analysis of serum proteomic patterns for early cancer diagnosis: drawing attention to potential problems. J Natl Cancer Inst. 2004;96:353–6. doi: 10.1093/jnci/djh056. [DOI] [PubMed] [Google Scholar]
- 8.Ioannidis JP. Microarrays and molecular research: noise discovery? Lancet. 2005;365:454–5. doi: 10.1016/S0140-6736(05)17878-7. [DOI] [PubMed] [Google Scholar]
- 9.Dupuy A, Simon RM. Critical review of published microarray studies for cancer outcome and guidelines on statistical analysis and reporting. J Natl Cancer Inst. 2007;99:147–57. doi: 10.1093/jnci/djk018. [DOI] [PubMed] [Google Scholar]
- 10.Simon R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J Clin Oncol. 2005;23:7332–41. doi: 10.1200/JCO.2005.02.8712. [DOI] [PubMed] [Google Scholar]
- 11.Taylor JM, Ankerst DP, Andridge RR. Validation of biomarker-based risk prediction models. Clin Cancer Res. 2008;14:5977–83. doi: 10.1158/1078-0432.CCR-07-4534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Somorjai RL, Dolenko B, Baumgartner R. Class prediction and discovery using gene microarray and proteomics mass spectroscopy data: curses, caveats, cautions. Bioinformatics. 2003;19:1484–91. doi: 10.1093/bioinformatics/btg182. [DOI] [PubMed] [Google Scholar]
- 13.Yousefi MR, Hua J, Sima C, et al. Reporting bias when using real data sets to analyze classification performance. Bioinformatics. 2010;26:68–76. doi: 10.1093/bioinformatics/btp605. [DOI] [PubMed] [Google Scholar]
- 14.Boulesteix AL, Strobl C. Optimal classifier selection and negative bias in error rate estimation: an empirical study on high-dimensional prediction. BMC Med Res Methodol. 2009;9:85. doi: 10.1186/1471-2288-9-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jelizarow M, Guillemot V, Tenenhaus A, et al. Over-optimism in bioinformatics: an illustration. Bioinformatics 10 AD. 26:1990–8. doi: 10.1093/bioinformatics/btq323. [DOI] [PubMed] [Google Scholar]
- 16.Ambroise C, McLachlan GJ. Selection bias in gene extraction on the basis of microarray gene-expression data. Proc Natl Acad Sci USA. 2002;99:6562–6. doi: 10.1073/pnas.102102699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Molinaro AM, Simon R, Pfeiffer RM. Prediction error estimation: a comparison of resampling methods. Bioinformatics. 2005;21:3301–07. doi: 10.1093/bioinformatics/bti499. [DOI] [PubMed] [Google Scholar]
- 18.Varma S, Simon R. Bias in error estimation when using cross-validation for model selection. BMC Bioinformatics. 2006;7:91. doi: 10.1186/1471-2105-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wood IA, Visscher PM, Mengersen KL. Classification based upon gene expression data: bias and precision of error rates. Bioinformatics. 2007;23:1363–70. doi: 10.1093/bioinformatics/btm117. [DOI] [PubMed] [Google Scholar]
- 20.Zervakis M, Blazadonakis ME, Tsiliki G, et al. Outcome prediction based on microarray analysis: a critical perspective on methods. BMC Bioinformatics. 2009;10:53. doi: 10.1186/1471-2105-10-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McShane LM, Altman DG, Sauerbrei W, et al. Reporting recommendations for tumor marker prognostic studies (REMARK) J Natl Cancer Inst. 2005;97:1180–4. doi: 10.1093/jnci/dji237. [DOI] [PubMed] [Google Scholar]
- 22.Dobbin KK. A method for constructing a confidence bound for the actual error rate of a prediction rule in high dimensions. Biostatistics. 2009;10:282–96. doi: 10.1093/biostatistics/kxn035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feiveson AH. Power by Simulation. Stata J. 2002;2:107–24. [Google Scholar]
- 24.Bland JM, Altman DG. Statistics notes. The odds ratio. Br Med J. 2000;320:1468. doi: 10.1136/bmj.320.7247.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altman DG, Bland JM. Interaction revisited: the difference between two estimates. Br Med J. 2003;326:219. doi: 10.1136/bmj.326.7382.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glas AS, Lijmer JG, Prins MH, et al. The diagnostic odds ratio: a single indicator of test performance. J Clin Epidemiol. 2003;56:1129–35. doi: 10.1016/s0895-4356(03)00177-x. [DOI] [PubMed] [Google Scholar]
- 27.Lusa L, McShane LM, Radmacher MD, et al. Appropriateness of some resampling-based inference procedures for assessing performance of prognostic classifiers derived from microarray data. Stat Med. 2007;26:1102–13. doi: 10.1002/sim.2598. [DOI] [PubMed] [Google Scholar]
- 28.DerSimonian R, Laird N. Meta-analysis in clinical trials. Control Clin Trials. 1986;7:177–88. doi: 10.1016/0197-2456(86)90046-2. [DOI] [PubMed] [Google Scholar]
- 29.Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–58. doi: 10.1002/sim.1186. [DOI] [PubMed] [Google Scholar]
- 30.Cochran W. The combination of estimates from different experiments. Biometrics. 1954:101–29. [Google Scholar]
- 31.Ioannidis JP, Patsopoulos NA, Evangelou E. Uncertainty in heterogeneity estimates in meta-analyses. Br Med J. 2007;335:914–6. doi: 10.1136/bmj.39343.408449.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knapp G, Hartung J. Improved tests for a random effects meta-regression with a single covariate. Stat Med. 2003;22:2693–710. doi: 10.1002/sim.1482. [DOI] [PubMed] [Google Scholar]
- 33.Thompson SG, Higgins JP. How should meta-regression analyses be undertaken and interpreted? Stat Med. 2002;21:1559–73. doi: 10.1002/sim.1187. [DOI] [PubMed] [Google Scholar]
- 34. Stata Statistical Software: Release 11. 2009. College Station, TX, StataCorp, LP. [Google Scholar]
- 35. R Development Core Team. R: A Language and Environment for Statistical Computing. 2010. Vienna, Austria, R Foundation for Statistical Computing. [Google Scholar]
- 36.ISI Web of Knowledge. Philadelphia: Thompson Reuters; 2010. Thompson Reuters. 6-30-2010. [Google Scholar]
- 37.Xi L, Junjian Z, Yumin L, et al. Serum biomarkers of vascular cognitive impairment evaluated by bead-based proteomic technology. Neurosci Lett. 2009;463:6–11. doi: 10.1016/j.neulet.2009.07.056. [DOI] [PubMed] [Google Scholar]
- 38.Ayers M, Symmans WF, Stec J, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–93. doi: 10.1200/JCO.2004.05.166. [DOI] [PubMed] [Google Scholar]
- 39.Zhang Z, Bast RC, Jr, Yu Y, et al. Three biomarkers identified from serum proteomic analysis for the detection of early stage ovarian cancer. Cancer Res. 2004;64:5882–90. doi: 10.1158/0008-5472.CAN-04-0746. [DOI] [PubMed] [Google Scholar]
- 40.Weber F, Shen L, Aldred MA, et al. Genetic classification of benign and malignant thyroid follicular neoplasia based on a three-gene combination. J Clin Endocrinol Metab. 2005;90:2512–21. doi: 10.1210/jc.2004-2028. [DOI] [PubMed] [Google Scholar]
- 41.Hughes SJ, Xi L, Raja S, et al. A rapid, fully automated, molecular-based assay accurately analyzes sentinel lymph nodes for the presence of metastatic breast cancer. Ann Surg. 2006;243:389–98. doi: 10.1097/01.sla.0000201541.68577.6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hess KR, Anderson K, Symmans WF, et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006;24:4236–44. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- 43.Reddy A, Wang H, Yu H, et al. Logical Analysis of Data (LAD) model for the early diagnosis of acute ischemic stroke. BMC Med Inform Decis Mak. 2008;8:30. doi: 10.1186/1472-6947-8-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xi L, Luketich JD, Raja S, et al. Molecular staging of lymph nodes from patients with esophageal adenocarcinoma. Clin Cancer Res. 2005;11:1099–109. [PubMed] [Google Scholar]
- 45.Engwegen JY, Mehra N, Haanen JB, et al. Identification of two new serum protein profiles for renal cell carcinoma. Oncol Rep. 2009;22:401–8. [PubMed] [Google Scholar]
- 46.Ziober AF, Patel KR, Alawi F, et al. Identification of a gene signature for rapid screening of oral squamous cell carcinoma. Clin Cancer Res. 2006;12:5960–71. doi: 10.1158/1078-0432.CCR-06-0535. [DOI] [PubMed] [Google Scholar]
- 47.Glas AM, Kersten MJ, Delahaye LJ, et al. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood. 2005;105:301–7. doi: 10.1182/blood-2004-06-2298. [DOI] [PubMed] [Google Scholar]
- 48.Folgueira MA, Carraro DM, Brentani H, et al. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin Cancer Res. 2005;11:7434–43. doi: 10.1158/1078-0432.CCR-04-0548. [DOI] [PubMed] [Google Scholar]
- 49.Helleman J, Jansen MP, Span PN, et al. Molecular profiling of platinum resistant ovarian cancer. Int J Cancer. 2006;118:1963–71. doi: 10.1002/ijc.21599. [DOI] [PubMed] [Google Scholar]
- 50.Hayashida Y, Honda K, Osaka Y, et al. Possible prediction of chemoradiosensitivity of esophageal cancer by serum protein profiling. Clin Cancer Res. 2005;11:8042–7. doi: 10.1158/1078-0432.CCR-05-0656. [DOI] [PubMed] [Google Scholar]
- 51.Munro NP, Cairns DA, Clarke P, et al. Urinary biomarker profiling in transitional cell carcinoma. Int J Cancer. 2006;119:2642–50. doi: 10.1002/ijc.22238. [DOI] [PubMed] [Google Scholar]
- 52.Aivado M, Spentzos D, Germing U, et al. Serum proteome profiling detects myelodysplastic syndromes and identifies CXC chemokine ligands 4 and 7 as markers for advanced disease. Proc Natl Acad Sci USA. 2007;104:1307–12. doi: 10.1073/pnas.0610330104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dressman HK, Berchuck A, Chan G, et al. An integrated genomic-based approach to individualized treatment of patients with advanced-stage ovarian cancer. J Clin Oncol. 2007;25:517–25. doi: 10.1200/JCO.2006.06.3743. [DOI] [PubMed] [Google Scholar]
- 54.Otu HH, Can H, Spentzos D, et al. Prediction of diabetic nephropathy using urine proteomic profiling 10 years prior to development of nephropathy. Diabetes Care. 2007;30:638–43. doi: 10.2337/dc06-1656. [DOI] [PubMed] [Google Scholar]
- 55.Chen G, Wang X, Yu J, et al. Autoantibody profiles reveal ubiquilin 1 as a humoral immune response target in lung adenocarcinoma. Cancer Res. 2007;67:3461–7. doi: 10.1158/0008-5472.CAN-06-4475. [DOI] [PubMed] [Google Scholar]
- 56.Wang Y, Barbacioru CC, Shiffman D, et al. Gene expression signature in peripheral blood detects thoracic aortic aneurysm. PLoS One. 2007;2:e1050. doi: 10.1371/journal.pone.0001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Patz EF, Jr, Campa MJ, Gottlin EB, et al. Panel of serum biomarkers for the diagnosis of lung cancer. J Clin Oncol. 2007;25:5578–83. doi: 10.1200/JCO.2007.13.5392. [DOI] [PubMed] [Google Scholar]
- 58.Maki J, Robinson K, Reguly B, et al. Mitochondrial genome deletion aids in the identification of false- and true-negative prostate needle core biopsy specimens. Am J Clin Pathol. 2008;129:57–66. doi: 10.1309/UJJTH4HFEPWAQ78Q. [DOI] [PubMed] [Google Scholar]
- 59.Watanabe H, Mogushi K, Miura M, et al. Prediction of lymphatic metastasis based on gene expression profile analysis after brachytherapy for early-stage oral tongue carcinoma. Radiother Oncol. 2008;87:237–42. doi: 10.1016/j.radonc.2007.12.027. [DOI] [PubMed] [Google Scholar]
- 60.Hoffmann K, Firth MJ, Beesley AH, et al. Prediction of relapse in paediatric pre-B acute lymphoblastic leukaemia using a three-gene risk index. Br J Haematol. 2008;140:656–64. doi: 10.1111/j.1365-2141.2008.06981.x. [DOI] [PubMed] [Google Scholar]
- 61.Teh SK, Zheng W, Ho KY, et al. Diagnosis of gastric cancer using near-infrared Raman spectroscopy and classification and regression tree techniques. J Biomed Opt. 2008;13:034013. doi: 10.1117/1.2939406. [DOI] [PubMed] [Google Scholar]
- 62.Petrovski S, Szoeke CE, Sheffield LJ, et al. Multi-SNP pharmacogenomic classifier is superior to single-SNP models for predicting drug outcome in complex diseases. Pharmacogenet Genomics. 2009;19:147–52. doi: 10.1097/FPC.0b013e32831d1dfa. [DOI] [PubMed] [Google Scholar]
- 63.Den Boer ML, van SM, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome-wide classification study. Lancet Oncol. 2009;10:125–34. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee NP, Chen L, Lin MC, et al. Proteomic expression signature distinguishes cancerous and nonmalignant tissues in hepatocellular carcinoma. J Proteome Res. 2009;8:1293–303. doi: 10.1021/pr800637z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Howrylak JA, Dolinay T, Lucht L, et al. Discovery of the gene signature for acute lung injury in patients with sepsis. Physiol Genomics. 2009;37:133–9. doi: 10.1152/physiolgenomics.90275.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andre F, Michiels S, Dessen P, et al. Exonic expression profiling of breast cancer and benign lesions: a retrospective analysis. Lancet Oncol. 2009;10:381–90. doi: 10.1016/S1470-2045(09)70024-5. [DOI] [PubMed] [Google Scholar]
- 67.Caron J, Mange A, Guillot B, et al. Highly sensitive detection of melanoma based on serum proteomic profiling. J Cancer Res Clin Oncol. 2009;135:1257–64. doi: 10.1007/s00432-009-0567-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kashani-Sabet M, Rangel J, Torabian S, et al. A multi-marker assay to distinguish malignant melanomas from benign nevi. Proc Natl Acad Sci USA. 2009;106:6268–72. doi: 10.1073/pnas.0901185106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kistler AD, Mischak H, Poster D, et al. Identification of a unique urinary biomarker profile in patients with autosomal dominant polycystic kidney disease. Kidney Int. 2009;76:89–96. doi: 10.1038/ki.2009.93. [DOI] [PubMed] [Google Scholar]
- 70.Julia A, Erra A, Palacio C, et al. An eight-gene blood expression profile predicts the response to infliximab in rheumatoid arthritis. PLoS One. 2009;4:e7556. doi: 10.1371/journal.pone.0007556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Showe MK, Vachani A, Kossenkov AV, et al. Gene expression profiles in peripheral blood mononuclear cells can distinguish patients with non-small cell lung cancer from patients with nonmalignant lung disease. Cancer Res. 2009;69:9202–10. doi: 10.1158/0008-5472.CAN-09-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson K, Creed J, Reguly B, et al. Accurate prediction of repeat prostate biopsy outcomes by a mitochondrial DNA deletion assay. Prostate Cancer Prostatic Dis. 2010;13:126–31. doi: 10.1038/pcan.2009.64. [DOI] [PubMed] [Google Scholar]
- 73.Lee JK, Coutant C, Kim YC, et al. Prospective comparison of clinical and genomic multivariate predictors of response to neoadjuvant chemotherapy in breast cancer. Clin Cancer Res. 2010;16:711–8. doi: 10.1158/1078-0432.CCR-09-2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shibru D, Hwang J, Khanafshar E, et al. Does the 3-gene diagnostic assay accurately distinguish benign from malignant thyroid neoplasms? Cancer. 2008;113:930–5. doi: 10.1002/cncr.23703. [DOI] [PubMed] [Google Scholar]
- 75.Braga-Neto UM, Dougherty ER. Is cross-validation valid for small-sample microarray classification? Bioinformatics. 2004;20:374–80. doi: 10.1093/bioinformatics/btg419. [DOI] [PubMed] [Google Scholar]
- 76.Ntzani EE, Ioannidis JP. Predictive ability of DNA microarrays for cancer outcomes and correlates: an empirical assessment. Lancet. 2003;362:1439–44. doi: 10.1016/S0140-6736(03)14686-7. [DOI] [PubMed] [Google Scholar]
- 77.Ioannidis JP. Is molecular profiling ready for use in clinical decision making? Oncologist. 2007;12:301–11. doi: 10.1634/theoncologist.12-3-301. [DOI] [PubMed] [Google Scholar]
- 78.Dwan K, Altman DG, Arnaiz JA, et al. Systematic review of the empirical evidence of study publication bias and outcome reporting bias. PLoS One. 2008;3:e3081. doi: 10.1371/journal.pone.0003081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lau J, Ioannidis JP, Terrin N, et al. The case of the misleading funnel plot. Br Med J. 2006;333:597–600. doi: 10.1136/bmj.333.7568.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lijmer JG, Mol BW, Heisterkamp S, et al. Empirical evidence of design-related bias in studies of diagnostic tests. JAMA. 1999;282:1061–6. doi: 10.1001/jama.282.11.1061. [DOI] [PubMed] [Google Scholar]
- 81.Rutjes AW, Reitsma JB, Di NM, et al. Evidence of bias and variation in diagnostic accuracy studies. CMAJ. 2006;174:469–76. doi: 10.1503/cmaj.050090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ransohoff DF, Feinstein AR. Problems of spectrum and bias in evaluating the efficacy of diagnostic tests. N Engl J Med. 1978;299:926–30. doi: 10.1056/NEJM197810262991705. [DOI] [PubMed] [Google Scholar]
- 83.Lumbreras B, Parker LA, Porta M, et al. Overinterpretation of clinical applicability in molecular diagnostic research. Clin Chem. 2009;55:786–94. doi: 10.1373/clinchem.2008.121517. [DOI] [PubMed] [Google Scholar]
- 84.Mostly, your results matter to others. Nat Genet. 2009;41:135. doi: 10.1038/ng0209-135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.