

SUMMARY
In the companion paper (Martin et al., 2011) we reported that Bacillus subtilis requires three proteins for lipoic acid metabolism, all of which are members of the lipoate protein ligase family. Two of the proteins, LipM and LplJ, have been shown to be an octanoyltransferase and a lipoate:protein ligase, respectively. The third protein, LipL, is essential for lipoic acid synthesis, but had no detectable octanoyltransferase or ligase activity either in vitro or in vivo. We report that LipM specifically modifies the glycine cleavage system protein, GcvH, and therefore another mechanism must exist for modification of other lipoic acid requiring enzymes (e.g., pyruvate dehydrogenase). We show that this function is provided by LipL which catalyzes the amidotransfer (transamidation) of the octanoyl moiety from octanoyl-GcvH to the E2 subunit of pyruvate dehydrogenase. LipL activity was demonstrated in vitro with purified components and proceeds via a thioester-linked acyl-enzyme intermediate. As predicted, ΔgcvH strains are lipoate auxotrophs. LipL represents a new enzyme activity. It is a GcvH:[lipoyl domain] amidotransferase that probably employs a Cys-Lys catalytic dyad. Although the active site cysteine residues of LipL and LipB are located in different positions within the polypeptide chains, alignment of their structures show these residues occupy similar positions. Thus, these two homologous enzymes have convergent architectures.
Keywords: Post-Translational Modification, Lipoic Acid, Bacillus subtilis
Introduction
Comparative genomics allows for detection of incomplete metabolic models. Analysis of these discrepancies in our knowledge provides opportunities for new discoveries. Here we describe the analysis of an “extra” protein required for lipoic acid metabolism in Bacillus subtilis; homologues of which are found in nearly all sequenced Firmicutes genomes. The Firmicutes, also known as the low G+C gram-positive bacteria, are a phylum present throughout the environment that includes many pathogens. B. subtilis is the best characterized of this group and has been subject to intense genetic study since the discovery of its natural competence in 1958 (Spizizen, 1958). B. subtilis is a facultative spore-forming bacterium that grows in chemically defined medium lacking lipoic acid (Spizizen, 1958). In general, lipoic acid is required for aerobic, energy-conserved metabolism of 2-oxoacids and glycine; pyruvate dehydrogenase being the canonical lipoate-requiring enzyme (Perham, 2000). In B. subtilis, pyruvate dehydrogenase activity is required for growth under both aerobic and anaerobic conditions (Nakano et al., 1997) and thus it is clear that B. subtilis must synthesize the cofactor.
The current model for lipoic acid metabolism is based on Escherichia coli (Cronan et al., 2005, Rock, 2009). Enzymes that require lipoic acid for activity become modified by covalent attachment of the coenzyme to the ε-amino group of a specific lysine residue of conserved ~80 residue domains called lipoyl domains (LDs). Lipoate biosynthesis begins by transfer of octanoate from octanoyl-acyl carrier protein (ACP), a transient intermediate of fatty acid biosynthesis, to the LD by the ACP:LD octanoyltransferase LipB (Zhao et al., 2003). After the LD has been octanoylated, hydrogen atoms attached to the octanoate C6 and C8 carbons atoms are replaced with two sulfur atoms by lipoyl synthase, an S-adenosyl-L-methionine radical enzyme (Booker et al., 2007) to give lipoic acid. Exogenous free lipoic acid can also be scavenged by lipoyl ligase, LplA (Green et al., 1995, Morris et al., 1995), which is also active with octanoate (Green et al., 1995, Jordan & Cronan, 1997). Octanoate can also come from intracellular sources and thereby bypass a biosynthesis requirement at the expense of ATP (Hermes & Cronan, 2009).
The lipoyl domain modifying enzymes are grouped with those that modify biotinyl domains into Pfam PF03099. There are very few residues that are absolutely conserved within the family, a lysine residue being the sole conserved residue (Reche, 2000). However, there is sufficient transitive homology throughout the sequences to allow grouping (Reche, 2000). Despite this lack of sequence conservation the enzymes have considerable structural similarity including a conserved folding pattern (Ma et al., 2006). Lipoyl ligases have an additional accessory domain, which has been shown to be required for adenylation but not for acyltransfer (Christensen & Cronan, 2009). Comparisons of the crystal structures of LplA obtained with and without the acyl adenylate intermediate show that the accessory domain rotates away from the active site following adenylate formation, thereby allowing interaction with the substrate lipoyl domain (Fujiwara et al., 2010). Interestingly, many proteins of the LplA family lack a C-terminal accessory domain. This can be due to the domain being a separate protein, as is the case in archaea (Christensen & Cronan, 2009). More recently, one of these single domain LplA homologues, B. subtilis LipM, have been shown to be a new ACP:GcvH octanoyltransferase isozyme (Christensen & Cronan, 2010).
GcvH is a lipoyl acceptor protein that consists of a single lipoyl domain and is a key component of the glycine cleavage system. Although the glycine cleavage system is lipoic acid dependent, its inactivation is not known to cause a growth defect in any bacterium. The glycine cleavage system is well characterized in Arabidopsis and other plants, where it is known as glycine decarboxylase (Bauwe & Kolukisaoglu, 2003). There it serves a vital role in the C2 pathway (dark cycle), where it concentrates the carbon dioxide released by glycine degradation. In E. coli the glycine cleavage system is one of two synthetic routes to serine and 5, 10-methylenetetrahydrofolate. Disruption of the glycine cleavage system in E. coli has no growth phenotype, although gcv mutants accumulate and excrete glycine (Plamann et al., 1983). Disruption results in an inability to make serine from glycine, which creates a serine auxotrophy in the presence of glycine when the primary serine synthetic pathway is inactivated as well (Vanden Boom et al., 1991). The well-characterized glycine cleavage systems consist of four proteins, GcvH, GcvT, GcvP and GcvL. In B. subtilis expression of only the T and P subunits is under control of a glycine-activated riboswitch (Mandal et al., 2004). No dedicated GcvL subunit is apparent, but this function (oxidation of the lipoate moiety) may be provided by the E3 subunit that is shared among the 2-oxoacid dehydrogenases, as is the case in E. coli (Steiert et al., 1990). Given its key role in transfer of the methylamine group of glycine from the P subunit to the T subunit, it is surprising that GcvH is not encoded together with the T and P subunits, but rather at a remote genomic location where no recognizable riboswitch is detected (Mandal et al., 2004). Recently, the glycine cleavage system H protein of yeast, Gcv3, has been shown to be required for lipoic acid biosynthesis and RNA processing (Schonauer et al., 2009). In this paper we demonstrate that B. subtilis GcvH is both a substrate and a required component of the lipoic acid biosynthesis pathway.
Based on the E. coli model, the characterized LipA lipoyl synthase (Martin et al., 2009) and the newly described LipM octanoyltransferase (Christensen & Cronan, 2010) should suffice for lipoic acid synthesis and attachment in B. subtilis. However, as reported in the companion paper (Martin et al., 2011) a third gene, lipL, is also required for lipoyl domain modification. LipL belongs to the Pfam PF03099 family and lacks an accessory domain. In this paper we describe the novel biosynthetic requirement for LipL and the unexpected reaction catalyzed by this new enzyme. Based on these data we present a novel pathway for lipoic acid biosynthesis.
RESULTS
Expression, purification and modification of LipL
LipL (YwfL) was readily expressed in E. coli and purified as a soluble hexahistidine-tagged protein which lacked precedented catalytic activites (Martin et al., 2011). Mycobacterium tuberculosis LipB and B. subtilis LipM have been shown to become modified by attachment of acyl moieties in E. coli (Christensen & Cronan, 2010, Ma et al., 2006), so we wanted to determine if LipL also became modified. To this end the protein was subjected to trypsin digestion and the resulting peptides were analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS). LipL was unusually amenable to this analysis, giving 88% coverage (Fig. 1A). Two sites of modification were found: cysteine residues C39 and C150. Lipoyl and octanoyl modifications were present at both sites (Fig. 1B) indicating that LipL accepted octanoate and lipoate from E. coli sources. Assuming that the reaction catalyzed by this enzyme remains the same in B. subtilis, the substrates must be common to both organisms. The two modified cysteine residues are located on opposite sides of the LipL active site cleft (see below). Unlike LipM and M. tuberculosis LipB, no decanoyl Michael adduct was found when LipL was expressed in a wild type strain of E. coli.
Fig. 1.
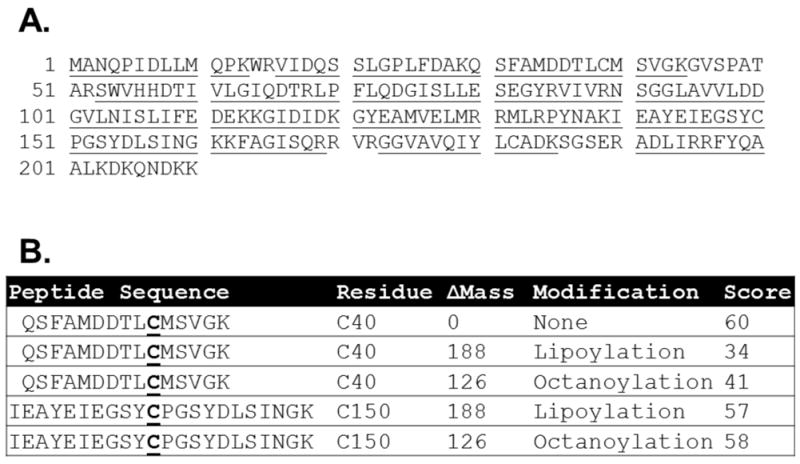
Panel A: Sequence coverage of LipL by LC-MS/MS analysis of LipL derived peptdides. The LipL peptide sequences detected are underlined. Panel B: Modifications of LipL detected after trypsin digestion followed by LC-MS/MS analysis. The theoretical peptides are shown with the modified cysteine residues in bold and underlined. The mass differences of the modification are given. The ion score is equal to −10log(P) where P is the probability the result is random chance
LipM is specific for GcvH
Although LipM readily catalyzed octanoyltransfer from ACP to GcvH (Christensen & Cronan, 2010), we were puzzled by the finding that the lipoyl domain of the B. subtilis pyruvate dehydrogenase PdhC subunit (E2PdhC) was a poor octanoyl acceptor substrate (Fig. 2A). This was first considered to be an artifact of use of overly truncated lipoyl domains. However, the full length PdhC subunit was also a poor substrate. PdhC was purified in good yield and eluted slightly before catalase upon size exclusion chromatography consistent with an octahedral complex (data not shown). This indicates that the C-terminal hexahistidine tag does not interfere with multimerization of the protein. Moreover, E. coli LipB was able to modify all of the B. subtilis lipoyl domains tested indicating that the acceptor proteins were properly folded. However, the enigmatic biotin lipoyl acceptor protein (BLAP), a protein previously reported to be biotinylated and lipoylated when expressed in E. coli (Cui et al., 2006)(Fig. 2) was not modified by any enzyme tested.
Fig. 2.
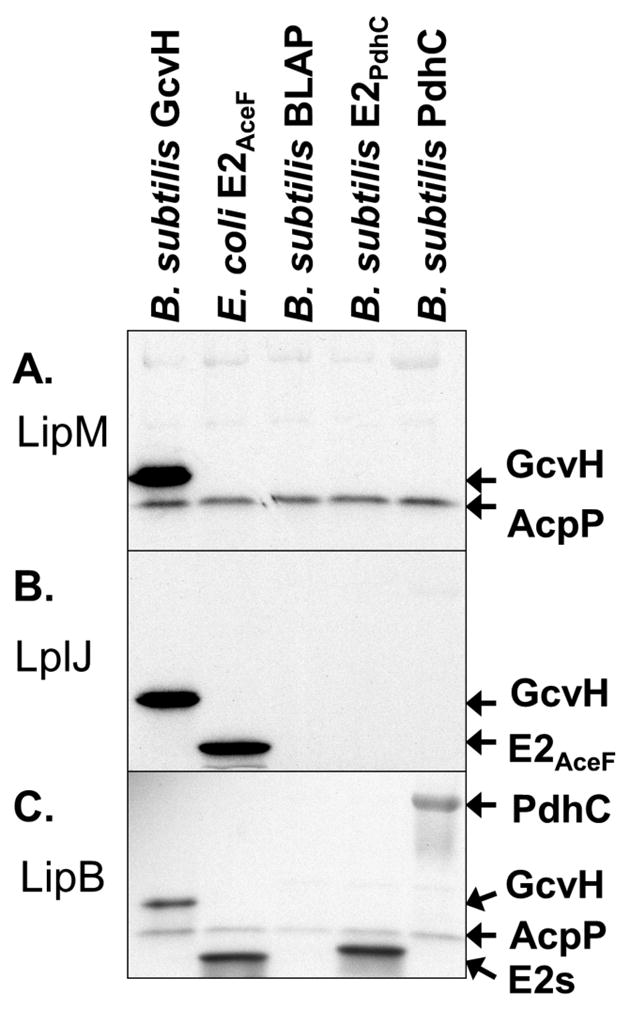
Specificity of B. subtilis LipM, B. subtilis LplJ, and E. coli LipB for various lipoyl domains. In panels A and C the lipoyl domain tested as a substrate is indicated at the top of each lane and by an arrow. The [1-14C]octanoyl-ACP substrate is indicated by an arrow. Autoradiograms of SDS-PAGE gel separations of lipoyl domain substrate specificity assays are shown. Sodium [1-14C]octanoate was used as a substrate for LplJ. Panel A: LipM octanoyltransfer to lipoyl domains from [1-14C]octanoyl-ACP generated using AasS. Panel B: Ligation of [1-14C]octanoate by LplJ to lipoyl domains. Panel C: LipB octanoyltransfer to lipoyl domains from [1-14C]octanoyl-ACP generated using Vibrio harveyi acyl-ACP synthetase (AasS). The two E2 domains migrate similarly and are denoted by E2s.
These data led to the hypothesis that LipL is essential for PdhC modification and hence growth. To test this scenario octanoyltransfer assays were done with octanoyl-ACP (generated in situ) as the acyl donor substrate (Christensen & Cronan, 2010). Similar to previous results (Fig. 2) addition of only LipM and LipL failed to result in octanoylation of either E2PdhC or LipL (Fig. 3A, lane 3). It was only in the presence of GcvH that octanoylation of LipL and E2PdhC proceeded (Fig. 3A, lane 6).
Fig. 3.

Panel A: Component controls for a coupled AasS/LipM/LipL assay starting with [1-14C]octanoate which becomes ligated to ACP by AasS. Following the reaction the proteins were resolved by SDS-PAGE and visualized by autoradiography. The identities of the [1-14C]octanoylated proteins are indicated. Panel B: Component controls for a coupled LplJ plus LipL assay as in Panel A. The starting substrates for LplJ–catalyzed ligation reactions were [1-14C]octanoate and ATP.
A similar phenomenon was also seen for the LplJ, which was identified and demonstrated to be a lipoyl ligase in the companion paper (Martin et al., 2011). Although LplJ was able to ligate octanoate to the E. coli lipoyl domain (E2AceF), no modification of either B. subtilis E2PdhC or of full-length B. subtilis PdhC was seen in vitro (Fig. 2B). Addition of LipL to the LplJ reaction failed to give modification of E2PdhC (Fig. 3B, lane 3). Only when GcvH, LipL and LplJ were all present did modification of E2PdhC proceed (Fig. 3B, lane 5). These observations argued that modified GcvH was an obligate intermediate in the modification of E2PdhC. It also showed that LipL was required for PdhC modification.
LipL is a novel amidotransferase
Since the octanoyl moiety of octanoyl-GcvH is attached by an amide linkage, LipL seemed to catalyze a novel amidotransfer reaction in which the active site cysteine thiol of LipL would attack the octanoyl-GcvH amide linkage to form octanoyl-S-LipL and would subsequently transfer the thioester-bound octanoyl moiety to E2PdhC to give a new amide-linked moiety (Fig. 4D). To test if LipL was a indeed a GcvH:E2 amidotransferase, LipL activity was assayed using purified [1-14C]octanoyl-GcvH as the donor substrate. Transfer of the octanoyl moiety to each of the PDH domains tested was observed whereas BLAP was not modified (Fig. 4A). Transfer from GcvH to LipL was also observed (Fig. 4C).
Fig. 4.

Autoradiograms of SDS-PAGE gels of assays for LipL-catalyzed amidotransfer from purified [1-14C]octanoyl-GcvH to lipoyl domains. Panel A: Amidotransfer of the [1-14C]octanoyl moiety from purified octanoyl-GcvH to the unmodified lipoyl domain indicated. Each reaction contained purified wild type (WT) LipL. The two E2 domains migrate similarly and are denoted by E2s. Panel B: Amidotransfer from purified [1-14C]octanoyl-GcvH to the E2PdhC. The purified wild type LipL or point mutant proteins indicated were used as enzyme sources. Panel C: Additional enzyme was added to allow detection of the octanoyl-LipL intermediate. Wild type (WT), C150A point mutant, C150S point mutant LipL were assayed in addition to a control (NE) lacking LipL. Panel D: Schematic of the LipL amidotransfer reaction is shown with the acyl-LipL intermediate.
To explore the LipL reaction mechanism site-directed mutagenesis of residues conserved in the PF03099 family was performed. LipM K162 is a strictly conserved residue in PF03099 whereas Y180 is conserved in the LipL amidotransferase subfamily (Fig. 5). Unfortunately, when expressed in E. coli both K162 substitutions (K162R and K162A) resulted in low levels of insoluble proteins whereas substitutions of alanine or phenylalanine for Y180 had no effect on catalysis. We also mutated residues C39 and C150, both of which had been found to be modified by LC-MS/MS (Fig. 1B). Replacement of C39 with alanine or serine had no effect on catalysis whereas replacement of C150 with either alanine or serine resulted in loss of transfer to E2PdhC (Fig. 4B) and the inability to form an octanoyl-enzyme intermediate (Fig. 4C). Given the above observations, we propose a mechanism for amidotransfer by LipL (Fig. 4D).
Fig. 5.

Alignment of members of the LipL clade. Positions having 50% or greater similarity are highlighted in grey. The catalytic cysteine residue (C150 in B. subtilis LipL), the other modified cysteine residue (C39 in B. subtilis LipL), and the conserved PF03099 lysine residue are highlighted in black. BACSU is Bacillus subtilis 168, BACHD is Bacillus halodurans, LISMO is Listeria monocytogenes, LACBA is Lactobacillus brevis and STRPC is Streptococcus pyogenes serotype M12 (strain MGAS9429). This alignment is part of the larger one used to create Fig. 8A.
Amidotransfer was also assayed in crude extracts of various B. subtilis mutant strains. Although LipM activity was observed, no amidotransfer by LipL was seen unless the extracts were supplemented with purified LipL and LipM (Fig. 6). The reason for this discrepancy is not clear. Perhaps higher concentrations of octanoyl-GcvH are required to detect LipL activity, although transfer was not seen using purified [1-14C]octanoyl-GcvH as a substrate (data not shown). Additionally, LipL may be present in low amounts under the growth conditions used or LipL may become inactivated during extract preparation.
Fig. 6.

Panel A: Autoradiograph of an SDS-PAGE gel of octanoyltransfer assays using AasS generated [1-14C]octanoyl-ACP as a substrate. Extracts of the mutant strains indicated were used as a source of enzyme for transfer to GcvH and PdhC. All strains also carried a disruption in lplJ. When purified LipM and LipL were added to replace the missing protein(s) this is indicated with a plus sign.
GcvH is required for protein lipoylation in vivo
Our in vitro results argued that GcvH was an essential intermediate in lipoylation of PdhC and possibly BkdB.. To test the model for lipoic acid biosynthesis in B. subtilis, we disrupted gcvH. The ΔgcvH strain was auxotrophic for lipoic acid, the same phenotype shown by ΔlipA and ΔlipM strains. As observed in the other lipoate auxotrophs, the lipoate requirement of the ΔgcvH strain was bypassed by addition of acetate and BCFA precursors. In vivo results showing that bypass of a ΔlipL mutation requires both acetate and branched chain fatty acid (BCFA) precursors indicates that LipL is also required for lipoic acid modification of the BkdB subunit of the essential branched chain dehydrogenase. Lipoic acid also restored growth suggesting that LplJ ligated lipoic acid to E2PdhC in the absence of GcvH (Fig. 7A). Expression of a functional ectopic copy of the gcvH gene under the control of an inducible promoter allowed growth of the ΔgcvH strain when induced with IPTG (Fig. 7B). This demonstrates that the growth deficiency was due to the absence of a functional copy of the gcvH gene. Residual growth of the uninduced cells was observed, which can be attributed to the known basal expression of the spac promoter. Moreover, the expression of the E. coli lipB gene in a ΔgcvH mutant strain restored growth in minimal medium (data not shown). These results indicated that the LipB octanoyl transferase activity was sufficient to overcome the absence of GcvH, confirming its role in the octanoyl transfer from ACP to the lipoyl domains.
Fig. 7.

Growth and lipoylation phenotypes of a B. subtilis ΔgcvH strain. Growth of strains on minimal glucose media plates with the supplements indicated after 48 h at 37°C. LA denotes lipoic acid. Panel A: Growth of strains JH642 (wild type) and NM20 (ΔgcvH). Panel B: Growth of strains JH642 (wild type) and NM21 (ΔgcvH amyE:: Pspac-gcvH). Panel C: Lipoylation of GcvH in B. subtilis crude extracts. SDS-PAGE and immunoblotting with anti-LA antibody was done as described in Experimental Procedures. Cells were grown 22 h in minimal media with or without lipoate added as indicated. Crude extracts of B. subtilis wild type (WT) and ΔgcvH strains were used as indicated.
LipM octanoyltransferase activity with GcvH as the acceptor was previously demonstrated in vitro (Christensen & Cronan, 2010). Western blotting analysis with anti-lipoic acid antibodies showed a lipoylated protein of apparent mass of 20 kDa (Fig. 7C). As shown in the companion paper, the GcvH band was detected in extracts of strains in which lipM was active. The gel migration of the band is consistent with the previously reported migration rate of GcvH (Christensen & Cronan, 2010) and disruption of gcvH resulted in loss of the 20 kDa band (Fig 7C) thereby confirming its identity. Disruption of gcvH resulted in a complete loss of biosynthetic lipoic acid assembly (Fig. 7C), confirming that GcvH was required in B. subtilis lipoic acid biosynthesis. Upon supplementation with lipoic acid the ΔgcvH strain showed strong lipoylation of the dehydrogenase E2 subunits (Fig. 7C) consistent with the robust growth seen in this medium (Fig. 7A). These results confirm the physiological relevance of the in vitro biochemical assays and show GcvH is required not only for biosynthetic lipoylation of PdhC, but other E2 subunits as well.
LipL and LipB are mechanistically convergent
Phylogenetic analysis of LipL compared to other members of PF03099 shows significant, albeit modest, support for B. subtilis LipL as part of a deeply branching clade related to lipoate protein ligases (Fig. 8A). LipL is also closely related to LipM octanoyltransferases, which branch off later from the same lipoate protein ligase clade (Christensen & Cronan, 2010). Indeed, the unpublished B. halodurans LipL structure (PDB entry 2P5I) is the closest available structure to LipM. The positions of the catalytic cysteine residues (C150 in both proteins) are very similar in LipM and LipL. Upon structural alignment of LipL with M. tuberculosis LipB (PDB 1W66) the catalytic cysteine residues are located only 2.34 Å apart (Fig. 9). This was unexpected because the residues are contributed by different loops that are ~45 residues removed in sequence alignments (Christensen & Cronan, 2010). Therefore, it appears that LipL and LipB have undergone mechanistic convergence within their active sites using the same protein scaffold.
Fig. 8.

Minimum evolution analysis of alignments with bootstrap percentage values shown for each branch. The trees are drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The scale represents a 50% difference in compared residues per length. Branches representing Bacillus proteins are indicated in bold. Panel A: Analysis of selected catalytic domains from PF03099 the structurally related biotin ligases are used as the out-group. Panel B: Selected lipoyl domains involved in lipoic acid metabolism are shown.
Fig. 9.

Structure of LipL and comparison with that of LipB. Panel A: The unpublished structure of B. halodurans LipL (PDB 2P5I). The modified cysteine residues, C39 and C150, detected by LC-MS/MS of the B. subtilis protein are shown in red. Panel B: Structural alignment of LipL from B. halodurans with M. tuberculosis LipB (PDB 1W66) (Ma et al., 2006). The active site adduct is shown in purple. Panel C: Close up view of the structural overlay with the LipB decanoyl adduct removed. The active site cysteine sulfur atoms are colored orange whereas carbon atoms are white. The distance between the two sulfur atoms is 2.4 Å.
DISCUSSION
We report delineation of a new pathway of lipoic acid synthesis in which octanoyl moieties are transferred from the ACP of fatty acid synthesis to the GcvH protein of the glycine cleavage system by LipM. The octanoyl moieties are then transferred by LipL, a novel GcvH:E2 amidotransferase, from GcvH to E2 subunits to form active dehydrogenase complexes (Fig. 10). Note that at this stage of our investigations it is not clear at what step(s) LipA acts. It is possible that LipA inserts the sulfur groups before LipL transfer, after LipL transfer or at either step. Note that although we characterized the B. subtilis pathway due to the sophisticated genetics available in this bacterium, paralogous genes are present in other Firmicutes proficient in lipoic acid synthesis.
Fig. 10.

Current models for lipoic acid biosynthesis in E. coli and B. subtilis. Lipoic acid synthesis in E. coli is accomplished by two enzymes whereas B. subtilis requires three enzymes. B. subtilis also requires GcvH as an intermediate carrier whereas E. coli does not. We lack data to determine if LipA acts before or after LipL. We propose that B. subtilis, LipA uses octanoyl-LD (both octanoyl-GcvH and octanoyl-E2) as substrates, and therefore can act before or after LipL.
The requirement for LipL is due to the specificity of the LipM for GcvH. We find significant in vitro modification of the E2AceF by LplJ, but not by LipM (Fig. 2) suggesting LplJ may be less specific than LipM. However, LipM complements an E. coli lipB lipoic acid auxotroph (Christensen & Cronan, 2010) which demonstrates modification of E2AceF. These data indicate that restoration of growth is a much more sensitive assay than the in vitro radiolabel assay. This is expected since growth results from protein lipoylation which in turn results in dehydrogenase catalysis with a concomitant amplification of the signal. Another difference is that lipoylation of PdhC may occur in vivo after the protein has been assembled into the PDH complex rather than as the free subunit we tested in vitro. The E2AceF and E2PdhC domains clearly form different clades indicating different conserved residues (Fig. 8B). This may explain why LplJ is able to modify E2AceF but not E2PdhC. Identification of the residues responsible for the observed differences in substrate recognition will require further study.
The amidotransfer reaction (also called transamidation) catalyzed by LipL was unexpected, but not unprecedented. The sortase transpeptidases that anchor proteins to the outer surfaces of various gram-positive bacteria use a Cys-His-Asn triad in their amidotransfer reaction (Clancy et al., 2010). Moreover, peptide bond synthesis catalyzed by cysteine proteases (Morihara & Oka, 1981) employs this same catalytic triad. The DD-transpeptidase/carboxypeptidases of bacterial cell wall biosynthesis employ a Ser-Lys-Tyr catalytic triad (Silvaggi et al., 2003) and substitution of cysteine for the nucleophilic serine residue in closely related enzymes often results in active enzymes (Hadonou et al., 1992). In our work, LipL residues C39 and C150 were found to be acylated. However, C39 was not essential for catalysis (Fig. 4b) and is not conserved in more diverse members of the LipL clade (Fig. 5). (Acylation of C39 may have resulted by trans-thioesterification from C150 during the overnight trypsin digestion.) Mutagenesis of LipL C150 resulted in loss of enzymatic activity and the inability to form an acyl-enzyme intermediate (Fig. 4) which is the same result seen upon mutagenesis of the active site LipM cysteine residue (Christensen & Cronan, 2010). Based on the B. halodurans LipL crystal structure K162 and Y180 are less than 3 Å from C150 (Fig. 10). We found that these residues are conserved in all members of the LipL clade (Fig. 9). From these data, it seemed likely that LipL employed a Cys-Lys-Tyr catalytic triad similar to those seen in other amidotransferases. However, we found that Y180 was not required for amidotransfer as the Y180F and Y180A proteins was active. Therefore, we propose that LipL amidotransferases utilize a Cys-Lys dyad as do the LipB and LipM ACP-octanoyltransferases(Christensen & Cronan, 2010, Ma et al., 2006). If so, this would a novel active site configuration for an amidotransferase (transamidase). Note that LipL shares no detectable overall sequence or structural similarity to other known amidotransferases.
The B. subtilis biotin-lipoyl acceptor protein (BLAP) was reported to be at least partially modified with lipoate when expressed in E. coli (Cui et al., 2006). In that work the concentration of lipoic acid added to the culture medium corresponded to >60,000-fold excess over that needed for half-maximal growth of a lipA strain (Reed & Cronan, 1993). As previously reported (Cui et al., 2006), we found that most of the BLAP was in the unmodified form after two hours of expression. It was thought that BLAP might a good candidate for intermediate carrier in lipoic acid biosynthesis. However, we assayed BLAP as an octanoyl acceptor and found it was not modified by any enzyme tested (Fig. 2, 4A). The role of BLAP in B. subtilis remains unknown, but it does not appear to be involved in lipoic acid metabolism.
Although the data presented in these papers provide a good working model of lipoic acid biosynthesis, a conflicting view of lipoic acid scavenging emerges. As shown in the companion paper, addition of external lipoic acid only partially restored growth of a ΔlipL mutant and modification of E2PdhC by LplJ was not seen in vitro (Fig. 2B) whereas growth of the ΔgcvH strain was fully restored by lipoic acid (Fig. 5A) and the dehydrogenase E2 subunits became highly modified (Fig. 5C). One possibility is LplJ activity may be lower in a lipL strain or higher in a gcvH strain. If there is an allosteric or regulatory effect, it would have to be large, as we find the phenotypes are the same over a wide range of lipoic acid concentrations (0.5 nM-0.5 mM) (data not shown). An alternative explanation would be that deletion of lipL results in global changes in gene expression making lipoyl scavenging insufficient to restore the required enzymatic activities. This may be why an unusual E2 lipoylation pattern was seen in the western blot of a ΔlipL strain in the companion paper (Martin et al., 2011).
A similar mechanism for lipoic acid synthesis based on genetic analyses has been proposed for the yeast Saccharomyces cerevisiae where it was found that the glycine cleavage system H protein Gcv3 is not only a target for lipoylation but also is necessary for the lipoylation of the other lipoate-modified proteins, the 2-oxoacid dehydrogenases (Schonauer et al., 2009). These workers favor a model of complex formation and allosteric interactions, although a model in which Lip2, the yeast LipB homolog, transfers the octanoyl moiety from octanoyl-ACP to Gcv3, where two sulfur atoms are inserted into the acyl chain by Lip5 (the yeast LipA homolog) was subsequently proposed (Hiltunten et al 2010). The lipoyl moiety would then be transferred to the 2-oxoacid dehydrogenases by a LplA homologue called Lip3. It is tempting to speculate that Lip3 might perform the same reaction as B. subtilis LipL.
It is not clear why B. subtilis routes the synthesis of the essential lipoate cofactor through GcvH. Given that this bacterium has a predominately aerobic life style, if routing were needed, PDH would seem more the likely intermediate especially since given the obscure role of the glycine cleavage system in bacterial metabolism. It should be noted that GcvH lipoyl domains are highly divergent from the dehydrogenase E2 lipoyl domains (Fig. 8B). This divergence may explain how GcvH behaves as a distinct acceptor protein. In B. subtilis, strains carrying a gcvH disruption grow well when provided with lipoic acid or the products of the inactivated dehydrogenases (Fig. 7A) which indicates that GcvH does not play an essential role in amino acid or single carbon metabolism. In the companion paper we show that E. coli lipB complements both ΔlipM and ΔlipL mutant strains. Some evolutionary pressure may exist for B. subtilis to maintain the specificity of LipM for GcvH. However, instead of a phenomenon that can be rationalized, constructive neutral evolution (Gray et al., 2010) may be involved. LipM may have coevolved with the glycine cleavage system and LipL provided a means for it to be used for other lipoate dependent enzymes. A more extensive bioinformatic analysis may reveal the ancestral states of the pathway.
B. subtilis is predicted to encode all five known lipoic acid dependent complexes; pyruvate dehydrogenase, 2-oxoacid dehydrogenase, branched-chain keto acid dehydrogenase, acetoin dehydrogenase and glycine cleavage system. Amidotransfer of octanoyl/lipoyl moieties may provide B. subtilis with the ability to quickly adapt to changing environments by transferring lipoic acid among different enzymes. Alternatively, the extra step in the biosynthetic pathway may provide additional opportunities for regulatory control. B. subtilis has previously been shown to utilize lipoic acid as a sulfur source (Mansilla & de Mendoza, 1997). Although it is unlikely that lipoic acid would be a significant source of sulfur in most environments, lipoylation could be used as an indirect readout of sulfur availability. No transcriptional regulation of lipoic acid biosynthesis has been found in E. coli (Cronan, unpublished) or Salmonella enterica (Smith et al., 1991). However, B. subtilis lplJ has been shown to be induced during spore outgrowth (Keijser et al., 2007) and lipM and lplJ are induced in response to valine (Ye et al., 2009). Constitutive expression of GcvH would allow it to be always available to act as a lipoate transfer protein regardless of glycine supplementation. Clearly, there is much to be learned about the physiological and ecological implications of this pathway.
EXPERIMENTAL PROCEDURES
Media and Chemicals
Escherichia coli strains were grown on LB rich or M9 minimal media (Sambrook & Russell, 2001) under aerobic conditions at 37°C unless otherwise indicated. Antibiotics (Sigma Chemical Co) were used at the following concentrations for E. coli (in μg/ml): sodium ampicillin, 100; kanamycin sulfate, 50; spectinomycin sulfate, 50; chloramphenicol, 12; gentamycin sulfate, 25 and tetracycline hydrochloride, 12. Difco Vitamin-Assay Casamino Acids was obtained from Becton, Dickenson. [1-14C]Octanoic acid and [1-14C]octanoyl-CoA were purchased from Moravek. American Radiolabeled Chemicals provided [α-32P]ATP. The pH of buffers and solutions is reported at room temperature.
Bacterial Strains and Plasmids
Strains and plasmids used are shown on Table 1. Primers used in this study are given in Table 2. Standard techniques for DNA manipulation and cloning were employed unless otherwise indicated (Sambrook & Russell, 2001). PCR amplification was performed using Taq (New England Biolabs) or Pfu (Stratagene) polymerase according to the manufacturer’s recommendation, except with the addition of 5% dimethyl sulfoxide to Pfu reactions.
Table 1.
| Strain | Genotype | Source |
|---|---|---|
| Escherichia coli: | ||
| MG1655 | rph-1 | CGSCa |
| RMK98 | MC1061 fadK::his6-cat ΔompT::FRT | this study |
| QC146 | rph-1 ΔlplA::FRT ΔlipB::FRT | (Christensen & Cronan, 2009) |
| QC152 | rph-1 ΔlplA::FRT ΔlipB::FRT/pTARA, pQC026 | this study |
| QC172 | rph-1 ΔlplA::FRT ΔlipB::FRT ΔompT::FRT | this study |
| QC191 | rph-1 ΔlplA::FRT ΔlipB::FRT ΔompT::FRT/pQC047, pTARA | this study |
| QC222 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC077, pCY598 | this study |
| QC223 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC078, pCY598 | this study |
| QC224 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC079, pCY598 | this study |
| QC225 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC080, pCY598 | this study |
| QC255 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC086, pCY598 | this study |
| QC256 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC087, pCY598 | this study |
| QC257 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC088, pCY598 | this study |
| QC258 | rph-1 IN(rrnD-rrnE)1 ϕ(fabA-lacZ)1(Hyb)cat/pQC089, pCY598 | this study |
| Bacillus subtilis: | ||
| JH642 | trpC2 pheA1 | Laboratory stock |
| NM20 | JH642 gcvH:: Km | This study |
| NM21 | NM20 amyE::Pspac-gcvH | This study |
| Plasmids | Relevant Properties | Source |
| pTARA | pACYC origin, arabinose inducible T7 polymerase | (Wycuff & Matthews, 2000) |
| pCY598 | RSF origin, arabinose inducible T7 polymerase | (Cronan, 2003) |
| pCR2.1 | TOPO TA cloning vector | Invitrogen |
| pSJ120 | E. coli LipB expression vector | (Jordan & Cronan, 2003) |
| pQC023 | B. subtilis E2p lipoyl domain expression vector | this study |
| pQC026 | C-terminal hexahistidine BLAP expression vector | this study |
| pQC033 | N-terminal hexahistidine LipL expression vector | (Martin et al., 2011) |
| pQC047 | C-terminal hexahistidine PdhC expression vector | this study |
| pQC077 | N-terminal hexahistidine LipL K162R mutant expression vector | this study |
| pQC078 | N-terminal hexahistidine LipL K162A mutant expression vector | this study |
| pQC079 | N-terminal hexahistidine LipL C150A mutant expression vector | this study |
| pQC080 | N-terminal hexahistidine LipL C150S mutant expression vector | this study |
| pQC086 | N-terminal hexahistidine LipL C39A mutant expression vector | this study |
| pQC087 | N-terminal hexahistidine LipL C39S mutant expression vector | this study |
| pQC088 | N-terminal hexahistidine LipL Y180A mutant expression vector | this study |
| pQC089 | N-terminal hexahistidine LipL Y180F mutant expression vector | this study |
| pGES485 | Integrational vector; Sp | (GE Schujman, unpublished) |
| pJM114 | Integrational vector; Km | (Perego, 1993) |
| pNM67 | pJM114 containing gcvH interrupted with a kanamycin cassette | This study |
| pNM70 | pGES485 containing gcvH cloned into HindIII-BamHI sites | This study |
CGSC, E. coli Genetic Stock Center
Table 2.
| Oligonucleotides | Sequence 5′-3′ |
|---|---|
| Q030 | ttaaccatggcatttgaatttaaacttcc |
| Q031 | aatggatccttaaaattgaagatcttcgtaacc |
| Q034 | gaaacagcacctataaaggaggca |
| Q042 | ttaatgatgatgatgatgatgttgagtggaattgctcagttcg |
| Q095 | taaggtaccaaggagatataccatggcatttgaatttaaacttc |
| Q096 | attggatccttagtgatggtgatggtgatgcgcctccattaaaattaattg |
| Q113 | aacggataagacgggcataa |
| Q114 | gggatgaaggaacgtcattt |
| Q147 | ctatgacctcagtataaatggcaaagcgttcgccggcatct |
| Q148 | agatgccggcgaacgctttgccatttatactgaggtcatag |
| Q149 | cagctatgacctcagtataaatggcaaacgtttcgccggcatctct |
| Q150 | agagatgccggcgaaacgtttgccatttatactgaggtcatagctg |
| Q151 | ctatgaaattgaagggtcttattctcccggcagcta |
| Q152 | tagctgccgggagaataagacccttcaatttcatag |
| Q153 | agcctatgaaattgaagggtcttatgctcccggcagctat |
| Q154 | atagctgccgggagcataagacccttcaatttcataggct |
| Q0171 | gcaatggatgatacgctagccatgtccgtcggaaaagg |
| Q0172 | ccttttccgacggacatggctagcgtatcatccattgc |
| Q0173 | cgcaatggatgatacgctatccatgtccgtcggaaa |
| Q0174 | tttccgacggacatggatagcgtatcatccattgcg |
| Q0175 | gcggcggagtcgctgttcaaattgctctctgtgcagata |
| Q0176 | tatctgcacagagagcaatttgaacagcgactccgccgc |
| Q0177 | cggcggagtcgctgttcaaatttttctctgtgcaga |
| Q0178 | tctgcacagagaaaaatttgaacagcgactccgccg |
| L | agtggatccaagagcatgggaaag |
| LI | ttgtcgactcgtgttctcctgagtaa |
| LII | gagtcgacacaagaagactaaaacag |
| LIII | aaagactgcagtgaaattcaccgcca |
The genes encoding proteins containing lipoyl domains were placed under the control of a T7 promoter. The lipoyl domain from B. subtilis PdhC (E2PdhC) was identified by alignment with the B. stearothermopolis lipoyl domain isolated by V8 protease digestion (Packman et al., 1988) and this portion of the pdhC gene was PCR amplified using primers Q030 and Q031(the latter primer added a termination codon). After digestion with NcoI and BamHI this product was inserted into pET28b+ cut with the same enzymes to give pQC023. The full-length pyruvate dehydrogenase E2p subunit (pdhC) was PCR amplified using primers Q095 and Q096 (which added a C-terminal hexahistidine tag) and inserted into pCR2.1 using the TA cloning kit. The pdhC insert was then moved into pRSF-1b using the NcoI and BamHI sites to give pQC047. The biotin/lipoic acid protein (BLAP) was PCR amplified using primers Q034 and Q042 (which added a C-terminal hexahistidine tag) and inserted into pCR2.1 using the TA cloning kit. The BLAP insert was then moved into pET28b+ using the NcoI and BamHI sites to give the pQC026.
In order to minimize proteolysis of the full length PdhC, the protein was expressed in a strain lacking the OmpT protease, which cleaves the flexible linker region (Lessard et al., 1998). The ompT::Km mutation was constructed as described using the primers described (Baba et al., 2006) to give strain RMK98. Strain QC146 was transduced with a phage P1 stock grown on strain RMK98 to give strain QC171. The kanamycin cassette was removed (Datsenko & Wanner, 2000) to give strain QC172 and the deletion was confirmed by PCR using primers Q113 and Q114.
Mutants of LipL were produced by modification of pQC033 using QuickChange mutagenesis as per the manufacturers recommendations (Agilent). Primers were used for the corresponding mutations were: Q147/Q148 for K148A, Q149/Q150 for K148R, Q151/Q152 for C150A, Q153/Q154 for C150R, Q171/Q172 for C39A, Q173/Q174 for C39S, Q175/Q176 for Y180A, Q177/Q178 for Y180F. Mutant LipL proteins were expressed and purified as described in the companion paper (Martin et al., 2011).
The gcvH deletion mutant, strain NM20, was obtained by transformation of strain JH642 with plasmid pNM67. This plasmid was constructed as follows. A 418 bp fragments from the 5′ end and upstream region of the gene was PCR amplified with oligonucleotides L and LI (Table 2) and inserted into BamHI and EcoRI sites of pJM114 (Perego, 1993). In the same manner a 434 bp fragment containing the 3′ end and downstream region of the gene was PCR amplified with oligonucleotides LII and LIII (Table 2) and cloned into XhoI and KpnI sites from the previously generated plasmid, yielding plasmid pNM67. This plasmid was linearized and used to transform JH642.
Strain NM20 was complemented with an ectopic wild type copy of gcvH under an IPTG dependent promoter (Pspac). To this end plasmid pNM70 was constructed by cloning of a 456 bp PCR-amplified fragment generated with primers XLVI and XLVII (Table 2) into the HindIII and BamH sites of pGES485 (Schujman GE, unpublished). Plasmid pNM70 was linearized and used to transformed strain NM20, yielding strain NM21. Transformants were selected on kanamycin plus spectinomycin plates and screened for amyE phenotype.
Purification of Lipoyl Domains
The E. coli E2p(1,3) hybrid lipoyl domain (LDAceF) was purified by acid treatment followed by ion exchange chromatography as described previously (Zhao et al., 2003). Mass spectroscopy analysis was performed as described for GcvH (Christensen & Cronan, 2010). The E2AceF was in the unmodified form and lacked the N-terminal methionine residue. The E2AceF lacks tryptophan and tyrosine residues, it was quantified using the Bradford assay reagent (Bio-Rad) with bovine gamma globulin (Pierce) as a standard.
To purify E2PdhC strain QC063 was grown in LB with kanamycin to an OD600 of 0.6. Protein expression was induced with 1 mM IPTG and the culture was incubated for another 2 h before the cells were pelleted by centrifugation and frozen at −80C. The cells were lysed by passage through a French pressure cell and the E2PdhC was purified by pH precipitation and anion exchange chromatography and prepared for mass spectrometry as described (Zhao et al., 2003). The anion exchange protocol allows resolution of the unmodified and modified forms of the domain as shown by 20% native PAGE. The modification state of the E2PdhC was verified to be in the unmodified form without the N-terminal methionine by electrospray mass spectrometry as described above for ACP (Christensen & Cronan, 2010). E2PdhC was quantified by absorbance at 280 nm using an extinction coefficient of 6,990 M−1 cm−1.
The B. subtilis BLAP protein was expressed using strain QC152 and purified by nickel affinity and anion exchange chromatographic steps as described for LplA (Christensen & Cronan, 2009). BLAP was verified to be in the unmodified form lacking the N-terminal methionine by electrospray mass spectrometry as described for ACP (Christensen & Cronan, 2010) and was quantified by absorbance at 280 nm (extinction coefficient of 5,500 M−1 cm−1). B. subtilis PdhC was expressed using strain QC191 and purified by nickel affinity and anion exchange as described for LplA (Christensen & Cronan, 2009). PdhC was quantified by absorbance at 280 nm using an extinction coefficient of 18,450 M−1 cm−1. To determine if the C-terminal hexahistidine tag interfered with complex formation, we subjected pure PdhC to size exclusion chromatography as described for LipM (Christensen & Cronan, 2010).
Trypsin digestion and LC-MS/MS analysis of LipL
Trypsin digestion and LC-MS/MS analysis of LipL was performed as previously described for LipM (Christensen & Cronan, 2010). Analysis was performed on LipL from QC083 grown without triclosan and from the fabA strain QC143 grown with triclosan.
Lipoic acid ligation Assay
Octanoyl ligase activity was assayed to determine the LD substrate specificity of LplJ. The reaction contained 100 mM sodium phosphate (pH 7.0), 50 mM sodium chloride, 5 mM TCEP, 0.25 mM sodium [1-14C]octanoate, 1 mM magnesium chloride, 1 mM ATP, 20 μM lipoyl domain and 1 μM LplJ. The reaction was analyzed using a modification of the method of Laskey and Mills (Laskey & Mills, 1975 )in which 10 μl of the reactions were subjected to SDS-PAGE on 4–20% gradient gels which were soaked in Amplify fluorographic reagent (GE Healthcare), dried, and exposed to preflashed Biomax XAR film (Kodak) at −70°C for 24 h.
Assay of octanoyl-[Acyl Carrier Protein]:Protein N-octanoyltransfer
Octanoyltransfer by LipM and LipB was assayed in order to determine the LD substrate specificity of these enzymes. Assays using AasS were performed as previously described (Christensen & Cronan, 2010) with different lipoyl domains substituted for GcvH. Reactions were analyzed as described above with LplJ.
Assay of amidotransfer
Amidotransfer was assayed coupled to lipoyl ligation by LplJ. The reactions contained 100 mM sodium phosphate (pH 7.0), 50 mM sodium chloride, 5 mM TCEP, 0.25 mM sodium [1-14C]octanoate, 1 mM magnesium chloride, 1 mM ATP, 20 μM GcvH, 20 μM PdhC, 1 μM LipL, and 1 μM LplJ. The reactions were analyzed as described above for the LplJ assays.
Amidotransfer was also assayed by coupling to octanoyltransfer by LipM. The reactions contained 100 mM sodium phosphate (pH 7.0), 50 mM sodium chloride, 5 mM TCEP, 0.25 mM sodium [1-14C]octanoate, 1 mM magnesium chloride, 1 mM ATP, 50 μM holo-AcpP, 2 μM AasS, 20 μM GcvH, 20 μM PdhC, 10 μM LipL and 10 μM LipM. Reactions were analyzed as described for the LplJ assays above.
B. subtilis crude extracts were also assayed for LipM and LipL activities. The cultures were grown in Spizizens minimal salts (Spizizen, 1958) supplemented with 0.5% glucose, 10 mM sodium acetate, 10 mM sodium succinate, 0.1 mM sodium isovalerate, 0.1 mM sodium isobutyrate, and 0.1 mM sodium 2-methylbutyrate. Cultures were grown to an OD600 of 0.6, pelleted by centrifugation, frozen at −80°C, and lysed in 100 mM sodium phosphate (pH 7.0) by sonication. The reaction conditions were the same as the LipM coupled assay with enzyme or 50 μg of extract protein added where indicated. Reactions were analyzed as described for the LplJ assays above.
In order to directly assay amidotransfer [1-14C]octanoyl labeled GcvH was prepared using LplJ. The reaction contained 100 mM sodium phosphate (pH 7.0), 50 mM sodium chloride, 5 mM TCEP, 1 mM sodium [1-14C]octanoate, 2 mM ATP, 0.5 mM GcvH, and 10 μM LplJ. The reaction was allowed to proceed at 37°C for 2 h. Then the reaction was diluted 20-fold in 25 mM sodium MES buffer (pH 6.1) and purified using anion exchange chromatography using an AKTA purifier10 (GE Healthcare) with a 1.8ml POROS QE anion exchange column with a flow rate of 5 ml per minute. Protein was bound with 25 mM sodium MES buffer (pH 6.1) and eluted with a 0–1 M gradient of lithium chloride, where octanoyl GcvH eluted at 480 mM. Purified lipoyl domains were stored as above except 5 mM TCEP was the reducing agent. Amidotransfer was directly assayed from purified 14C-octanoyl GcvH to various LDs. The reactions contained 100 mM sodium phosphate (pH 7.0), 50 mM sodium chloride, 5 mM TCEP, 20 μM [1-14C]octanoyl-GcvH, 20 μM of different lipoyl domains, and 1 μM LipL. Amidotransfer reactions with mutant LipL proteins were performed with 10 μM LipL or mutant LipL plus E2PdhC domain as an additional lipoyl domain. The LipL acyl-enzyme intermediate was visualized using the same assay conditions lacking an acceptor domain and with 10 μM LipL or mutant LipL. The reactions were analyzed as described for the LplJ assays above.
Immunoblotting analysis
Lipoylation of crude extracts of B. subtilis strains was assayed with anti-lipoic acid antibody (Calbiochem) as described in the companion paper (Martin et al., 2011)
Phylogenetic Analysis and Bioinformatics
We determined the phylogeny of select amino acid sequences of the LipB/LplA/BirA superfamily (PF03099), the biotin_lipoyl_domain superfamily (PF00364), and the glycine cleavage system H protein family (PF01597) (Finn et al., 2010). Alignment was performed using Dialign-TX with low scoring region length 4, low quality region length 40, and unlimited sensitivity (Subramanian et al., 2008). The edges of the alignment were trimmed using Jalview (Waterhouse et al., 2009) so only the relevant domains remained. A minimum evolution tree was constructed with bootstrap analysis of 1000 replicates using Mega4 (Tamura et al., 2007) using the default settings. All positions containing gaps and missing data were eliminated from the dataset (complete deletion option). There were a total of 63 positions in the final dataset comparing lipoyl domains. There were a total of 57 positions in the final dataset that compared catalytic domains.
Display of protein structures and structural alignments were performed using the Swiss PDB viewer (Kaplan & Littlejohn, 2001). Protein extinction coefficients and theoretical masses were calculated using Protparam at the EXPASY website (Gasteiger et al., 2003) and used to determine molarity. The extinction coefficients were calculated on the assumption that all cysteine residues were in the reduced state.
Acknowledgments
This investigation was supported by the National Institutes of Health under Ruth L. Kirschstein National Research Service Award 5 T32 GM070421 from the National Institute of General Medical Sciences, grant AI15650 from the National Institute of Allergy and Infectious Disease, the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Agencia Nacional de Promoción Científica y Tecnológica (FONCYT) and Fundación Josefina Prats. N. Martin is a Fellow of CONICET and M. C. Mansilla and D. de Mendoza are Career Investigators of the same institution. D. de Mendoza is an International Research Scholar of the Howard Hughes Medical Institute. We thank Drs. Yanfang Jiang, Sean Jordan, Gustavo Schujman, and Xin Zhao for research materials. We thank the New York Structural Genomics Research Consortium and Dr. Subramanyam Swaminathan for making the LipL structural coordinates available.
References
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauwe H, Kolukisaoglu U. Genetic manipulation of glycine decarboxylation. J Exp Bot. 2003;54:1523–1535. doi: 10.1093/jxb/erg171. [DOI] [PubMed] [Google Scholar]
- Booker SJ, Cicchillo RM, Grove TL. Self-sacrifice in radical S-adenosylmethionine proteins. Curr Opin Chem Biol. 2007;11:543–552. doi: 10.1016/j.cbpa.2007.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen QH, Cronan JE. The Thermoplasma acidophilum LplA-LplB complex defines a new class of bipartite lipoate-protein ligases. J Biol Chem. 2009;284:21317–21326. doi: 10.1074/jbc.M109.015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen QH, Cronan JE. Lipoic acid synthesis: A new family of octanoyltransferases generally annotated as lipoate protein ligases. Biochemistry. 2010 doi: 10.1021/bi101215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy KW, Melvin JA, McCafferty DG. Sortase transpeptidases: insights into mechanism, substrate specificity, and inhibition. Biopolymers. 2010;94:385–396. doi: 10.1002/bip.21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE. Cosmid-based system for transient expression and absolute off-to-on transcriptional control of Escherichia coli genes. J Bacteriol. 2003;185:6522–6529. doi: 10.1128/JB.185.22.6522-6529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronan JE, Zhao X, Jiang Y. Function, attachment and synthesis of lipoic acid in Escherichia coli. Adv Microb Physiol. 2005;50:103–146. doi: 10.1016/S0065-2911(05)50003-1. [DOI] [PubMed] [Google Scholar]
- Cui G, Nan B, Hu J, Wang Y, Jin C, Xia B. Identification and solution structures of a single domain biotin/lipoyl attachment protein from Bacillus subtilis. J Biol Chem. 2006;281:20598–20607. doi: 10.1074/jbc.M602660200. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer ELL, Eddy SR, Bateman A. The Pfam protein families database. Nucl Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara K, Maita N, Hosaka H, Okamura-Ikeda K, Nakagawa A, Taniguchi H. Global conformational change associated with the two-step reaction catalyzed by Escherichia coli lipoate-protein ligase A. J Biol Chem. 2010;285:9971–9980. doi: 10.1074/jbc.M109.078717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucl Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MW, Lukes J, Archibald JM, Keeling PJ, Doolittle WF. Cell biology. Irremediable complexity? Science. 2010;330:920–921. doi: 10.1126/science.1198594. [DOI] [PubMed] [Google Scholar]
- Green D, Morris T, Green J, Cronan J, Jr, Guest J. Purification and properties of the lipoate protein ligase of Escherichia coli. Biochem J. 1995;309:853. doi: 10.1042/bj3090853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadonou AM, Wilkin JM, Varetto L, Joris B, Lamotte-Brasseur J, Klein D, Duez C, Ghuysen JM, Frere JM. Site-directed mutagenesis of the Streptomyces R61 DD-peptidase. Catalytic function of the conserved residues around the active site and a comparison with class-A and class-C beta-lactamases. Eur J Biochem. 1992;207:97–102. doi: 10.1111/j.1432-1033.1992.tb17025.x. [DOI] [PubMed] [Google Scholar]
- Hermes F, Cronan J. Scavenging of cytosolic octanoic acid by mutant LplA lipoate ligases allows growth of Escherichia coli strains lacking the LipB octanoyltransferase of lipoic acid synthesis. J Bacteriol. 2009;191:6796. doi: 10.1128/JB.00798-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan SW, Cronan JE., Jr A new metabolic link. The acyl carrier protein of lipid synthesis donates lipoic acid to the pyruvate dehydrogenase complex in Escherichia coli and mitochondria. J Biol Chem. 1997;272:17903–17906. doi: 10.1074/jbc.272.29.17903. [DOI] [PubMed] [Google Scholar]
- Jordan SW, Cronan JE., Jr The Escherichia coli lipB gene encodes lipoyl (octanoyl)-acyl carrier protein:protein transferase. J Bacteriol. 2003;185:1582–1589. doi: 10.1128/JB.185.5.1582-1589.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan W, Littlejohn TG. Swiss-PDB Viewer (Deep View) Brief Bioinform. 2001;2:195–197. doi: 10.1093/bib/2.2.195. [DOI] [PubMed] [Google Scholar]
- Keijser BJ, Ter Beek A, Rauwerda H, Schuren F, Montijn R, van der Spek H, Brul S. Analysis of temporal gene expression during Bacillus subtilis spore germination and outgrowth. J Bacteriol. 2007;189:3624–3634. doi: 10.1128/JB.01736-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskey RA, Mills AD. Quantitative film detection of 3H and 14C in polyacrylamide gels by fluorography. Eur J Biochem. 1975;56:335–341. doi: 10.1111/j.1432-1033.1975.tb02238.x. [DOI] [PubMed] [Google Scholar]
- Lessard IA, Domingo GJ, Borges A, Perham RN. Expression of genes encoding the E2 and E3 components of the Bacillus stearothermophilus pyruvate dehydrogenase complex and the stoichiometry of subunit interaction in assembly in vitro. Eur J Biochem. 1998;258:491–501. doi: 10.1046/j.1432-1327.1998.2580491.x. [DOI] [PubMed] [Google Scholar]
- Ma Q, Zhao X, Eddine AN, Geerlof A, Li X, Cronan JE, Kaufmann SHE, Wilmanns M. The Mycobacterium tuberculosis LipB enzyme functions as a cysteine/lysine dyad acyltransferase. Proc Natl Acad Sci U S A. 2006;103:8662–8667. doi: 10.1073/pnas.0510436103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Lee M, Barrick JE, Weinberg Z, Emilsson GM, Ruzzo WL, Breaker RR. A glycine-dependent riboswitch that uses cooperative binding to control gene expression. Science. 2004;306:275–279. doi: 10.1126/science.1100829. [DOI] [PubMed] [Google Scholar]
- Mansilla MC, de Mendoza D. L-cysteine biosynthesis in Bacillus subtilis: identification, sequencing, and functional characterization of the gene coding for phosphoadenylylsulfate sulfotransferase. J Bacteriol. 1997;179:976–981. doi: 10.1128/jb.179.3.976-981.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N, Christensen Q, Mansilla M, Cronan J, de Mendoza D. A novel two-gene requirement for the octanoyltransfer reaction of Bacillus subtilis lipoic acid biosynthesis. Mol Microbiol Companion paper. 2011 doi: 10.1111/j.1365-2958.2011.07597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N, Lombardia E, Altabe SG, de Mendoza D, Mansilla MC. A lipA (yutB) mutant, encoding lipoic acid synthase, provides insight into the interplay between branched-chain and unsaturated fatty acid biosynthesis in Bacillus subtilis. J Bacteriol. 2009;191:7447–7455. doi: 10.1128/JB.01160-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morihara K, Oka T. Peptide bond synthesis catalyzed by subtilisin, papain, and pepsin. J Biochem. 1981;89:385–395. doi: 10.1093/oxfordjournals.jbchem.a133213. [DOI] [PubMed] [Google Scholar]
- Morris T, Reed K, Cronan J. Lipoic acid metabolism in Escherichia coli: the lplA and lipB genes define redundant pathways for ligation of lipoyl groups to apoprotein. J Bacteriol. 1995;177:1–10. doi: 10.1128/jb.177.1.1-10.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano MM, Dailly YP, Zuber P, Clark DP. Characterization of anaerobic fermentative growth of Bacillus subtilis: identification of fermentation end products and genes required for growth. J Bacteriol. 1997;179:6749–6755. doi: 10.1128/jb.179.21.6749-6755.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packman LC, Borges A, Perham RN. Amino acid sequence analysis of the lipoyl and peripheral subunit-binding domains in the lipoate acetyltransferase component of the pyruvate dehydrogenase complex from Bacillus stearothermophilus. Biochem J. 1988;252:79–86. doi: 10.1042/bj2520079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego M. Integrational vectors for genetic manipulation in Bacillus subtilis. In: Sonenshein JAHAL, Losick R, editors. Bacillus subtilis and other gram-positive bacteria: biochemistry, physiology, and molecular genetics. Washington DC: American Society for Microbiology; 1993. pp. 615–624. [Google Scholar]
- Perham RN. Swinging arms and swinging domains in multifunctional enzymes: catalytic machines for multistep reactions. Annu Rev Biochem. 2000;69:961–1004. doi: 10.1146/annurev.biochem.69.1.961. [DOI] [PubMed] [Google Scholar]
- Plamann MD, Rapp WD, Stauffer GV. Escherichia coli K12 mutants defective in the glycine cleavage enzyme system. Mol Gen Genet. 1983;192:15–20. doi: 10.1007/BF00327641. [DOI] [PubMed] [Google Scholar]
- Reche PA. Lipoylating and biotinylating enzymes contain a homologous catalytic module. Protein Science. 2000;9:1922–1929. doi: 10.1110/ps.9.10.1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: sequencing and functional characterization of the lipA and lipB genes. J Bacteriol. 1993;175:1325–1336. doi: 10.1128/jb.175.5.1325-1336.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock CO. Opening a new path to lipoic acid. J Bacteriol. 2009;191:6782–6784. doi: 10.1128/JB.01151-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell D. Molecular cloning: a laboratory manual. CSHL Press; 2001. [Google Scholar]
- Schonauer MS, Kastaniotis AJ, Kursu VA, Hiltunen JK, Dieckmann CL. Lipoic acid synthesis and attachment in yeast mitochondria. J Biol Chem. 2009;284:23234–23242. doi: 10.1074/jbc.M109.015594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvaggi NR, Anderson JW, Brinsmade SR, Pratt RF, Kelly JA. The crystal structure of phosphonate-inhibited D-Ala-D-Ala peptidase reveals an analogue of a tetrahedral transition state. Biochemistry. 2003;42:1199–1208. doi: 10.1021/bi0268955. [DOI] [PubMed] [Google Scholar]
- Smith RL, Pelley JW, Jeter RM. Characterization of lip expression in Salmonella typhimurium: analysis of lip::lac operon fusions. J Gen Microbiol. 1991;137:2307–2312. doi: 10.1099/00221287-137-10-2307. [DOI] [PubMed] [Google Scholar]
- Spizizen J. Transformation of biochemically deficient strains of Bacillus subtilis by deoxyribonucleate. Proc Natl Acad Sci U S A. 1958;44:1072–1078. doi: 10.1073/pnas.44.10.1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiert PS, Stauffer LT, Stauffer GV. The lpd gene product functions as the L protein in the Escherichia coli glycine cleavage enzyme system. J Bacteriol. 1990;172:6142–6144. doi: 10.1128/jb.172.10.6142-6144.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian AR, Kaufmann M, Morgenstern B. DIALIGN-TX: greedy and progressive approaches for segment-based multiple sequence alignment. Algorithms Mol Biol. 2008;3:6. doi: 10.1186/1748-7188-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) Software Version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Vanden Boom TJ, Reed KE, Cronan JE., Jr Lipoic acid metabolism in Escherichia coli: isolation of null mutants defective in lipoic acid biosynthesis, molecular cloning and characterization of the E. coli lip locus, and identification of the lipoylated protein of the glycine cleavage system. J Bacteriol. 1991;173:6411–6420. doi: 10.1128/jb.173.20.6411-6420.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wycuff DR, Matthews KS. Generation of an araC-araBAD promoter-regulated T7 expression system. Anal Biochem. 2000;277:67–73. doi: 10.1006/abio.1999.4385. [DOI] [PubMed] [Google Scholar]
- Ye BC, Zhang Y, Yu H, Yu WB, Liu BH, Yin BC, Yin CY, Li YY, Chu J, Zhang SL. Time-resolved transcriptome analysis of Bacillus subtilis responding to valine, glutamate, and glutamine. PLoS One. 2009;4:e7073. doi: 10.1371/journal.pone.0007073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Miller JR, Jiang Y, Marletta MA, Cronan JE. Assembly of the covalent linkage between lipoic acid and its cognate enzymes. Chem Biol. 2003;10:1293–1302. doi: 10.1016/j.chembiol.2003.11.016. [DOI] [PubMed] [Google Scholar]