

Abstract
The effects of 3-position substitution of 9-aminomethyl-9,10-dihydroanthracene (AMDA) on 5-HT2A receptor affinity were determined and compared to a parallel series of DOB-like 1-(2,5-dimethoxyphenyl)-2-aminopropanes substituted at the 4-position. The results were interpreted within the context of 5-HT2A receptor models that suggest that members of the DOB-like series can bind to the receptor in two distinct modes that correlate with the compounds’ functional activity. Automated ligand docking and molecular dynamics suggest that all of the AMDA derivatives, the parent of which is a 5-HT2A antagonist, bind in a fashion analogous to that for the sterically demanding antagonist DOB-like compounds. The failure of the F3406.52L mutation to adversely affect the affinity of AMDA and the 3-bromo derivative is consistent with the proposed modes of orientation. Evaluation of ligand-receptor complex models suggest that a valine/threonine exchange between the 5-HT2A and D2 receptors may be the origin of selectivity for AMDA and two substituted derivatives.
Keywords: Serotonin receptors, 5-HT2A, 9-Aminomethyl-9, 10-dihydroanthracene, AMDA, Phenylethylamines, Homology Modeling, Structure-Affinity Relationship, Structure-Activity Relationship
INTRODUCTION
Serotonin has been implicated in a large number of processes including the regulation of sleep, appetite, mood, aggression, perception, memory, and anxiety.1 At least 14 distinct 5-HT G protein-coupled receptors (GPCRs) have evolved that are divided into seven main families.2 Not surprisingly, alterations of 5-HT receptor activity have been shown to occur in many psychiatric diseases including anxiety, depression, eating disorders, schizophrenia, personality disorders, and many drug-induced psychotic states.2 Additionally, a number of effective psychopharmacologic agents for diseases as diverse as depression, schizophrenia, and anxiety have been developed that either specifically alter brain levels of serotonin or bind to 5-HT receptor subtypes.1, 3 Over the last few years, all of the 5-HT receptor subtypes have been cloned and sequenced.3, 4 Among the first to be studied were the 5-HT2A and 5-HT2C receptors, and a significant body of reliable information has been accumulated regarding these 5-HT2 receptors. Nevertheless, it is still not known with certainty how serotonergic agents (or, for that matter, how the endogenous ligand 5-HT itself) interact at 5-HT receptors. Crucial to an understanding of how serotonergic agents act, whether agonists, partial agonists or antagonists, is some understanding of this drug-receptor interaction. A novel class of high-affinity 5-HT2 agents5–9 has been described, the parent structure of which (1a, 9-aminomethyl-9,10-dihydroanthracene, AMDA) is a 5-HT2 selective6 antagonist8 that appears to bind with the 5-HT2A receptor in a fashion distinct from classical tricyclic agents.6, 9 As an anthracene derivative, AMDA is a fairly conformationally restrained molecule. Its 9-aminomethyl group is preferentially oriented in a pseudoaxial10 conformation, and its tricyclic ring system exhibits a fold angle of about 147 degrees.5 Since AMDA (1a) shares a phenylethylamine skeleton with phenylethylamine agonists such as DOB (2b), potential binding mode relationships between AMDA and phenylethylamines were evaluated by exploring the effects of substitution at the 3-position of AMDA and the effects of the structurally analogous 4-position substitution of DOB-like phenylethylamine derivatives upon the binding affinity. Possible modes of binding of AMDA analogs and phenylethylamines were identified using 5-HT2A receptor models constructed from the crystal structure of bovine rhodopsin. These studies have allowed us to formulate some useful generalizations about binding modes of agonists versus antagonists as well as to identify potential explanations for the observed receptor selectivity in the AMDA series.
RESULTS AND DISCUSSION
Chemistry
The structures of the target compounds 1a–h, 3a, 3b, 3d, 4a, 4b, and 4d are shown in Table 1. Compound 1a was prepared using a literature procedure.8 Compounds 2a–e and 2g have also been previously reported in the literature, and their syntheses are discussed elsewhere.11–13 Compounds 3a and 4a were obtained from commercial sources. Compound 4a was purchased as the free base and subsequently converted to the HCl salt14 using ethereal HCl. Compound 3c was prepared as previously described.15
Table 1.
The effects of aromatic substitution on 5-HT2A receptor affinity.
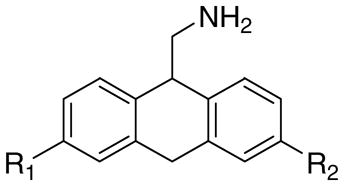 1a–h |
 2a–e, g |
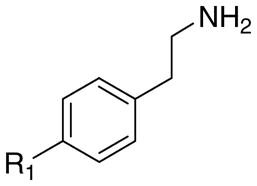 3a–d |
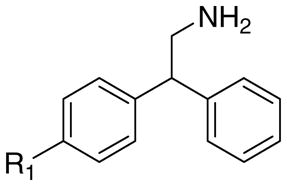 4a,b,d |
||||||
|---|---|---|---|---|---|---|---|---|---|
| R1 | R2 | Cpd. | Ki, nMa | Cpd. | Ki, nMb | Cpd. | Ki, nMa | Cpd. | Ki, nMa |
| -H | -H | 1a | 20 | 2a | 5200 | 3a | 16,800 | 4a | 4610 |
| -Br | -H | 1b | 1.3 | 2b | 41 | 3b | 1770 | 4b | 260 |
| -(CH2)3Ph | -H | 1c | 3.2 | 2c | 10 | 3c | 60c | - | - |
| -C6H13 | -H | 1d | 7.0 | 2d | 2.5 | 3d | 78 | 4d | 200 |
| -OCH3 | -H | 1e | 7.5 | 2e | 1200 | - | - | - | - |
| -O(CH2)4CH3 | -H | 1f | 23 | - | - | - | - | - | - |
| -OH | -H | 1g | 107 | 2g | >50,000 | - | - | - | - |
| -C6H13 | -OCH3 | 1h | 43 | - | - | - | - | - | - |
The synthesis of compounds 1b–h was not without difficulty. The initial plan was to convert 3-substituted anthrones to the desired aminomethanes in a straightforward fashion. This route proved unsuccessful due to the rapid isomerization of anthrone to 9-anthrol under acidic and basic conditions. Under most nucleophilic conditions, anthrone was converted to the 9-alkyl anthracenes by dehydration of the intermediate 9-alkyl-9-hydroxy anthracene. Treatment of anthrone with the Tebbe reagent16 did, however, provide 9-methylene-9,10-dihydroanthracene in modest yields. It was expected that 9-methylene-9,10-dihydroanthracene could then be oxidized to the 9-aminomethyl-9,10-dihydroanthracene with BH3 and Chloramine T. Unfortunately, we were unable to generate large enough quantities of the 9-methylene-9,10-dihydroanthracene for this route to prove practical. Our focus changed following a review of the synthetic strategy employed by the Nichols group.17 The synthetic utility of an oxazoline as an ortho lithiating agent provided the key intermediates essential to the synthesis of compounds 1d–h (Scheme 1). 4-Substituted phenyloxazolines were used to provide compounds 1e and 1f. In these cases, it was reasoned that strongly electron donating groups (methoxy and pentyloxy) in the para position would facilitate the Friedel-Crafts cyclodehydration reaction (i, Scheme 1). Performing the cyclodehydration reaction of the aminoalcohol intermediates 20 with weakly electron donating substituents (R2 = hexyl, phenylpropyl) could lead to a mixture of two regioisomers. Separation of the resulting 1- and 3-substituted isomers would prove difficult since the free bases of compounds 1a–h are remarkably unstable. Exposure to air and/or aqueous conditions causes a rapid (30 min–1 hour) degradation to unknown highly colored compounds. Thus, 3-substituted benzaldehydes were used to generate compounds 1e, 1f, and 1g.
Scheme 1.

Reagents and Conditions: (a) 1; 2-amino-2-methyl-1-propanol, CH2Cl2. 2; SOCl2, Toluene; (b) sec-butyllithium, THF −78 °C; (c) 5% HCl 10 h; (d) 10% Pd/Charcoal, HClO4 (cat), 2-PrOH; (e) BH3-THF; (f) PCC, CH2Cl2; (g) Trimethylsilyl cyanide, CH2Cl2; (h) LiAlH4, THF; (i) Eaton’s Reagent, PPA or Methane sulfonic acid; (j) BBr3, CH2Cl2.
The general synthetic approach17 (Scheme 1) began with ortho lithiation of 16 using sec-butyl lithium. The appropriate benzaldehydes were then added to the lithium anion at 0 °C. The crude reaction mixtures were subjected to acidic hydrolysis giving rise to the lactones 17d,e,f,h in moderate overall yield. The lactones were hydrogenated in 2-PrOH with a catalytic amount of HClO4 to give the acids 18d,e,f,h in good yields. The acids were then reduced with BH3-THF and reoxidized with PCC to give the aldehydes 19d,e,f,h in excellent yields. The aldehydes were converted to the appropriately substituted 2-amino-1-hydroxy-(2-benzylphenyl)ethanols 20d,e,f,h using TMSCN with a catalytic amount of ZnI2 followed by LiAlH4 reduction in THF. The target compounds 1d–h were then prepared through a cyclodehydration reaction using either PPA, Eaton’s Reagent, or methanesulfonic acid. Compound 1g was prepared from the hydrobromide salt of compound 1e with BBr3 in CHCl3.17
An alternate route was chosen for the synthesis of 1b due to the presence of a bromine capable of undergoing lithium insertion (Scheme 1). The Grignard of 5 reacted in a 1,4 manner with 4-bromophenyl nitrostyrene (Scheme 2) to provide 6 by the method of Ashwood et al.18 Deprotection of the alcohol 6 with HCl in methanol followed by the PPA mediated cyclodehydration provided 8 in very low yield. The poor yield of the cyclodehydration reaction can be attributed to the deactivating nature of the bromo substituent. However, this route provided 8 in a regiochemically unambiguous manner. SnCl2 reduction of the nitro group in 8 was chosen to eliminate any potential halogen loss.19
Scheme 2.

Reagents and Conditions: (a) THF, 10 °C; (b) Conc. HCl, MeOH; (c) PPA, room temp; (d) SnCl2, EtOH.
Compound 14, the precursor of 1c, was prepared as shown in Scheme 3. Compound 10 was obtained by benzylic bromination (NBS in CCl4) of 4-bromo-2-methylbenzonitrile followed by Friedel-Crafts alkylation with benzene. Conversion of the cyano group 10 to the aldehyde was carried out in a stepwise manner using KOH in ethylene glycol20 followed by reduction of the acid with BH3-THF and reoxidation to the aldehyde 13 with PCC in CH2Cl2. This method was found to be superior to DIBAL reduction of the nitrile due to difficulties encountered in the separation of the aldehyde from the starting materials even with the use of aldehyde conjugation reagents such as sodium hydrogen sulphite. A modified Suzuki coupling reaction was employed to introduce the phenylpropyl substituent using allylbenzene/9-BBN followed by PdCl2(dppf) and NaOH in THF.21 The synthesis of 1c from 14 was identical to that previously described for 1d,e,f,h (Scheme 1) by conversion of the aldehyde to the 2-amino-1-hydroxy-[2-benzyl-4-(3-n-phenylpropyl)phenyl]ethanol followed by cyclodehydration using Eaton’s Reagent.
Scheme 3.

Reagents and Conditions: (a) NBS, CCl4; (b) AlCl3, Benzene; (c) KOH, Ethylene glycol; (d) BH3-THF; (e) PCC, CH2Cl2; (f) 1; Allylbenzene, 9-BBN 2; PdCl2(dppf), NaOH, THF.
4-Bromophenylethylamine (3b) was prepared by BH3-THF reduction of 4-bromophenyl acetonitrile. 4-n-Hexylbenzoyl cyanide 26 was prepared as per the method of Olah et al22 using SnCl4 and TMSCN. The benzoyl cyanide was reduced to the target 4-n-hexylphenylethylamine (3d) using catalytic hydrogenation (10% Pd/C in acetic acid).23 Friedel-Crafts alkylation of 2-bromo-2-(4-bromophenyl)acetonitrile with benzene24 provided the diphenyl acetonitrile 27 which was reduced with BH3-THF complex to provide the target 4b. Compound 4d was prepared by the reaction of 4-n-hexylbenzoyl chloride with benzene and AlCl3 to provide 28 (4-n-hexylphenyl)(phenyl)methanone. Trimethylsilyl cyanide was then employed in the preparation of the cyanohydrin 29 followed by reduction of the hydroxy group with NaBH4 in TFA25 to provide 30 (2-(4-n-hexylphenyl)-2-phenylacetonitrile). Reduction of the nitrile was then carried out using Raney Nickel under a hydrogen atmosphere to provide 4d in moderate yield.
Biological Evaluation/5-HT2A Receptor Affinities
Radioligand binding data (5-HT2A receptor affinities) were obtained for each of the target compounds (Table 1). The 5-HT2A receptor can accommodate a wide range of substituents associated with the 3-position of AMDA (1a–1g; Table 1). Affinities varied only about 80-fold (1b, Ki = 1.3 nM; 1g, Ki = 107 nM) within the series. With the exception of the 3-hydroxy compound (1g, Ki = 107 nM), monosubstitution of AMDA (1a, Ki = 20 nM) either does not change (1f, Ki = 23 nM) or increases affinity to a maximum of 15-fold (1b, Ki = 1.3) regardless of steric bulk or electronic character of the substituent. The effects of 4-position substitution on the affinities of 1-(2,5-dimethoxy)-2-aminopropanes (DOX; 2a–2e) are qualitatively similar in that each of these, with the exception of the hydroxy substituent (2g, Ki > 50,000 nM), retains or enhances affinity. However, in the DOX series, the range of affinity enhancement is much greater (2d, Ki = 2.5 nM; 2a, Ki = 5,200 nM) than for the AMDA series with a maximum range of about 2,000-fold, excluding the 4-hydroxy compound (2g) that shows no measurable affinity. Consistent with these observations, the lipophilic character of the 4-position substituent of DOX has been shown to modulate affinity over a broad range.13 These results suggest that the AMDA and DOX series may interact differently with the 5-HT2A receptor. The principal structural feature distinguishing AMDA from other phenylethylamines is the presence of a second, fused aromatic group. Introduction of a second non-fused phenyl group to phenylethylamine, (i.e., 2,2-diphenylethylamine) slightly increases affinity (3a, Ki = 16,800 nM; 4a, Ki = 4,610 nM). Introduction of 4-substituents can enhance the affinity of phenylethylamine by about 280-fold (3a, Ki = 16,800 nM; 3c, Ki = 60 nM; Table 1) and 2,2-diphenylethylamine by about 23-fold (4a, Ki = 4,610 nM; 4d, Ki = 200 nM). These increases in affinity (particularly with respect to the phenylethylamines) are greater than the increases seen in the AMDA series (1a, 1b; 15-fold), suggesting that there are differences in the modes of receptor interaction. Thus, it appears that DOB-like compounds, AMDA derivatives and ring-opened AMDA derivatives (i.e. 3 and 4) behave differently with respect to their binding at the 5-HT2A receptor. This is perhaps not surprising given the fact that DOB is an agonist15 whereas AMDA is an antagonist.8 At the very least, even if the two series bind in a comparable fashion, they must interact preferentially with functionally and conformationally distinct forms of the receptor. An alternative possibility is that the binding sites of agonists and antagonists only share a common ammonium ion binding site with the remaining bulk of each type of agent occupying completely different domains within the receptor.
Receptor Complex Models
There are numerous examples of similar compounds binding quite differently to a common receptor as well as ligands with multiple binding modes at a single receptor.9, 26 Analysis of early 5-HT2A receptor models led us to consider two general areas of steric accessibility, as depicted in Figure 1: Site 1 (TM3 flanked by TM4, TM5, and TM6) and Site 2 (TM3 flanked by TM1, TM2, TM6, and TM7). The presence of two distinct binding sites for GPCRs has been noted in the literature.27 Previously, consideration of ligand SAR and receptor mutagenesis data prompted us to provisionally consider Site 1 the “agonist site” and Site 2 the “antagonist site.”28–30 Similar suggestions have also been made for the 5-HT1A receptor.31 Subsequently, the method used here to select “agonist-biased” and “antagonist-biased” receptor models from a population of conformationally distinct receptor models (described in detail in the Experimental Section) has identified Site 1 as an agonist binding site and Site 2 as an antagonist binding site. The selected models identified in this way are thus referred to in this work as the agonist and antagonist receptor models, respectively. The computational methodology used in this work was designed to mimic the current model of protein-ligand binding in which the ligand selects a particular receptor conformation from an ensemble of metastable states.32 Site 1 and Site 2 overlap, and the shared region between these sites includes residues that are a part of helices TM3 (D1553.32 and S1593.36) and TM6 (W3366.48 and F3396.51).33
Figure 1.

Figure 1a. Schematic representation of sterically accessible binding sites within the 5-HT2A receptor provisionally considered to be the agonist site (Site 1) and the antagonist site (Site 2).28, 29 1b. Connolly channel depicting Site 1 and Site 2 within the 5-HT2A receptor model. The GOLD-generated docking mode for AMDA is also shown (CPK space-filling model) to highlight the complementarity between the shape of Site 2 and the fold angle of the AMDA ring system. D1553.32 is shown for reference (ball-and-stick model).
Whenever receptor homology models are generated whose purpose is to model the interaction of an agonist with the receptor, the accuracy of these models is called into question (more so than for antagonist-interaction models) since until very recently only inactive or ground-state rhodopsin crystal structures were available as homology modeling templates. More precisely, the additional requirement of an agonist to activate or trigger the receptor is thought to involve large-scale movements of at least part of the secondary structure of the receptor, making the activated receptor’s conformation significantly different from the inactive state’s conformation. Currently, such large-scale changes in conformation are not routinely incorporated into homology models. Recent crystallographic evidence, however, suggests that the conformation of the activated form of bovine rhodopsin does not significantly change in the ligand binding region: Salom et al.34 have obtained a crystal structure, at 4.15 Å resolution, of the deprotonated form of Metarhodopsin II (Meta II), the fully activated state of rhodopsin. This structure is strikingly similar to the structure of ground-state rhodopsin in the transmembrane and extracellular loop regions, where ligand binding sites are located. The authors thus conclude that “rhodopsin is a good template for homology models of other GPCRs used in docking calculations of both agonists and antagonists, because ground-state and photoactivated rhodopsin are structurally similar”.34 Other studies have proposed that a cluster of residues on TM6 in Site 1 form a molecular “toggle switch” that is responsible for the activation of rhodopsin-like GPCRs.35 This work utilizes the ground-state conformation of bovine rhodopsin (A chain of 1U19; 2.2 Å resolution) as a homology modeling template. However, the conformations of the sidechains (and the backbone to a lesser degree) are allowed to vary from that of the template rhodopsin structure.
Viewed from the perspective of the ligand, in the most general terms it is usually observed that structures of antagonists differ from the endogenous neurotransmitters and other agonists in that they either lack key functional groups or present molecular features in areas of space not occupied/utilized by any portion of the agonist (i.e., an “accessory site”), or both.36 For example, while 5-methoxytryptamine is a serotonin agonist, tryptamine is a partial agonist (see the review by Glennon, Westkaemper and Bartyzel37); also, it has been shown that 2-phenyltryptamines are high-affinity 5-HT2A receptor antagonists.38 Similarly, LSD is an agonist or partial agonist whereas 2-bromo LSD is an antagonist.39 In the DOX series, compounds with small substituents at the 4-position are agonists and those with bulky substituents such as phenylpropyl are antagonists.13, 15 In the latter case, the 2,5-dimethoxy groups of 1-(2,5-dimethoxy-4-(3-phenylpropyl)phenyl)-2-aminopropane (2c), functional groups characteristically required for agonist activity, are no longer required for binding and, in fact, the desmethoxy parent 3c has comparable affinity to the 2,5-dimethoxy substituted derivative.13, 15 A similar observation can be made for 2d and 3d. It has been hypothesized that phenylalkylamines with small 4-position substituents (e.g. 2a, 2b, 2e) bind differently from those with bulky 4-position substituents (e.g. 2c, 2d). Models of complexes of the 5-HT2A receptor and DOB support the notion that there may be limited bulk tolerance at the 4-position for some modes of binding. Bound within Site 1, substituents at the 4-position of DOX project into the interfacial region between TM5 and TM6 (see Figure 2a for an example). Preliminary modeling studies have indicated that whereas 4-methyl and 4-ethyl substituents appear to be tolerated in the DOB-like series, successively adding methylene units to the 4-position of 1-(2,5-dimethoxy-4-ethylphenyl)-2-aminopropane bound to the receptor actually causes a displacement of the of the aromatic ring (2.3 Å) from the initial site on minimization. The bound ligand 2d is rapidly displaced from its initial site during dynamics simulations (100 ps, 300° K, range constraint NH-OD155, 1.3–2.6Å, helix backbone constrained) whereas DOB (2b) is not. Another possible binding mode would place large 4-position substituents in Site 2 (Figure 1). A 5-HT2A receptor model with the phenylethylamine 3d bound in Site 2 did not show displacement of the aromatic ring and the ligand remained in the binding site on dynamics simulation.
Figure 2.

The proposed binding mode of selected compounds docked and energy-minimized in the 5-HT2A receptor models. Carbon atoms of the ligand are colored green. Residues whose heavy atoms fall within 4 Å of the bound ligand heavy atoms are displayed. A light blue transparent trace indicates the position of the receptor backbone. Hydrogen bonding interactions are indicated with a thin black line and H-bond donor-acceptor distances (in Å) are indicated in blue. The antagonist models are displayed from a common point of view in which TMs 6, 7 and 1 are closest to the viewer. A. R(−)-DOB (R(−)-2b); agonist model. B. ketanserin; antagonist model. C. AMDA (1a); antagonist model. D. (S)-3-hydroxy-AMDA (1g); antagonist model. E. (S)-3-phenylpropylAMDA (1c); antagonist model. F. (R)-2c; antagonist model.
The effects of N-alkylation and N-benzylation appear to support the notion that DOB and AMDA interact with the receptors differently. In the case of both 5-methoxytryptamine and DOB, successive N-methylation decreased affinity but N-benzyl DOB and N-benzyl-5-methoxytryptamine have slightly higher affinities (2- to 6-fold) than their parents.40 In the AMDA series, successive N-methylation also decreased affinity but, unlike the DOB and 5-methoxytryptamine series, N-benzylation decreased affinity (36-fold).6
Information from mutagenesis experiments further suggests that AMDA and phenylalkylamines (i.e. 3) or DOX analogs (i.e. 2) with small 4-position substituents (e.g. DOB, DOI) bind differently, at least with respect to F3406.52. In the current models, the sidechain of F3406.52 is at the interface between TM5 and TM6, pi-stacked with the sidechain of F2435.47 (Figure 2a). Any effect that an F340 mutation might have on ligand affinity could either be due to changes in a direct, ligand-receptor van der Waals interaction, or an indirect effect caused by a change in the shape of the helical bundle. The mutation F340L has been shown to decrease affinity of agonists, but generally hasno effect on the binding of classical antagonists.41 AMDA (1a) and the bromo analog 1b both bind to the mutant receptor equally well (3-fold decrease and no change in affinity, respectively) compared to their binding at the wild type receptor (Table 2). The same mutation has little effect on ketanserin affinity but essentially abolishes DOI binding (an approximately 14,000-fold decrease).41 This is entirely consistent with AMDA and AMDA derivatives binding in a completely different fashion from DOI, at least with respect to the F3406.52 position in the receptor structure. The affinities of the two 2,2-diphenylaminoethane compounds (Table 2) are either unchanged or increased by F340L mutation (4a, Ki = 4,140 nM; 4b, Ki = 3.5 nM) relative to the wild type (4a, Ki = 4,610 nM; 4b, Ki = 260 nM). Again, these results support the notion that, with respect to F340, the diphenyl compounds (4a, 4b) behave differently from phenylethylamines and most likely bind in a mode distinct from that of the analogous tricyclic compounds 1a and 1b. Changes in the sidechain conformation of F340 have been previously invoked to explain affinity enhancement for some classes of compounds with the F340L mutant.30
Table 2.
The effects of the 5-HT2A receptor F3406.52L mutation on ligand affinity.
| Compound | KI (nM)a |
|
|---|---|---|
| Wild type | F340L | |
| 1a | 20 | 57 |
| 1b | 1.3 | 1.8 |
| 4a | 4,610 | 4,140 |
| 4b | 260 | 3.5 |
| DOI | 0.92 | 13,700 |
| ketanserin | 0.4 | 0.23 |
[3H]Ketanserin labeled cloned 5-HT2A sites.
Ki values at the wild type receptors are from Table 1.
Standard errors typically range between 15–25% of the Ki value.
Model Construction
In the following subsections, computationally-derived 5-HT2A GPCR models are described that separately model the binding characteristics of selected agonists and antagonists. ‘Agonist-biased’ and ‘antagonist-biased’ receptor models were generated in the following way: Using the MODELLER software package, a population of 100 5-HT2A conformationally distinct receptor models derived from bovine rhodopsin was generated. The automated docking program GOLD was then used to separately dock both stereoisomers of a high-affinity agonist (DOB, 2b) and an antagonist (ketanserin) into each of the 100 5-HT2A receptor models (Chart 1). Based on the quality of the docked receptor-ligand complexes and information from site-directed mutagenesis, one of the 100 models was selected to be the ‘agonist’ 5-HT2A receptor model and another was selected as the ‘antagonist’ 5-HT2A model (Supplemental Figure 1). Both stereoisomers of each of the compounds listed in Table 1 were then docked into both the agonist and antagonist receptor models. Docking scores and information from mutagenesis data were then used to select the most appropriate receptor (agonist or antagonist) for each ligand.
Chart 1.

Structures of AMDA (1a), DOB (2b) and ketanserin.
Agonist Receptor Complex Models
Both isomers of DOB were found to favorably interact with the selected agonist receptor model, and in a nearly identical fashion (Supplemental Figure 2). The most significant difference in the binding modes of the stereoisomers is in the position of the protonated amine; however, both isomers are able to form a salt bridge with D1553.32. The proposed binding pocket for R(−)-DOB (R(−)-2b) is shown in Figure 2a, and residues that can potentially interact with it are reported (Supplemental Table 1). The aromatic ring is associated most closely with W3366.48, F3396.51 and F3406.52. The 4-bromo substituent is oriented toward the interfacial region between TM5 and TM6 and the 2-methoxy group of DOB accepts a hydrogen bond from N3436.55. The 5-methoxy group is near S1593.36, T1603.37 and S2425.46, and can potentially form hydrogen bonds with these residues to further stabilize the receptor-ligand complex. Additionally, a lipophilic interaction occurs between the methyl of the 5-methoxy group and W3366.48. Other nearby aromatic residues that can potentially interact with the aromatic ring of DOB include F2435.47 and F3406.52. F3396.51 is in a position to further stabilize the ammonium-D1553.32-S1593.36 complex via a π-cation interaction.
Recently, it was reported42 that R(−)-DOB and S(+)-DOB both have high affinity for the 5-HT2A receptor, but with the R(−)-isomer showing a somewhat lower Ki than the S(+)-isomer (R(−)-DOB, Ki = 0.29 nM; S(+)-DOB, Ki = 1.9 nM). Employing modeling techniques, the authors showed that the R and S isomers of DOX phenylethylamines can bind in a very similar fashion, but that the orientation of S2395.43, F2405.44, F2435.47, F2445.48 and F3406.52 differed depending on which isomer was docked into the receptor. Although our modeling technique places DOB in the same location as the previous authors, the model described here features a pi-stacked interaction between F3406.52 and F2435.47. Disruption of this association (either through a point mutation or via interactions with a ligand) may alter the location and orientation of the helices within the helix bundle. As mentioned by Parrish, et al.,42 the cognate residues in bovine rhodopsin, F2125.47 and A2696.52, have been shown to have highly coupled evolution as part of a physically connected network that links distant functional sites in the tertiary structure of G-protein coupled receptors (GPCRs).43 The close association of the DOX ligands with F3406.52 would also explain why the F340L mutation nearly abolished44 the affinity of DOI for the 5-HT2A receptor. Both enantiomers of DOB and DOI have a high affinity for the 5-HT2A receptor42 and were docked into the agonist receptor model in a nearly identical manner, the only difference being the position of the β-methyl group (Supplemental Figure 2). In our agonist receptor model, DOB occupies the same binding pocket that has been predicted for the endogenous ligand 5-HT.45–47
The proposed binding mode for DOB features the ligand accepting a hydrogen bond from N3436.55. An analysis of the primary sequences of the known human 5-HT receptor subtypes reveals that only 5-HT2A, 5-HT2B, 5-HT2C, 5-HT4 and 5-HT6 receptors feature asparagine at 6.55, although other subtypes have sidechains capable of donating a hydrogen bond at this position (6.55 = Ser in 5-HT1B, 5-HT1D and 5-HT7). The Psychoactive Drug Screening Program (PDSP) Ki Database (http://pdsp.med.unc.edu/pdsp.php) contains entries for DOB as the test ligand for 5-HT1 and 5-HT2 receptor subtypes. In all cases, the affinity of DOB for the 5-HT2 subtypes was much greater (0.6 to 152 nM) than for the 5-HT1 subtypes (556 to 6327 nM). This is consistent with the hypothesis that N6.55 contributes significantly to the observed high affinity of DOB at the 5-HT2 subtypes. The 6.55 position has also been shown to be important for the binding of other agonists and antagonists at serotonergic and other closely related aminergic GPCR subtypes.48–53
The ligand-accessible and highly conserved residues W3366.48 and F3406.52 of the “aromatic cluster”54 in Site 1 have been proposed to be part of a rotameric “toggle switch”35, 55 in which the χ1 torsion angles of these residues determine, in conjunction with the proline kink in TM6, the proximity of the intracellular ends of TM3 and TM6 (i.e. the “ionic lock”56). The χ1 conformations that correspond to the “activated” receptor are trans for both 6.48 and 6.52, which is consistent with the putative agonist model presented here. Taken together, this information along with the examples given above provide additional evidence that the agonist model described here is accurate.
AMDA (1a) was successfully docked into Site 1 in the agonist model with the basic amine H-bonded to both D1553.32 and S1593.36. One of the aromatic rings is oriented toward the cluster of hydrophobic residues on TM6, interacting with I1633.40, F2435.47, W3366.48 and F3406.52; the second aromatic ring of AMDA interacts with V1563.33, I2064.56 and L229xl2.52. Importantly, it is shown here (Table 2) that mutation of F3406.52 to leucine has little effect on the binding of antagonists like AMDA (1a). In contrast, the F3406.52L mutation had dramatic effects on the binding of DOI, an agonist very similar to DOB (2b). This would indicate that 1a and 2b bind differently with respect to F3406.52.
AMDA analogs with small substituents at the 3-position (1b, 1e, 1g) are oriented in Site 1 in a manner analogous to that of AMDA. However, for 1e and 1g there are no nearby H-bond donors or acceptors to effectively interact with the polar functionality at the 3-position (W3366.48 is close, but with poor H-bond geometry). For the larger, more flexible hydrophobic analogs (1c, 1d, 1f), the 3-position substituent is either directed toward the opening of the receptor cavity (for S-isomers) or folds back onto the dihydroanthracene core (for R-isomers), a characteristic that is statistically unlikely based upon an analysis of crystal structures of ligand-receptor complexes.57, 58 In the phenylisopropylamine series, DOB analogs with small substituents at the 4-position (2a, 2e, 2g) dock into the receptor in a similar fashion as DOB, with the aromatic ring associated with W3366.48, F3396.51 and F3406.52 and the methoxy groups interacting with S1593.36, T1603.40, S2425.46 and N3436.55. The position of the aromatic ring for these compounds is close to the position of the aromatic ring in AMDA most closely associated with the aromatic cluster on TM6. As with the AMDA analogs substituted with small polar substituents at the 3-position, there is no H-bonding partner for the small polar substituents at the 4-position of the DOB analogs. This is consistent with the low observed binding affinities for these compounds. For DOB analogs 2c and 2d, the large 4-position substituents are folded back onto the ligand’s aromatic ring (both isomers), analogous to the AMDA analogs (R-isomer) with large 3-position substituents. In order to accommodate the bulk of the large substituent, the aromatic ring is displaced toward TM4, and the H-bonds with the methoxy groups are diminished or eliminated. The phenylethylamine analogs 3a–d dock with conformations that are similar to their corresponding DOB analogs, and the diphenylmethylamine analogs 4a, 4b and 4d dock in the receptor like their corresponding AMDA analogs 1a, 1b and 1d.
Antagonist Receptor Complex Models
The energy-minimized ketanserin-receptor model is depicted in Figure 2b (nearby residues are listed in Supplemental Table 1). In addition to the hydrogen bond formed between the ammonium ion in the ketanserin piperidine ring and the conserved D1553.32, three other hydrogen bonds are evident: S1312.61 bonds with the p-fluorobenzoyl carbonyl oxygen, S1593.36 bonds with the N1 quinazolinedione nitrogen atom, and S3737.46 bonds with the carbonyl oxygen at position 2 of the quinazolinedione ring system (Figure 2b). Hydrophobic residues surrounding the remainder of the ligand include W1513.28, I1523.29, V1563.33, L229xl2.52, W3366.48, V3667.39, W3677.40 and Y3707.43.
To provide an indication of the correctness of the docked solution, relevant mutagenesis binding data were collected from the literature; these are listed in Table 3. As [3H]ketanserin is often used as the radioligand in competitive binding assays involving the 5-HT2A receptor and its mutants, Kd values were frequently available. Many of the mutations listed in Table 3 involve residues that are not located in the binding crevice of the receptor. Others involve residues that are conserved across all GPCRs and are probably required to maintain the structural integrity and/or basic functioning of the receptor. Several of the mutations involve the conserved aspartate D1553.32, and the result of mutating this residue to something other than aspartate at this site is a near or complete loss of affinity for ketanserin. Presumably this mutation would also disrupt the binding of many other small basic amine-containing compounds, agonists and antagonists alike. Other mutations are more relevant to the binding of ketanserin itself. Mutation of serines S2395.43 and S2425.46 on TM5 to alanine has no significant effect upon the binding of ketanserin. This is consistent with the proposed model as ketanserin does not approach TM5. The F2435.47 and F3406.52 mutations each minimally decrease the binding of ketanserin. While neither F2435.47 nor F3406.52 are within van der Waals interaction distance with ketanserin, the small decreases in affinity at these mutated positions could be accounted for by indirect destabilization of the binding site. The S1593.36A and S1593.36C mutations were found to have almost no effect on the affinity of ketanserin. While this may seem to contradict our proposed model because S1593.36 participates in a hydrogen bond, the actual situation is probably more subtle. For example, it has been shown59 that the entire quinazolinedione ring system may be replaced with a with a phenylethyl fragment lacking H-bonding capability without significant loss of affinity (less that 2-fold decrease in Ki). If ketanserin binds as proposed, then this would suggest that the hydrogen-bonding capability of S1593.36 is not required, and thus consistent with the mutagenesis data. W3366.48A was found to have one of the largest effects on ketanserin binding (a 900-fold decrease). This is consistent with the model because there is a substantial amount of hydrophobic surface contact area between the quinazolinedione ring system and the indole ring of W3366.48. Similarly, mutation of F3396.51 to alanine or leucine results in moderate decrease (8- to 25-fold) in ketanserin’s binding affinity due to the loss of hydrophobic bulk in the region. The F3396.51Y mutation introduces a phenolic group into an area occupied by the fused phenyl ring of the quinazolinedione moiety, resulting in a moderate decrease in binding affinity. Mutation of F3406.52 to alanine or leucine has no significant effect. This is also consistent with the model because this residue is at a distant location in Site 1 and is not expected to interact with ketanserin. The effect of the F3406.52Y mutation is substantial with a 70-fold decrease in binding affinity. Mutation to tyrosine at this position would introduce a hydroxyl group into the lipid bilayer. This could possibly facilitate the disruption of the F2435.47-F3406.52 interaction and the binding cavity as a result, since the tyrosine OH group would presumably prefer to be located in the more polar interior of the receptor. The W3677.40L mutation abolished ketanserin binding and nearly abolished the binding of small agonists like 5-HT and DOM that would be expected to bind completely within Site 1.44 As well as providing a site of interaction for ketanserin, this would seem to indicate that W3677.40 forms part of an extended Site 1, as mentioned earlier. Alternatively, W3677.40 may also interact with W761.34. Such an interaction may serve to stabilize the helical bundle, at least for the serotonin receptor subtypes, in which tryptophan is uniformly conserved at the 7.40 position, and a hydrophobic residue (tryptophan for the 5-HT2 subtypes) appears at the 1.34 position in all but the 5-HT1D receptor (serine for 5-HT1D). Finally, the Y3707.43A mutation decreases ketanserin’s affinity by nearly 20-fold. This seems reasonable, considering the relatively close proximity of Y3707.43 to the piperidine ring of the ligand. In summary, these results are consistent with our proposed binding mode for the antagonist ketanserin.
Table 3.
The effect of various mutations on the binding affinity of ketanserin for 5-HT2A mutants.
| Mutation | Effect | Ref. | Commentsa |
|---|---|---|---|
| W761.34A | 10-fold ↓ in affinity | 44 | Interacts with W3677.40. |
| D1202.50N | 10-fold ↓ in affinity | 105 | Widely conserved across GPCRs. |
| F1252.55L | no effect | 104 | Not in binding pocket. |
| F1252.55L | no effect | 44 | “ |
| F1252.55L | no effect | 106 | “ |
| F1252.55S | 2-fold ↓ in affinity | 104 | “ |
| F1252.55S | no effect | 107 | “ |
| M1322.62L | no effect | 107 | Not in binding pocket. |
| T1342.64A | no effect | 107 | Inaccessible when e2 loop is in cavity. |
|
| |||
| D1553.32A | no detectable binding | 108 | Ammonium binding site. |
| D1553.32E | no detectable binding | 108 | “ |
| D1553.32N | 75-fold ↓ in affinity | 105 | “ |
| D1553.32N | no detectable binding | 108 | “ |
| D1553.32Q | no detectable binding | 108 | “ |
| S1593.36A | no effect | 45 | One turn below D1553.32. |
| S1593.36C | no effect | 45 | “ |
| D1723.49N | no effect | 105 | Conserved D/ERY motif. |
|
| |||
| W2004.50A | no effect | 44 | Widely conserved; not in binding site. |
|
| |||
| S2395.43A | < 2-fold ↑ in affinity | 47 | In Site 1. |
| F2405.44A | 2-fold ↑ in affinity | 47 | Not in binding pocket. |
| S2425.46A | ~2-fold ↑ in affinity | 109 | In Site 1. |
| F2435.47A | 4.5-fold ↓ in affinity | 47 | Interacts with F3406.52. |
| F2445.48A | 2-fold ↓ in affinity | 47 | Not in binding pocket. |
|
| |||
| W3366.48A | 900-fold ↓ in affinity | 44 | “Toggle switch”; Site 1/Site 2. |
| F3396.51A | 10-fold ↓ in affinity | 104 | One turn above W3366.48. |
| F3396.51L | 25-fold ↓ in affinity | 104 | “ |
| F3396.51L | 8-fold ↓ in affinity | 44 | “ |
| F3396.51L | 25-fold ↓ in affinity | 106 | “ |
| F3396.51L | 20-fold ↓ in affinity | 107 | “ |
| F3396.51Y | 7-fold ↓ in affinity | 104 | “ |
| F3406.52A | 2-fold ↓ in affinity | 104 | Interacts with F2435.47. |
| F3406.52L | no effect | 104 | “ |
| F3406.52L | 2-fold ↓ in affinity | 44 | “ |
| F3406.52L | no effect | 107 | “ |
| F3406.52L | 2-fold ↑ in affinity | 106 | “ |
| F3406.52Y | 70-fold ↓ in affinity | 104 | “ |
|
| |||
| F3657.38L | 4-fold ↓ in affinity | 44 | Not in binding site. |
| W3677.40L | no detectable binding | 44 | Interacts with W761.34. |
| Y3707.43A | 18-fold ↓ in affinity | 44 | In Site 2. |
| F383A | 3.5-fold ↓ in affinity | 44 | In the turn between TM7 and Helix 8. |
Comments refer to the antagonist model described here.
Our approach in selecting a receptor for antagonists involved choosing the most highly ranked (as measured by the ChemScore fitness function) receptor-ligand complex that exhibited reasonable conformations for both ligand and receptor; in this particular case the ketanserin test ligand adopted a twist-boat conformation. Twist-boat forms of cyclohexane are known to have energies that are about 5.5 kcal/mol higher than the corresponding “chair” forms;60 those for piperidine would be expected to exhibit a similar increase in energy. At first glance, then, this particular docked solution for ketanserin may seem unreasonable. However, it has been noted that in many cases, the conformation of the docked ligand is one that may not even be close to a local energy minimum.57, 58, 61, 62
Very recently, Dezi, et al.63 have generated a 5-HT2A model suited to the binding of butyrophenone antipsychotics using methodology that is quite similar to that described here. In their study, they present a ketanserin binding mode that is essentially “backwards” when compared to the ketanserin model proposed here (i.e. the p-fluorobenzoyl group is oriented toward TM5 instead of toward TM2). However, the authors go on to mention that there are likely to be multiple binding modes that contribute to the observed affinity for slender, roughly symmetric ligands (wherein a centrally positioned cation is flanked by two sets of roughly equivalent hydrogen bonding groups) such as ketanserin and the butyrophenones. Indeed, in our own experience, the GOLD-derived solutions for ketanserin usually dock in either of these two major orientations. Thus, it is possible that there exist alternate valid docked solutions for ketanserin.
Qualitatively, ketanserin and 3-phenylpropyl-AMDA (1c) are docked in much the same way (Figure 2e), with the p-fluorobenzoyl group occupying nearly the same region of space as the phenyl group of the phenylpropyl substituent of 1c, and the quinazolinedione ring system is in roughly the same area as the tricyclic ring system of 1c (the rings of 1c are oriented toward TM6; the ketanserin quinazolinedione rings are oriented toward TM5). Significantly, all four isomers of 1c (most likely acting as antagonists) received very high scores when docked into the ketanserin-selected antagonist model (Supplemental Figure 3), indicating that both ketanserin and 1c can recognize and engage the same receptor conformation.
The docked and minimized AMDA-5-HT2A model is depicted in Figure 2c (nearby residues are listed in Supplemental Table 1). When compared to 5-HT and traditional phenylethylamine-derived agonists, AMDA lacks both agonist-like functional groups (e.g. the 5-OH group of 5-HT or the 2,5-dimethoxy substituents of DOB) and presents an added feature, the “second” aromatic ring. The shape of the binding pocket in the antagonist receptor model exquisitely compliments the general shape of the AMDA molecule, and that of its tricyclic core in particular (Figure 1b). The AMDA molecule is situated in the receptor in a distinctly different location than DOB, although AMDA and DOB do interact with common residues on TM3 (D1553.32) and TM6 (Y3396.51). Effectively situated between TM3 and TM7, AMDA binds in the receptor such that one aromatic ring orients toward TM6 and the other is oriented toward TM1. The ammonium group of AMDA interacts with D1553.32 and also can interact (as suggested by molecular dynamics experiments) with the backbone carbonyl oxygen atom of C227xl2.50, one of the cysteines of the disulfide bridge anchoring the e2 loop to the extracellular end of TM3. The aromatic ring that is oriented toward TM1 is sandwiched between W1513.28 and Y3707.43; the ring oriented toward TM6 forms π-π interactions with D1553.32. The docked AMDA solution also forms close hydrophobic contacts with V3667.39, which could have implications for the selectivity of AMDA for 5-HT2A over the dopaminergic D2 receptor (vide infra).
Site 2 is lined most notably with several polar residues (T811.39, S1312.61, S1593.36, S3737.46) and hydrophobic residues (M1282.58, W1513.28, V3667.39, Y3707.43). The distribution of polar and hydrophobic residues is such that an amphiphilic cavity is created between the relatively polar faces of TM1 and TM2 and the lipophilic face of TM7. It is possible that the amphiphilic nature of the site is the characteristic that allows both relatively polar (e.g. 1e, 1g), non-polar (e.g. 1c, 1d), and mixed (e.g. 1f) groups to bind with reasonably high affinity almost without discrimination, as described in the following paragraphs.
The 3-position substituents of AMDA are directed either toward TM1 and TM2 (for S-isomers) or toward TM3 and TM6 (for R-isomers). The tricyclic core of the AMDA analogs with small substituents (1b, 1e, 1g) adopts a position in Site 2 that is the same as for the parent AMDA. For the small polar groups (1e, 1g), H-bonding takes place with either T811.39 and S1312.61 (S-isomers) or with S1593.36 and S3737.46 (R-isomers). The top-ranked GOLD-docked solution for (S)-1g features a hydrogen bond to both T811.39 and S1312.61 (Figure 2d). The more elongated shape of Site 2 relative to Site 1 allows for larger 3-position substituents (1c, 1d, 1f, 1h) to dock in a more fully extended conformation. For S-isomers, the substituent is directed toward and interacts with Y3707.43. In order to accommodate the large substituent, the tricyclic core is shifted away from the e2 loop and toward Site 1. This is possible due to the basic amine’s ready accessibility to both sidechain oxygen atoms on D1553.32. The docked and minimized (S)-3-phenlypropyl-AMDA (S-1c) interaction model is depicted in Figure 2e (nearby residues are listed in Supplemental Table 1). The tricyclic core of compound 1c interacts with residues on TM3 (D1553.32 and S1593.36) and on TM6 (M3356.47, W3366.48 and F3396.51). The bulky 3-position substituent is located in the cavity bounded by TM1, TM2 and TM7 (Site 2), with the aromatic portion anchored at Y3707.43. For R-isomers with large 3-position substituents, the R-group is either directed toward S1593.36, W3366.48 and M3656.47 (1d, 1f) or the AMDA core is inverted in an “upside-down” fashion, with the R-group interacting with Y3707.43 as described for the S-isomers (1c).
The bis-substituted compound 1h (Table 1) was initially evaluated in an attempt to bridge and interact simultaneously with both Sites 1 and 2. The 6-methoxy group was expected to interact with a hydrogen bond donating residue of Site 1, with the n-hexyl group anchored in Site 2. While monosubstitution with either an n-hexyl (Ki = 7.0 nM, 1d) or a methoxy group (Ki = 7.5 nM, 1e) enhances affinity relative to AMDA (Ki = 20 nM, 1a) to a small extent, the bis-substituted compound (Ki = 43 nM, 1h) has a lower affinity than either the unsubstituted compound (1a) or the monosubstituted derivatives (1c, 1d). At very least, the bifunctional nature of 1h does not greatly enhance affinity. The docked solution of (R)-1h (not shown) in the antagonist model reveals that the ligand primarily occupies Site 2 and that the n-hexyl group is situated in a similar fashion to the phenylpropyl group of (S)-1c. The methoxy group of (R)-1h is directed toward TM6 and interacts with M3356.47, but there are no H-bond donors nearby with which the methoxy oxygen can interact. For (S)-1h, the positions of the substituents are reversed: the methoxy group H-bonds with T811.39 and/or S1312.61 and the n-hexyl group is associated with W3366.48 and M3356.47.
DOB-like isopropylamines with small substitutions at the 4-position of the phenyl ring (2a, 2b, 2e, 2g) are docked into the antagonist model such that the aromatic ring is situated between W1513.28 and Y3707.43. Those that are nonpolar (2a, 2b) orient either the 2-methoxy group toward S226xl2.49 or the 5-methoxy group toward S771.35 (both isomers). Those that are polar (2e, 2g) orient the 4-substituent to interact with either T811.39 or S1312.61 (both isomers). DOB-like isopropylamines with large substituents at the 4-position place the phenyl ring between D1553.32 and V3667.39 (both isomers) as depicted in Figure 2f for compound (R)-2c. Such compounds are stabilized in the binding site via hydrogen bonds with D1553.32 and S1593.36 (ligand ammonium group) as well as W1513.28 and S226xl2.49 (5-methoxy group), though the latter H-bonds are far from ideal. An intramolecular hydrogen bond is also possible between the 2-methoxy group oxygen atom and the ammonium group. The large 4-position substituent is directed toward the same pocket in Site 2 as large 3-position substituents of the AMDA analogs, and is stabilized in an analogous manner. Supplemental Figure 4 shows the similarity in binding modes of 1c and 2c in the antagonist receptor binding site. The lack of H-bonding with the methoxy groups is consistent with the observation that they are not needed when the 4-position is phenylpropyl.15 The phenylethylamine derivatives 3a–d dock into Site 2 in the same way as do the isopropylamine analogs 2a–d, and the diphenylmethylamine derivatives (4a, 4b, 4d) dock similarly to their respective AMDA derivatives.
There is very little mutagenesis data available to lend insight into the nature of the interaction between the AMDA class of compounds and the 5-HT2A receptor specifically. However, residues that contribute to the amphiphilic nature of Site 2 and have been implicated to be important for the binding of ligands into the antagonist model have also been implicated to be important for ligands binding in other closely-related aminergic GPCRs. These include the residues at positions S1312.61,64, 65 W1513.28,65–68 Y3707.43,44, 69–71 and V3667.39.68, 72–78 Once again, when taken together, these examples provide evidence that Site 2 is accessible in the 5-HT2A receptor and that the antagonist model described here is accurate.
The results of the docking experiments may be summarized as follows: Each of the tested compounds containing a phenylmethyl, n-hexyl or n-pentyloxy group exhibited significant binding affinity (Ki <= 200 nM), and it was these compounds that tended to show the greatest preference for Site 2 (antagonist model) over Site 1 (agonist model) based on the ChemScore fitness function used by the GOLD docking program (Supplemental Table 2). Compound 2c has been shown to have antagonist character,15 which is consistent with its preference for the antagonist model. The relatively high affinity of AMDA derivatives with small polar substituents (1e, 1g) is likely due to the stabilization of both isomers in the amphiphilic Site 2. The high-affinity compounds with small nonpolar R-groups (1a, 1b, 2b) tended to favor the agonist model to a greater degree. This makes sense for the agonist 2b, where there is more extensive and effective hydrogen bonding with the methoxy groups, which have been shown to be necessary for high affinity in these compounds.13, 15 In addition, the F3406.52L mutation has been shown to abolish the affinity of DOI, a compound closely related to 2b (DOB). The preference of the antagonist 1a for Site 1 may be an anomaly, however, since the AMDA core is in close proximity to F3406.52, yet the F3406.52L mutation has little effect on its binding affinity. Similarly, 4b may actually bind in Site 1 rather that in Site 2 as predicted from the fitness function. Further mutagenesis testing and functional assays will be necessary, however, in order to unequivocally resolve the binding modes of these compounds.
Receptor Selectivity
As shown in Table 4, AMDA and two of its high affinity analogs are quite selective for 5-HT2 receptors. 5-HT2A affinity is between 900- and 7000-fold higher than D2 receptor affinity. There is little selectivity for 5-HT2A versus 5-HT2C receptors (2- to 9-fold). Selectivity for the 5-HT2A receptor over the serotonin and norepinephrine transporters is pronounced for 1a and 1b (between 500- to 3,000-fold) and less pronounced for 1d (60- and 120-fold). Since selectivity against the D2 receptor is not strongly influenced by the nature of the 3-position substituent, the observed selectivity is probably attributable to the AMDA nucleus itself. Examination of the docked AMDA structure (Figure 2c) along with an alignment of the 5-HT2A and D2 receptor sequences (Figure 3) allows the identification of residues that are likely responsible for the selectivity of AMDA for the 5-HT2A/C receptors. Residues in the antagonist model that possess a heavy atom within a 4.5-Å radius of any heavy atom in AMDA are highlighted in yellow boxes in Figure 3. Within this set of residues, one variant position in TM1 (S771.35), one position in TM2 (S1312.61), three in TM3 (W1513.28, I1523.29 and S1593.36), one in the e2 loop (S226xl2.49) and one in TM7 (V3667.39) that face the central cavity of the 5-HT2A receptor model can be identified. It is possible that each of these residues contribute to the selectivity of ligands for 5-HT2A over D2. The cognate residue of S771.35 in the 5-HT2A receptor is Y371.35 in the D2 receptor. The additional steric bulk of the tyrosine sidechain (compared to serine) would be sufficient to displace AMDA from its preferred binding site. The cognate residue of W1513.28 in the D2 receptor is F1103.28. Although tryptophan and phenylalanine are both aromatic, the close association of the sidechain at this position with the aromatic ring system of AMDA (3.6 Å) would suggest that even small differences in sidechain topology could take on greater significance for binding. S226xl2.49, whose equivalent residue in D2 is E181xl2.49, is adjacent to the disulfide bond anchoring the e2 loop to the top of TM3. In this position, the sidechain of a glutamic acid residue could extend into AMDA’s proposed binding site, placing its polar carboxyl group in approximately the same location as one of the aromatic rings of AMDA. Perhaps the most influential residue in determining 5-HT2A/C/D2 selectivity is V3667.39, which is equivalent to position to T4127.39 of the D2 receptor, and whose sidechain heavy atoms are within 4 Å of three heavy atoms (3.54, 3.85 and 3.93 Å) of the Site 2 aromatic ring system of AMDA in the antagonist model. As mentioned earlier, in adrenergic receptors, the presence of an alanine or threonine instead of the asparagine residue at this position has been shown to be responsible for subtype selectivity within the serotonin receptor family, particularly with respect to the ability to bind β-adrenergic antagonists such as propranolol.72, 73 It is possible that placement of a polar threonine near the AMDA aromatic ring may be unfavorable enough to account for the lower affinity of AMDA and AMDA derivatives with the D2 receptor. This hypothesis could be tested by evaluation of the V366T mutant of the 5-HT2A receptor.
Table 4.
Receptor and transporter selectivity for compounds 1a, 1b, and 1d.
| Compound | Ki, nM (±SEM)
|
||||
|---|---|---|---|---|---|
| 5-HT2Aa | 5-HT2Cb | D2c | SERTd | NETe | |
| 1af | 20 | 43 (5) | >10,000 | >10,000 | >10,000 |
| 1b | 1.3 | 3.3 (0.4) | >10,000 | 1,200 (160) | 4490 (1080) |
| 1d | 7.0 | 62 (6) | 6,280 (820) | 490 (9) | 845 (270) |
Radioligands:
[3H]ketanserin,
[3H]mesulergine,
[3H]spiperone,
[3H]paroxitine,
[3H]nisoxitine.
Data from ref. 6.
Figure 3.

Alignment of the bovine rhodopsin, 5-HT2A and D2 receptor sequences. Sequence positions highlighted in red indicate highly conserved amino acids among the Class A GPCR family that serve as reference positions in the general Ballesteros-Weinstein110 numbering system. The traditional numbering is also given for the 5-HT2A (top) and D2 (bottom) sequences. Bovine rhodopsin residues highlighted in green indicate positions that are within 12.0 Å of bound retinal; these were mutated to alanine prior to the 3-D model building phase. Bovine rhodopsin residues highlighted in purple indicate positions in the third intracellular loop that were mutated to glycine in the 5-HT2A sequence and in subsequent 5-HT2A models. Residues highlighted in yellow boxes in the 5-HT2A sequence represent those that are closest to AMDA (within 4.5 Å heavy atom to heavy atom) in the antagonist model. Note: The D3, M1, V1a, β2 and δ-opioid GPCR sequences have been omitted for brevity and clarity (see text for details). The figure was created using ALSCRIPT.111
CONCLUSION
Previous investigations have provided evidence that phenylalkylamines can be agonists or antagonists depending on the nature of the 4-position substituent.13, 15 It has been speculated that the difference in functional behavior is a reflection of the possibility that agonist and antagonist phenylalkylamines bind in a different fashion with the 5-HT2A receptor. A comparison of the effects of a parallel series of aromatic substituents based on the tricyclic 5-HT2A antagonist AMDA suggests that the AMDA-series may bind in a fashion similar to that of antagonist phenylalkylamines with bulky aromatic substituents. Differential effects of the F340L mutation observed for the AMDA series and phenylethylamine agonists supports this hypothesis. Automated docking studies with ligands docked into 5-HT2A models are consistent with the hypothesis that agonists bind in a fashion such that the aromatic rings are oriented toward the fifth and sixth transmembrane helices (Site 1), a region of limited bulk tolerance, whereas antagonists place the substituted aromatic ring near the seventh and toward the first, second and seventh transmembrane helices (Site 2) in a region of greater bulk tolerance. AMDA and two substituted derivatives were found have a high degree of selectivity against the D2 receptor and the serotonin (SERT) and norepinephrine (NET) transporters, but were non-selective with respect to the 5-HT2C receptor. Analysis of the putative binding modes of AMDA and related derivatives indicate that a valine/threonine exchange between 5-HT2A/C and D2 receptors contributes significantly to the observed selectivity of these compounds.
EXPERIMENTAL METHODS
Synthesis
Melting points were determined using a Thomas-Hoover melting point apparatus and are uncorrected. Proton magnetic resonance (1H NMR and 13C NMR) spectra were obtained with a Varian Gemini 300 spectrometer, using tetramethylsilane as an internal standard. Infrared spectra were recorded on a Nicolet Avatar 360 E.S.P. FT-infrared spectrometer. Elemental analysis was performed by Atlantic Microlab, Inc., and determined values are within 0.4% of theory (Supplemental Table 3). Thin-layer chromatography (TLC) was performed using silica gel-coated GHLF plates (250 μm, 2.5 × 10 cm, Analtech, Inc., Newark, DE). Anhydrous solvents were purchased and stored under nitrogen over molecular sieves. Medium pressure column chromatography was carried out using Silica gel 60, 0.040–0.063 mm, (230–400 mesh), Lancaster Synthesis.
3-Bromo-9-aminomethyl-9,10-dihydroanthracene hydrochloride (1b)
Tin(II) chloride dihydrate (1.06 g, 4.70 mmol) was added to a well stirred solution of 3-bromo-9-nitromethyl-9,10-dihydroanthracene (8, 0.300 g, 0.940 mmol) in absolute EtOH (3 mL). The suspension was heated at 70 °C on an oil bath (5 h). The resulting bright yellow solution was allowed to cool to room temperature and the solvent removed under reduced pressure to provide a yellow oil. The oil was dissolved in EtOAc (10 mL), sat. NaHCO3 solution (5 mL) was added, the suspension was filtered and the filter cake was washed with EtOAc (5 × 30 mL). The filtrate was collected, H2O (20 mL) was added and the mixture was extracted with EtOAc. The EtOAc portion was washed with H2O (2 ×20 mL), brine (20 mL), dried (MgSO4) and concentrated under reduced pressure to provide a viscous yellow oil. The resulting yellow oil was purified using medium pressure column chromatography (CH2Cl2/MeOH, 9:1) to provide an opaque semisolid. The semisolid was dissolved in EtOAc (10 mL) and ethereal HCl was added until no further precipitate formed. The resulting suspension was filtered and washed with EtOAc to provide a white solid that was recrystallized from MeOH/CHCl3 to provide 1b (0.10 g, 33%) as white crystals; mp 282–284 °C dec. 1H NMR (DMSO-d6) δ 2.92–2.95 (d, J=8 Hz, 2H, CH2-NH3), 3.92–3.98 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.12–4.19 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.28–4.34 (t, J=8 Hz, 1H, Ar-CH). Anal. (C15H14N • HCl • 0.25 H2O) C, H, N.
3-(3-Phenylpropyl)-9-aminomethyl-9,10-dihydroanthracene fumarate (1c)
Compound 1c was prepared from 15 in a manner analogous to 1e. The fumarate salt was recrystallized from acetone/CHCl3 to provide 1c (0.042 g, 13%) as pale yellow crystals; mp 172–175 °C. 1H NMR (DMSO-d6) δ 1.83–1.93 (m, 2H, CH2), 2.56–2.63 (m, 4H, Ar-CH2), 2.82–2.85 (d, J=8 Hz, 2H, CH2-NH2), 3.83–3.89 (d, J=18 Hz, 1H, Ar-CH2-Ar), 4.06–4.12 (d, J=18 Hz, 1H, Ar-CH2-Ar), 4.12–4.17 (t, J=8 Hz, 1H, Ar-CH-Ar), 6.48 (s, 2H, Fumarate), 7.05–7.43 (brm, 12H, Ar-H). Anal. (C24H25N • C4H4O4•0.25 acetone) C, H, N.
3-n-Hexyl-9-aminomethyl-9,10-dihydroanthracene fumarate (1d)
2-Amino-1-(2-benzyl-4-n-hexylphenyl)-1-ethanol oxalate (20d, 0.350 g, 1.13 mmol) was added to PPA (10.0 g) and the viscous mixture was stirred at room temperature by hand for 30 min. Water (100 mL) was slowly added and the mixture was made basic to pH 12 with sat. NaHCO3. The aqueous solution was extracted with EtOAc (3 × 70 mL) and the organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a brown oil. The oil was purified using medium pressure column chromatography (CH2Cl2/MeOH, 9:1) to provide (0.100 g, 30%) as a pale orange oil. The oil was dissolved in anhydrous acetone (20 mL) and anhydrous fumaric acid (0.030 g, 0.340 mmol) was added. The mixture was heated until the solid dissolved and the mixture was then cooled and filtered to provide 1d (0.070 g, 15%) as a white powder; mp 185–186 °C. 1H NMR (DMSO-d6) δ 0.85–0.86 (s, 2H, CH3), 1.24 (brs, 6H, CH2), 1.55–1.62 (m, 2H, CH2), 2.52–2.57 (t, J=8 Hz, 2H, CH3), 2.83–2.85 (d, J=8 Hz, 2H, CH2-NH2), 3.82–3.88 (d, J=18.5 Hz, 1H, Ar-CH2-Ar), 4.06–4.12 (d, J=18.5 Hz, 1H, Ar-CH2-Ar), 4.12–4.19 (t, J=7 Hz, 1H, Ar-CH-Ar), 6.45 (s, 2H, Fumarate), 7.05–7.38 (brm, 7H, Ar-H). Anal. (C21H27N • C4H4O4 • 0.25 H2O) C, H, N.
3-Methoxy-9-aminomethyl-9,10-dihydroanthracene fumarate (1e)
2-Amino-1-[2-(3-methoxybenzyl)phenyl]-1-ethanol oxalate (20e, 0.130 g, 0.370 mmol) was added to a well stirred solution of Eaton’s Reagent (20 mL) under N2. The mixture was allowed to stir (1 h) and water (50 mL) was added. The suspension was made basic to pH 13 with 10% NaOH and extracted with EtOAc (3 × 35 mL). The extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a pale yellow oil (0.080 g, 0.330 mmol). The oil was dissolved in anhydrous 2-PrOH and fumaric acid (0.0420 g, 0.360 mmol) was added. The suspension was heated until all of the solid dissolved, allowed to cool to room temperature and concentrated under reduced pressure. The solid residue was then recrystallized (EtOAc/2-PrOH) to provide 1e (0.060 g, 45%); mp 195–197 °C. 1H NMR (CD3OD) δ 2.79–2.82 (d/d, J=8 Hz, J=8 Hz, 2H, CH2-NH2), 3.58 (s, 3H, OCH3), 3.65–3.71 (d, J=19 Hz, 1H, Ar-CH2-Ar), 3.88–3.94 (d, J=19 Hz, 1H, Ar-CH2-Ar), 3.98–4.2 (t, J=8 Hz, 1H, Ar-CH-Ar), 6.47 (s, 2H, Fumarate), 6.62–6.64 (d, J=8 Hz, 1H, Ar-H), 6.72 (s, 1H, Ar-H), 7.04–7.12 (brm, 5H, Ar-H). 13C NMR (CD3OD) δ 36.49, 45.51, 46.08, 56.30, 114.05, 115.26, 128.46, 129.0, 129.16, 129.78, 129.98, 130.83, 136.79, 137.56, 138.51, 140.08. Anal. (C16H17NO • C4H4O4) C, H, N.
3-n-Pentyloxy-9-aminomethyl-9,10-dihydroanthracene fumarate (1f)
2-Amino-1-{2-[3-(n-pentyloxy)benzyl]phenyl}-1-ethanol oxalate 20f (0.500 g, 1.16 mmol) was added to methanesulfonic acid (20 mL) under N2. The suspension was allowed to stir at room temperature (2 h) and water (50 mL) was added. The suspension was made basic to pH 13 with 10% NaOH and extracted with EtOAc (3 × 50 mL). The combined extracts were washed with water, brine, dried (MgSO4) and concentrated to provide a brown oil. The oil was purified using medium pressure column chromatography (CH2Cl2\MeOH, 9:1) to provide a yellow oil (0.190 g, 55%). The fumarate salt was prepared by adding fumaric acid (0.750 g, 0.650 mmol) to the amine in 2-PrOH (30 mL). The suspension was heated until the solid dissolved, cooled and filtered. The yellow solid was recrystallized from EtOAc/2-PrOH to provide 1f (0.210 g, 44%) as pale yellow crystals; mp 178–180 °C. 1H NMR (CD3OD) δ 0.70–0.75 (t, J=7 Hz, 3H, CH3), 1.16–1.26 (m, 4H, CH2), 1.50–1.57 (m, 2H, CH2), 2.84–2.87 (d, J=8 Hz, 2H, CH2-NH2), 3.10–3.17 (m, 2H, CH2-O), 3.64–3.70 (d, J=19 Hz, 1H, Ar-CH2-Ar), 3.91–3.97 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.11–4.16 (t, J=8 Hz, 1H, Ar-CH-Ar), 6.49 (s, 2H, fumarate), 6.59–6.62 (d, J=8 Hz, 1H, Ar-H), 6.72 (s, 1H, Ar-H), 7.04–7.23 (brm, 5H, Ar-H). 13C NMR (CD3OD) δ 14.95, 19.38, 24.08, 29.96, 30.68, 36.63, 45.51, 48.71, 48.99, 50.7, 53.28, 69.62, 114.59, 115.91, 128.5, 129.98, 130.86, 136.85, 137.8, 138.75, 140.30, 160.58, 171.87. Anal (C20H25NO • C4H4O4) C, H, N.
3-Hydroxy-9-aminomethyl-9,10-dihydroanthracene hydrobromide (1g)
The hydrobromide salt of 1e was prepared by adding ethereal HBr to 3-methoxy-9-aminomethyl-9,10-dihydroanthracene in anhydrous Et2O until no further precipitate formed. The suspension was filtered and the filter cake was washed with anhydrous Et2O. The white powder was recrystallized from EtOAc\2-PrOH to provide 3-methoxy-9-aminomethyl-9,10-dihydroanthracene hydrobromide as a white powder; mp 258–261 °C dec. A 1.0 M solution of BBr3 in CH2Cl2 (1.0 mL, 0.960 mmol) was added under N2 in a dropwise manner to 3-methoxy-9-aminomethyl-9,10-dihydroanthracene hydrobromide (0.100 g, 0.320 mmol) in CHCl3 at −78 °C (dry ice/acetone). The suspension was allowed to warm to room temperature over 2 h and was then allowed to stir for 5 h. The suspension was cooled to −78 °C, anhydrous MeOH (7 mL) was added and the suspension was allowed to warm to room temperature. The suspension was concentrated under reduced pressure to provide a white solid that was recrystallized from 2-PrOH\CHCl3 to provide 1g (0.430 g, 43%) as a white powder; mp 278–281 °C dec. 1H NMR (DMSO-d6) δ 2.88–2.90 (d, J=6 Hz), 2H, CH2-NH2), 3.77–3.84 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.03–4.10 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.10–4.16 (t, J=6 Hz, 1H, Ar-CH-Ar), 6.64–6.67 (d, J=8 Hz, 1H, Ar-H), 6.77 (s. 1H, Ar-H), 7.17–7.39 (brm, 7H, Ar-H). Anal. (C15H15NO • HBr • 0.25 H2O) C, H, N.
3-n-Hexyl-6-methoxy-9-aminomethyl-9,10-dihydroanthracene fumarate (1h)
Compound 1h was prepared from 20h in a manner analogous to that of 1f. The resulting yellow oil was purified using medium pressure column chromatography (CH2Cl2/MeOH, 9:1) to provide 1h (0.0580 g, 20%) as a white powder; mp 169–171 °C. 1H NMR (DMSO-d6) δ 0.85–0.86 (t, J=6 Hz, 3H, CH3), 1.27 (m, 6H, CH2), 1.53–1.58 (m, 2H, CH2), 2.79–2.84 (d, J=7 Hz, 2H, CH2-NH2), 3.74 (s, 3H, OCH3), 3.78–3.84 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.04–4.08 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.08–4.12 (t, J=7 Hz, 1H, Ar-CH-Ar), 6.44 (s, 2H, Fumarate), 6.79–6.82 (d, J=8 Hz, 1H, Ar-H), 6.93 (s, 1H, Ar-H), 7.04–7.06 (d, J=8 Hz, 1H, Ar-H), 7.14 (s, 1H, Ar-H), 7.25–7.30 (m, 2H, Ar-H) Anal. (C22H30NO • C4H4O4) C, H, N.
2-(4-Bromophenyl)-1-aminoethane hydrochloride (3b)
A 1.0 M solution of borane-THF complex (30.6 ml, 30.6 mmol) was added to 4-bromophenyl acetonitrile (2.00 g, 10.2 mmol) in anhydrous THF (10 mL). The solution was heated at reflux for 8 h. The solution was allowed to cool to room temperature, 6 M HCl (10 mL) was cautiously added and the solution was heated at reflux for 30 min. The solution was allowed to cool to room temperature, 10% NaOH (45 mL) was added and the suspension was extracted with CH2Cl2 (3 × 50 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide (1.50 g) of a colorless oil. The oil was dissolved in anhydrous Et2O (50 mL) and ethereal HCl was added until no more precipitate formed. The suspension was filtered and washed with anhydrous Et2O to provide a white solid that was recrystallized from 2-PrOH to provide 3b (1.20 g, 58%) as white needles; mp 239–241 °C dec. (Lit47 mp 240–243 °C) 1H NMR (CD3OD) δ 2.71–2.76 (m, 2H, CH2), 2.93–2.98 (m, 2H, CH2), 7.00–7.03 (d, J=8 Hz, 2H, Ar-H), 7.27–7.30 (d, J=8 Hz, 2H, Ar-H). 13C NMR (CD3OD) δ 34.43, 42.19, 132.37, 133.59
2-(4-n-Hexylphenyl)-1-aminoethane hydrochloride (3d)
Pd on charcoal (10%, 0.50 g) and concentrated sulfuric acid (0.300 g) were added to 26 (0.300 g, 1.40 mmol) in glacial acetic acid (5 mL). The mixture was hydrogenated (10 h) at 50 psi. The mixture was filtered through a Celite pad and the filter cake was washed with glacial acetic acid (15 mL). The filtrate was concentrated under reduced pressure and made basic to pH 10 with sat. NaHCO3. The suspension was extracted with EtOAc (3 × 40 mL) and the organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide (0.210 g, 1.00 mmol) of a pale yellow oil. The oil was dissolved in anhydrous Et2O (25 mL) and a 1.0 M solution of HCl in Et2O (1 mL) was added. The solution was cooled and filtered to provide a white powder. The powder was recrystallized from 2-PrOH/Et2O to provide 3d (0.180 g, 53%) as white plates; mp 176–178 °C. (Lit.31 175–177 °C). 1H NMR (DMSO-d6) δ 0.85–0.87 (t, J=8 Hz, 3H, CH3), 1.12–1.14 (s, 6H, CH2) 1.61–1.63 (m, 2H, CH2), 2.49–2.54 (t, J=7.7 Hz, 2H, Ar-CH2), 2.83–2.87 (m, 2H, CH2), 2.95–2.99 (m, 2H, CH2), 7.13 (s, 4H, Ar-H). 13C NMR (DMSO-d6) δ 14.33, 22.44, 28.71, 31.34, 31.48, 32.91, 35.13, 40.27, 128.84, 134.37, 141.02.
2-(4-Bromophenyl)-2-phenyl-1-aminoethane hydrochloride (4b)
A 1.0 M solution of borane-THF complex (7.28 mL, 7.28 mmol) was added at room temperature to a well stirred solution of 27 2-(4-bromophenyl)-2-phenylacetonitrile (0.500 g, 1.82 mmol) in anhydrous THF (5 mL). The solution was then heated at reflux (5 h) and allowed to cool. A 6.0 M solution of HCl (7 mL) was cautiously added and the suspension was heated at reflux (30 min). The suspension was allowed to cool, made basic with 10% NaOH (≈ 35 mL) and extracted with Et2O (3 × 35 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide an oily solid. The solid was dissolved in anhydrous Et2O (40 mL) and ethereal HCl was added. The white suspension was filtered and washed with anhydrous Et2O (10 mL). The white solid was recrystallized from 2-PrOH to provide 4b (0.290 g, 51%) as white crystals; mp 216–218 °C. 1H NMR (DMSO-d6) δ 3.44–3.58 (m, 2H, CH2-NH2), 4.37–4.42 (t, J=8 Hz, 1H, CH), 7.23–7.53 (brm, 9H, Ar-H). Anal. (C14H14BrN • HCl) C, H, N.
2-(4-n-Hexylphenyl)-2-phenyl-1-aminoethane hydrochloride (4d)
A methanolic solution containing Raney nickel (≈ 0.500 g) was added to 2-(4-n-hexylphenyl)-2-phenylacetonitrile (30, 0.400 g, 1.44 mmol) in anhydrous MeOH (20 mL) and sat. NH3/MeOH solution (5 mL). The suspension was hydrogenated at 40 psi for 10 h. The suspension was filtered through a Celite pad and the filter cake was washed with anhydrous MeOH (25 mL). The filtrate was concentrated under reduced pressure to provide a colorless oil. The oil was dissolved in anhydrous Et2O (35 mL) and ethereal HCl was added until no further precipitate formed. The white suspension was filtered and washed with anhydrous Et2O (10 mL). The white solid was recrystallized from 2-PrOH/Et2O to provide 4d (0.200 g, 44%) as white crystals; mp 174–175 °C. 1H NMR (CD3OD) δ 0.65–0.69 (t, J=8 Hz, 2H, CH3), 1.09 (s, 6H, CH2), 1.32–1.39 (m, 2H, CH2), 2.34–2.39 (t, J=7.7 Hz, 2H, CH2), 3.38–3.41 (d, J=7.8 Hz, 2H, CH2), 4.02–4.07 (t, J=8.25 Hz, 1H, CH), 6.95–7.17 (brm, 9H, Ar-H). 13C NMR (DMSO-d6) δ 14.51, 20.41, 30.48, 33.17, 33.21, 37.02, 45.08, 50.87, 129.33, 129.42, 130.68, 130.73. Anal. (C20H27N • HCl): C, H, N.
1-Bromo-2-[(methoxymethoxy)methyl]benzene (5)
Chloromethylmethyl ether (1.61 g, 20.0 mmol) was added in a dropwise manner to a well stirred solution of 2-bromobenzyl alcohol (3.00 g, 16.0 mmol) in N,N-diisopropylethylamine (10 mL) at 0 °C under N2. The yellow solution was allowed to stir at 0 °C (2 h), allowed to warm to room temperature and stirred (5 h). The solvent was removed under reduced pressure to provide a yellow oil. The oil was dissolved in CHCl3 (30 mL) and washed with water (3 × 75 mL). The organic extract was dried (MgSO4) and concentrated under reduced pressure to provide a pale yellow oil that was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 5 (2.79 g, 75%) as a colorless oil. 1H NMR (CDCl3) δ 3.49 (s, 3H, CH3), 4.65 (s, 2H, CH2), 4.75 (s, 2H, CH2), 7.16–7.57 (brm, 4H, Ar-H). Compound 5 was used without further characterization in the preparation of 6.
1-[1-(4-Bromophenyl)-2-nitroethyl]-2-[(methoxymethoxy)methyl]benzene (6)
A crystal of I2 was added to clean, dry magnesium turnings (0.160 g, 6.58 mmol) in anhydrous THF (10 mL). 1-Bromo-2-[(methoxymethoxy)methyl]benzene (5, 1.58 g, 6.58 mmol) was slowly added (maintaining a gentle reflux) to the THF/Mg suspension and the mixture was heated at reflux (≈ 45 min. or until most of the Mg was dissolved). The suspension was allowed to cool to room temperature, the solvent was decanted from the unreacted Mg turnings, and slowly added in a dropwise manner (not allowing the temperature to rise above 10 °C) to an ice-cold well-stirred solution of trans-2-(4-bromophenyl)-1-nitroethene (1.50 g, 6.58 mmol) in anhydrous THF (15 mL). After the addition was complete the reaction was allowed to warm to room temperature (1 h) and stirring was continued (10 h). HCl (1.0 M, 15 mL) was added and the suspension was concentrated under reduced pressure. Water (25 mL) was added and the yellow suspension was extracted with EtOAc (3 × 20 mL), washed with sat. NaHCO3 (40 mL) and brine (40 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a viscous yellow oil. The resulting oil was purified by medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 6 (1.00 g, 40%) as a viscous yellow oil. 1H-NMR (CDCl3): δ 3.45 (s, 3H, OCH3), 4.56–4.98 (brm, 6H, CH2), 5.29–5.35 (t, J=8 Hz, 1H, Ar-CH-Ar), 7.1–7.46 (brm, 9H, Ar-H). 13C-NMR (CDCl3) δ 44.07, 56.25, 68.06, 79.35, 96.32, 127.68, 128.39, 129.46, 130.05, 131.55, 132.66. IR (Film) 1556, 1376 cm−1. Compound 6 was used in the preparation of 7.
{2-[1-(4-Bromophenyl)-2-nitroethyl]phenyl}methanol (7)
Concentrated HCl (4 drops) was added to 1-[1-(4-bromophenyl)-2-nitroethyl]-2- [(methoxymethoxy)methyl]benzene (6, 1.00 g, 2.63 mmol) in MeOH (15 mL). The reaction mixture was heated in an oil bath at 65 °C (5 h). The solution was allowed to cool to room temperature and concentrated under reduced pressure to provide a viscous yellow oil. The oil was dissolved in EtOAc (20 mL), the suspension was made basic with sat. NaHCO3 (≈ 50 mL) and the mixture was extracted with EtOAc (3 × 20 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a viscous yellow oil that was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 7 (0.800 g, 90%) as a viscous yellow oil. 1H NMR (CDCl3) δ 4.64–4.92 (m, 2H, Ar-CH2-OH), 4.93–5.06 (m, 2H, CH2-NO2), 5.3–5.36 (t, J=8 Hz, 1H, Ar-CH-Ar), 7.1–7.4 (brm, 8H, Ar-H). IR (Film) 3384, 1550, 1375 cm−1.
3-Bromo-9-nitromethyl-9,10-dihydroanthracene (8)
PPA (5ml) was added to {2-[1-(4-Bromophenyl)-2-nitroethyl]phenyl}methanol (7, 1.02 g, 3.04 mmol) and the viscous mixture was stirred by hand and heated on an oil bath at 65 °C (30 min). After the reaction was complete, ice (40.0 g) was added to the white/brown semisolid and the solution was made basic to pH 12 with sat. NaHCO3 solution. The suspension was extracted with EtOAc (3 × 25 mL) and the organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a brown semisolid (0.540 g). The crude solid was then recrystallized from methanol to provide 8 (0.480 g, 49%) as pale yellow crystals; mp 113–114 °C. 1H NMR (CDCl3): δ 3.80–3.95 (d, J=19 Hz, 1H, CH2), 4.07–4.13 (d, J=19 Hz, 1H, Ar-CH2-Ar), 4.37–4.49 (m, 2H, CH2-NO2) 4.76–4.82 (t, J=9 Hz, 1H, CH), 7.15–7.39 (brm, 7H, Ar-H). 13C NMR (CDCl3) δ 35.22, 45.5, 80.15, 122.27, 127.85, 128.56, 128.66, 128.85, 130.24, 130.61, 131.75, 133.91, 134.28, 136.05, 139.17. IR (KBr) 1537, 1382, cm−1.
4-Bromo-2-(bromomethyl)benzonitrile (9)
NBS (3.49 g, 19.6 mmol) was added to 4-bromo-2-methylbenzonitrile (3.50 g, 17.9 mmol) in CCl4 (25 mL) under N2. The reaction was slowly warmed with an IR lamp and heated at reflux (6 h). The suspension was cooled, filtered and the filtrate was concentrated under reduced pressure to provide an oily solid. The oily solid was purified using medium pressure column chromatography (petroleum ether/acetone 9:1) to provide a white solid that was recrystallized (toluene/petroleum ether) to provide 9 (3.45 g, 70%) as white needles; mp 88–91 °C. 1H NMR (CDCl3) δ 4.55 (s, 2H, CH2-Br), 7.54–7.58 (m, 2H, Ar-H), 7.73 (s, 1H, Ar-H). 13C NMR (CDCl3) δ 28.78, 128.72, 132.90, 133.81, 134.26, 134.78, 142. 67. The product was used in the preparation of 10.
2-Benzyl-4-bromobenzonitrile (10)
4-Bromo-2-(bromomethyl)benzonitrile (9, 3.25 g, 11.82 mmol) was added under N2 to AlCl3 (3.14 g, 23.6 mmol) in anhydrous benzene (30 mL) at 0 °C. The reaction mixture was allowed to warm to room temperature and heated at reflux (45 min). The reaction was allowed to cool, poured onto ice (50.0 g) and made acidic to pH 2 with 5% HCl. The suspension was extracted with CH2Cl2 (3 × 75 mL) and the combined extracts were washed with water, brine, dried (MgSO4) and concentrated under reduced pressure to provide a white solid. The white solid was recrystallized from toluene/petroleum ether to provide 10 (3.10 g, 96%) as white crystals; mp 73–75 °C. 1H NMR (CDCl3) δ 4.13 (s, 2H, Ar-CH2-Ar), 7.07–7.50 (brm, 8H, Ar-H). 13C NMR (CDCl3) δ 40.56, 127.63, 129.49, 129.55, 130.88, 133.82, 134.55. Compound 10 was used without further characterization in the preparation of 11.
2-Benzyl-4-bromobenzoic acid (11)
2-Benzyl-4-bromobenzonitrile (10, 0.90 g, 3.31 mmol) was added to KOH (1.15 g, 24.2 mmol) in ethylene glycol (7 mL) and water (0.5 mL). The solution was heated at reflux (3 h), allowed to cool to room temperature and made acidic to pH 2 with 5% HCl. The suspension was extracted with CHCl3 (3 × 50 mL) and the extracts were washed with water, brine, dried (MgSO4) and concentrated under reduced pressure to provide a white solid. The solid was recrystallized from formic acid/acetic acid to provide 11 (0.950 g, 100%) as white crystals; mp 131–134 °C. 1H NMR (DMSO-d6) δ 4.36 (s, 2H, Ar-CH2-Ar), 6.67 (s, 1H, Ar-H), 7.01–7.04 (d, J=8 Hz, 1H, Ar-H), 7.16–7.78 (brm, 6H, Ar-H).
2-Benzyl-4-bromobenzyl alcohol (12)
Compound 12 was prepared from 11 in a manner analogous to that of 21. The resulting oil (1.01 g, 96 %) was used in the next step without further purification.
2-Benzyl-4-bromobenzaldehyde (13)
Compound 13 was prepared from 12 in a manner analogous to that of 19d. The resulting oil was purified using medium pressure column chromatography (petroleum ether/acetone, 9:1) to provide 13 (0.750 g, 75%) as a colorless oil. 1H NMR (CDCl3) δ 4.27 (s, 2H, Ar-CH2-Ar), 7.02–7.29 (brm, 7H, Ar-H), 7.69–7.74 (d, J=6 Hz, 1H, Ar-H), 10.11 (s, 1H, COH).
2-Benzyl-4-(3-phenylpropyl)benzaldehyde (14)
A 0.5 M solution of 9-BBN in THF (8 mL, 4.00 mmol) was added under N2 in a dropwise manner to a solution of allylbenzene (0.470 g, 4.01 mmol) in anhydrous THF (2.5 mL) at 0 °C. The mixture was then allowed to stir for 12 h at room temperature. 2-Benzyl-4-bromobenzaldehyde (13, 1.00 g, 3.34 mmol) in THF (12 mL), PdCl2(dppf) (0.080 g, 0.100 mmol) and NaOH (3 M, 3.34 mL) were then added to the flask containing the 9-phenylpropyl-9-BBN. The mixture was heated at reflux (12 h), allowed to cool to room temperature and water (20 mL) was added. The suspension was extracted with EtOAc (3 × 50 mL) and the combined extracts were washed with water, brine, dried (MgSO4) and concentrated under reduced pressure. The resulting brown oil was purified using medium pressure column chromatography (petroleum ether/acetone, 9:1) to provide 14 (0.700 g, 66%) as a colorless oil. 1H NMR (CDCl3) δ 1.90–2.00m (m, 2H, CH2), 2.60–2.68 (m, 4H, Ar-CH2), 4.40 (s, 2H, Ar-CH2-Ar), 7.06–7.30 (brm, 12H, Ar-H), 7.76–7.79 (d, J=8Hz, 1H, Ar-H), 10.17 (s, 1H, COH). 13C NMR (CDCl3) δ 33.00, 35.90, 36.03, 38.62, 126.50, 126.80, 127.70, 128.97, 129.13, 129.29, 130.23, 132.49, 132.98, 141.13, 142.36, 143.60, 149.73, 192.55.
2-Amino-1-[2-benzyl-4-(3-n-phenylpropyl)phenyl]-1-ethanol oxalate (15)
Compound 15 was prepared from 14 in a manner analogous to that of 20d. The oxalate salt was recrystallized from 2-PrOH to provide 15 (0.630 g, 64%) as a white powder; mp 168–170 °C. 1H NMR (DMSO-d6) δ 1.78–1.88 (m, 2H, CH2), 2.45–2.57 (m, 4H, CH2-Ar), 2.70–2.79 (m, 2H, CH2-NH2), 3.92–3.97 (d, J=16 Hz, 1H, Ar-CH2-Ar), 4.02–4.07 (d, J=16 Hz, 1H, Ar-CH2-Ar), 5.05–5.08 (d, J=9 Hz, CH-OH), 6.96 (s, 1H, Ar-H), 7.15–7.53 (brm, 12H, Ar-H). Anal (C24H27NO • C2H2O4) C, H, N.
2-(4-n-Hexylphenyl)-4,4-dimethyl-2-oxazoline (16d)
A solution of 4-n-hexylbenzoyl chloride (3.00 g, 13.3 mmol) in CH2Cl2 (6 mL) was added under N2 to 2-amino-2-methyl-1-propanol (2.49 g, 27.9 mmol) in CH2Cl2 (6 mL) at such a rate as to maintain a temperature of 0 °C. The suspension was allowed to stir for 4 h and was filtered. The filter cake was washed with CH2Cl2 (2 × 20 mL) and the filtrate was washed with 3N HCl (100 mL) and water (100 mL). The solvent was removed under reduced pressure to provide (3.60 g, 97%) a yellow oil. Thionyl chloride (1.83 g, 15.31 mmol) was added slowly to the crude 4-n-hexyl-N-(2-hydroxy-1,1-dimethylethyl) benzamide (3.60 g, 13.0 mmol) in anhydrous toluene (25 mL). The solution was stirred at room temperature (10 h), poured into water (30 mL), made basic to pH 10 with sat. NaHCO3 and extracted with EtOAc (3 × 40 mL). The extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a pale orange oil. The oil was purified by Kuhgelrohr bulb to bulb distillation (bp 141 °C @ 0.10 mm Hg) to provide 16d (3.33 g, 98%) as a colorless oil. 1H NMR (CDCl3) δ 0.85–0.89 (t, J=7 Hz, 3H, CH3), 1.28 (s, 6H, CH2), 1.37 (s, 6H, CH3), 1.53–1.62 (m, 2H, CH2), 2.6–2.65 (t, J=7 Hz, 2H, Ar-CH2), 4.09 (s, 2H, CH2-O), 7.19–7.22 (d, J=8 Hz, 2H, Ar-H), 7.83 (d, J=8 Hz, 2H, Ar-H). 13C NMR (CDCl3) δ 14.64, 23.14, 28.99, 29.43, 31.76, 32.23, 36.48, 79.59, 128.74, 128.92, 147.09. IR (Film) 1655 cm−1.
5-n-Hexyl-3-phenyl-1,3-dihydro-1-isobenzofuranone (17d)
A well stirred solution of 2-(4-n-hexylphenyl)-4,4-dimethyl-2-oxazoline (16d, 1.00 g, 3.85 mmol) in anhydrous THF (5 mL) was cooled to −78 °C (dry ice/acetone) under N2. A 2.5 M solution of n-butyl lithium in cyclohexane (1.77 mL, 4.43 mmol) was added slowly to the solution so as to maintain the temperature at −78 °C. The solution was allowed to warm to room temperature and stirred (1 h). The red solution was then cooled to 0 °C and a solution of benzaldehyde (0.410 g, 3.85 mmol) in anhydrous THF (2 mL) was added over 30 min. The reaction mixture was allowed to stir for 5 h at room temperature and H2O (20 mL) was added. The mixture was extracted with EtOAc (3 × 30 mL). The combined fractions were dried (MgSO4) and concentrated under reduced pressure to provide a yellow oil. HCl (5%, 25 mL) was added to the oil and the suspension was heated at reflux (10 h). The suspension was allowed to cool and extracted with EtOAc (3 × 40 mL). The combined extracts were dried (MgSO4) and concentrated under reduced pressure to provide an orange oil that was purified by medium pressure column chromatography (petroleum ether/acetone, 9:1) to provide 17d (0.910 g, 80%) as a colorless oil that solidified on standing. The solid was recrystallized from EtOAc/petroleum ether; mp 69–70 °C. 1H NMR (CDCl3) δ 0.85 (s, 3H, CH3), 1.28 (s, 6H, CH2), 1.53–1.62 (m, 2H, CH2), 2.6–2.65 (s, 2H, Ar-CH2), 6.35 (s, 1H, Ar-CH-Ar), 7.26–7.86 (brm, 8H, Ar-H). IR (KBr) 1743 cm−1.
3-(3-Methoxyphenyl)-1,3-dihydro-1-isobenzofuranone (17e)
Compound 17e was prepared from 3-methoxybenzaldehyde and 2-phenyl-4,4-dimethyl-2-oxazoline in a manner analogous to 17d. The resulting yellow solid was recrystallized from EtOH/MeOH to provide 17e (5.60 g, 81%) as yellow needles; mp 112–114 °C. 1H NMR (CDCl3) δ 3.77 (s, 3H, OCH3), 6.37 (s, 1H, Ar-CH-Ar), 6.78–6.91 (m, 3H, Ar-H), 7.27–7.96 (brm, 5H, Ar-H). 13C NMR (CDCl3) δ 54.89, 82.08, 111.98, 114.20, 118.65, 122.41, 125.02, 125.20, 128.94, 129.63, 133.90, 137.50, 149.16, 159.57, 170.06.
3-[3-(n-Pentyloxy)phenyl]-1,3-dihydro-1-isobenzofuranone (17f)
Compound 17f was prepared from 21 (3-n-pentyloxybenzaldehyde) and 2-phenyl-4,4-dimethyl-2-oxazoline in a manner analogous to 17d. The resulting yellow solid was recrystallized from toluene/petroleum ether to provide 17f (4.78 g, 55%) as yellow needles; mp 94–96 °C. 1H NMR (CDCl3) δ 0.89–0.94 (t, J=7 Hz, 3H, CH3), 1.3–1.46 (m, 4H, CH2), 1.70–1.79 (m, 2H, CH2), 3.88–3.93 (t, J=7 Hz, 3H, CH2-O), 6.36 (s, 1H, Ar-CH-Ar), 6.77 (s, 1H, Ar-H), 6.85–6.90 (m, 2H, Ar-H) 7.25–7.67 (brm, 4H, Ar-H), 7.94–7.96 (d, J=8 Hz, 1H, Ar-H). 13C NMR (CDCl3) δ 14.56, 23.0, 28.71, 29.45, 68.63, 83.15, 113.55, 115.65, 119.46, 123.42, 126.04, 126.19, 129.91, 130.57, 134.88, 138.42, 150.23, 160.16.
5-n-Hexyl-3-(3-methoxyphenyl)-1,3-dihydro-1-isobenzofuranone (17h)
Compound 17h was prepared from 16d (2-(4-n-hexylphenyl)-4,4-dimethyl-2-oxazoline) and 3-methoxybenzaldehyde in a manner analogous to that of 17d. The resulting orange oil was purified using medium pressure column chromatography (petroleum ether/acetone, 9:1) to provide 17h (4.82 g, 77%) as a yellow oil. 1H NMR (CDCl3) δ 0.83–0.87 (t, J=6 Hz, 3H, CH3), 1.28 (m, 6H, CH2), 1.57–1.60 (m, 2H, CH2), 2.64–2.69 (t, J=8 Hz, 2H, CH2-Ar), 3.77 (s, 3H, OCH3), 6.31 (s, 1H, Ar-CH-Ar), 6.79–6.90 (m, 3H, Ar-H), 7.12 (s, 1H, Ar-H), 7.27–7.36 (m, 2H, Ar-H), 7.82–7.85 (d, J=8 Hz, 1H, Ar-H). 13C NMR (CDCl3) δ 14.59, 23.08, 29.44, 31.74, 32.12, 36.93, 55.88, 82.91, 112.99, 115.14, 119.73, 122.99, 123.68, 126.00, 130.52, 130.58, 138.78, 150.69, 151.24, 160.62. IR (Film) 1772 cm−1.
2-Benzyl-4-n-hexylbenzoic acid (18d)
Five drops of HClO4 (70% in water), 10% Pd/C (0.100 g) and 5-n-hexyl-3-phenyl-1,3-dihydro-1-isobenzofuranone 17d (0.400 g, 1.36 mmol) was hydrogenated in 2-PrOH (11 mL) at 55 psi for 12 h. The suspension was filtered through a Celite pad and concentrated under reduced pressure. The resulting oil was dissolved in CHCl3 (50 mL) and extracted with 10% NaOH (40 mL). The aqueous layer was made acidic to pH 3 with 5% HCl and extracted with CH3Cl (3 × 50 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a white solid. The solid was recrystallized from formic acid/acetic acid to provide 18d (0.300 g, 74%) as white needles; mp 67–68 °C. 1H NMR (CDCl3) δ 0.84–0.89 (t, J=7 Hz, 2H, CH3), 1.27 (brs, 6H, CH2), 1.53–1.58 (m, 2H, CH2), 2.56–2.61 (t, J=6 Hz, 2H, CH3), 4.43 (s, 2H, Ar-CH2-Ar), 7.03–7.28 (brm, 7H, Ar-H), 7.98–8.01 (d, J=8 Hz,1H, Ar-H). 13C NMR (CDCl3) δ 14.65, 23.15, 29.47, 31.51, 32.20, 36.43, 40.23, 126.42, 127.04, 128.84, 129.52, 132.56, 132.65, 149.22, 173.23. IR (KBr) 2924, 1687 cm−1.
2-(3-Methoxybenzyl)benzoic acid (18e)
Compound 18e was prepared from 17e in a manner analogous to 18d. The resulting white solid was recrystallized from formic acid/acetic acid to provide 18e (1.81 g, 89%) as white needles; mp 95–96 °C. 1H NMR (CDCl3) δ 3.77 (s, 3H, OCH3), 4.46 (s, 2H, Ar-CH2-Ar), 6.75–6.77 (d, J=7 Hz, 1H, Ar-H), 6.8 (s, 1H, Ar-H), 7.18–7.5 (brm, 5H, Ar-H), 8.08–8.10 (d, J=7.8 Hz, 1H, Ar-H). 13C NMR (CDCl3) 40.19, 55.67, 111.92, 115.48, 122.13, 126.97, 129.87, 132.29, 133.62, 142.89, 143.89, 173.79.
2-[3-(n-Pentyloxy)benzyl]benzoic acid (18f)
Compound 18f was prepared from 17f in a manner analogous to that of 18d. The resulting white solid was recrystallized from formic acid/acetic acid to provide 18f (4.18 g, 96%) as white needles; mp 89–91 °C. 1H NMR (DMSO-d6) δ 0.85–0.89 (t, J=6 Hz, 3H, CH3), 1.27–1.33 (m, 4H, CH2), 1.61–1.68 (m, 2H, CH2), 3.84–3.88 (t, J=6 Hz, 2H, CH2-O), 4.30 (s, 2H, Ar-CH2-Ar), 6.69 (m, 2H, Ar-H), 6.71 (s, 1H, Ar-H), 7.11–7.48 (brm, 4H, Ar-H), 7.79–7.82 (d, J=8 Hz, 1H, Ar-H). 13C NMR (DMSO-d6) 14.26, 22.26, 28.09, 28.76, 38.74, 67.53, 111.83, 115.44, 121.18, 126.65, 129.59, 130.57, 131.06, 131.70, 132.07, 141.80, 143.03, 159.03, 169.22.
4-n-Hexyl-2-(3-methoxybenzyl)benzoic acid (18h)
Compound 18h was prepared from 17h in a manner analogous to that of 18d. The resulting yellow semisolid was recrystallized from formic acid/acetic acid to provide 18h (2.21 g, 84%) as a white powder; mp 58–60 °C. 1H NMR (CDCl3) δ 0.86–0.91 (t, J=6 Hz, 3H, CH3), 1.29 (m, 6H, CH2), 1.55–1.62 (m, 2H, CH2), 2.58–2.63 (t, J=8 Hz, 2H, CH2-Ar), 3.76 (s, 3H, OCH3), 4.43, (s, 2H, Ar-CH2-Ar), 6.73–6.78 (m, 3H, Ar-H) 7.05–7.21 (brm, 3H, Ar-H), 7.99–8.0 (d, J=8 Hz, 1H, Ar-H). 13C NMR (CDCl3) δ 14.64, 23.14, 29.77, 31.50, 32.21, 36.43, 40.22, 55.63, 111.83, 115.32, 122.01, 126.23, 127.06, 129.75, 132.55, 132.60, 143.16, 143.84, 149.23, 160.00, 173.44.
2-Benzyl-4-n-hexylbenzaldehyde (19d)
A solution of 2-benzyl-4-n-hexylbenzyl alcohol (21, 0.400 g, 1.42 mmol) in anhydrous CH2Cl2 (5 mL) was added over 30 min at room temperature to a well stirred suspension of PCC (0.460 g, 2.13 mmol) and Celite (1.0 g in 50 mL CH2Cl2). The solution was allowed to stir at room temperature for 2 h, anhydrous Et2O (25 mL) was added and the dark brown suspension was filtered through a Florisil column. The solution was concentrated under reduced pressure to provide 19d (0.320 g, 80%) as a colorless oil. The product was used without further purification. 1H NMR (CDCl3) δ 0.85–0.87 (t, J=7 Hz, 2H, CH3), 1.28 (brs, 6H, CH2), 1.55–1.60 (m, 2H, CH2), 2.60–2.65 (t, J=8 Hz, 2H, CH3), 4.14 (s, 2H, Ar-CH2-Ar), 7.07–7.29 (brm, 7H, Ar-H), 7.75–7.78 (d, J=8 Hz, 1H, Ar-H), 10.17 (s, 1H, COH). 13C NMR (CDCl3) δ 14.65, 23.15, 29.50, 31.52, 32.21, 36.66, 38.65, 126.76, 127.68, 129.08, 129.28, 132.47, 132.93, 140.85, 143.48, 150.55, 192.59. IR (Film) 1693 cm−1.
2-(3-Methoxybenzyl)benzaldehyde (19e)
Compound 19e was prepared from 23 in a manner analogous to that of 19d. The resulting oil was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 19e (0.490 g, 98%) as a pale yellow oil. 1H NMR (CDCl3) δ 3.75 (s, 3H, OCH3), 4.42 (s, 2H, Ar-CH2-Ar), 6.68 (s, 1H, Ar-H), 6.72–6.75 (d, J=8.2 Hz, 1H, Ar-H), 7.17–7.53 (brm, 5H, Ar-H), 7.84–7.85 (d, J=3 Hz, 1H, Ar-H), 10.24 (s, 1H, COH). 13C NMR (CDCl3) 38.59, 55.69, 111.96, 115.34, 121.77, 127.60, 130.10, 132.21, 132.56, 134.50, 192.95. IR (Film) 1699 cm−1.
2-[3-(n-Pentyloxy)benzyl]benzaldehyde (19f)
Compound 19f was prepared from 24 in a manner analogous to that of 19d. The resulting oil was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 19f (3.56 g, 100%) as a pale yellow oil. 1H NMR (CDCl3) δ 0.85–0.93 (t, J=7 Hz, 3H, CH3), 1.32–1.42 (m, 4H, CH2), 1.69–1.76 (m, 2H, CH2), 3.86–3.90 (t, J=7 Hz, 2H, CH2-O), 4.40 (s, 2H, Ar-CH2-Ar), 6.68–6.73 (m, 3H, Ar-H), 7.14–7.54 (brm, 4H, Ar-H), 7.83–7.87 (d, J=8 Hz, 1H, Ar-H), 10.24 (s, 1H, COH).
4-n-Hexyl-2-(3-methoxybenzyl)benzaldehyde (19h)
Compound 19h was prepared from 25 in manner analogous to that of 19d. The resulting brown oil was purified using medium pressure column chromatography (petroleum ether/acetone, 9:1) to provide 19h (1.98 g, 92 %) as a pale yellow oil. 1H NMR (CDCl3) δ 0.85–0.89 (t, J=6 Hz, 3H, CH3), 1.26–1.29 (m, 6H, CH2), 1.58–1.63 (m, 2H, CH2), 2.59–2.65 (t, J=8 Hz, 2H, CH2-Ar), 3.75 (s, 3H, OCH3), 4.39 (s, 2H, Ar-CH2-Ar), 6.68–6.74 (m, 3H, Ar-H), 7.07–7.23 (m, 3H, Ar-H), 7.75–7.78 (d, J=8 Hz, 1H, Ar-H), 10.17 (s, 1H, COH). 13C NMR (CDCl3) δ 14.62, 23.12, 29.48, 31.50, 32.20, 36.64, 38.62, 55.66, 111.89, 115.25, 121.70, 127.69, 130.03, 132.44, 132.87, 142.68, 143.27, 150.41, 160.0, 192.54. IR (Film) 1700 cm−1.
2-Amino-1-(2-benzyl-4-n-hexylphenyl)-1-ethanol oxalate (20d)
Trimethylsilyl cyanide (0.270 g, 2.78 mmol) was added to a suspension of 2-benzyl-4-n-hexylbenzaldehyde (19d, 0.650 g, 2.32 mmol) and ZnI2 (catalytic amount) in anhydrous CH2Cl2 (3 mL). The solution was allowed to stir at room temperature (3 h) and heated at reflux (1 h). The solution was allowed to cool to room temperature and concentrated under reduced pressure to give a pale yellow oil. The oil in anhydrous THF (5 mL) was added under N2 to a suspension of LiAlH4 (0.260 g, 6.85 mmol) in anhydrous THF (10 mL) at 0 °C. The solution was warmed to room temperature, and heated at reflux (5 h). The solution allowed to cool to room temperature and water, (0.25 mL), 10% NaOH (0.25 mL) and Celite (1.5 g) were added. The suspension was filtered through a sintered glass filter and the filter cake was washed with CH2Cl2 (75 mL). The filtrate was dried (MgSO4) and concentrated under reduced pressure to provide (0.460 g) a colorless oil. The oil was dissolved in anhydrous acetone (20 mL) and oxalic acid (0.130 g, 1.38 mmol) was added. The solution was heated until the solid dissolved and the solution was cooled and filtered to provide 20d (0.420 g, 45%) as white crystals; mp 164–166 °C. 1H NMR (DMSO-d6) δ 0.85–0.87 (s, 2H, CH3), 1.28 (brs, 6H, CH2), 1.55–1.60 (brs, 2H, CH2), 2.48–2.53 (t, J=6 Hz, 2H, CH3), 2.69–2.78 (m, 2H, CH2-NH2), 3.91–3.96 (d, J=16 Hz, 1H, Ar-CH2-Ar), 4.00–4.06 (d, J=16 Hz, 1H, Ar-CH2-Ar), 5.03–5.06 (d, J=8.5 Hz, 1H, CH-OH), 6.97–7.44 (brm, 8H, Ar-H). 13C NMR (DMSO-d6) δ 14.67, 23.20, 29.60, 31.93, 32.27, 36.14, 38.79, 46.38, 67.06, 126.55, 126.91, 127.67, 129.01, 129.46, 131.11, 137.95, 136.09, 141.01, 143.45.
2-Amino-1-[2-(3-methoxybenzyl)phenyl]-1-ethanol oxalate (20e)
Compound 20e was prepared from 19e in a manner analogous to 20d. The oxalate salt was prepared and recrystallized from 2-PrOH to provide 20e (0.130 g, 33%) as a white powder; mp 159–161 °C. 1H NMR (CD3OD) δ 2.54–2.62 (m, 2H, CH2-NH2), 3.51 (s, 3H, OCH3), 3.86 (s, 2H, Ar-CH2-Ar), 4.93–4.98 (d, J=9 Hz, 1H, Ar-CH-OH), 6.47–6.54 (m, 2H, Ar-H), 6.94–7.10 (brm, 5H, Ar-H), 7.38–7.40 (m, 1H, Ar-H). 13C NMR (CD3OD) δ 39.65, 47.19, 56.11, 67.91, 113.02, 116.15, 122.61, 127.99, 128.75, 129.77, 131.13, 132.50, 139.18, 141.31, 144.10, 166.95.
2-Amino-1-{2-[3-(n-pentyloxy)benzyl]phenyl}-1-ethanol oxalate (20f)
Compound 20f was prepared from 19f in a manner analogous to that of 20d. The oxalate salt was prepared and recrystallized from 2-PrOH to provide 20f (2.68 g, 51%) as a white powder; mp 140–142 °C. 1H NMR (DMSO-d6) δ 0.85–0.90 (t, J=7 Hz, 3H, CH3), 1.13–1.18 (m, 2H, CH2), 1.28–1.34 (m, 2H, CH2), 2.7–2.84 (m, 2H, CH2-NH2), 3.83–3.92 (t, J= 7 Hz, 2H, CH2-O), 3.93–3.98 (d, J=16 Hz, 1H, Ar-CH2-Ar), 4.02–4.07 (s, J=16 Hz, 1H, Ar-CH2-Ar), 5.2–5.23 (d, J=8 Hz, 1H, CH-OH), 6.66–6.78 (m, 3H, Ar-H), 7.14–7.35 (brm, 4H, Ar-H), 7.53–7.67 (d, J=8 Hz, 1H, Ar-H).
2-Amino-1-[4-n-hexyl-2-(3-methoxybenzyl)phenyl]-1-ethanol fumarate (20h)
Compound 20h was prepared from 19h in a manner in a manner analogous to that of 20d. The fumarate salt was recrystallized from EtOAc/2-PrOH to provide 20h (0.510 g, 46%) as a white powder; mp 162–164 °C. 1H NMR (DMSO-d6) δ 0.81–0.85 (t, J=6 Hz, 3H, CH3), 1.24 (m, 6H, CH2), 1.49–1.53 (m, 2H, CH2), 2.64–2.83 (m, 2H, CH2-NH2), 3.69 (s, 3H, OCH3), 3.86–3.91 (d, J=16 Hz, 1H, Ar-CH2-Ar), 3.98–4.03 (d, J=16 Hz, 1H, Ar-CH2-Ar), 5.05–5.08 (d, J=10 Hz, 1H, CH-OH), 6.46 (s, 2H, Fumarate), 6.72–6.74 (m, 3H, Ar-H), 6.96 (s, 1H, Ar-H), 7.06–7.19 (brm, 2H, Ar-H), 7.41–7.43 (d, J=8 Hz, 1H, Ar-H). 13C NMR (DMSO-d6) δ 14.31, 22.43, 28.60, 31.25, 31.46, 35.10, 37.46, 45.97, 55.22, 66.21, 111.59, 114.80, 121.23, 126.54, 126.87, 129.66, 130.54, 135.68, 137.21, 138.04, 141.82, 142.62, 159.66, 168.84.
3-n-Pentyloxybenzaldehyde (21)
KOH (3.25 g, 49.1 mmol) was added to 3-hydroxybenzaldehyde (5.00 g, 40.9 mmol) in absolute EtOH (125 mL). The mixture was allowed to stir for 30 min at room temperature and 1-bromopentane (6.80 g, 45.0 mmol) was added. The reaction mixture was heated at reflux (12 h) and the suspension was allowed to cool to room temperature, water (75 mL) was added and the solution was extracted with Et2O (3 × 75 mL). The combined extracts were washed with water, brine, dried (MgSO4) and concentrated under reduced pressure to provide a pale orange oil. The oil was purified using medium pressure column chromatography (petroleum ether/acetone, 9:1) to provide 21 (5.82 g, 74%) as an orange oil. 1H NMR (CDCl3) δ 0.91–0.96 (t, J=7 Hz, 3H, CH3), 1.31–1.48 (m, 4H, CH2), 1.17–1.85 (m, 2H, CH2), 3.98–4.03 (t, J=6 Hz, 2H, CH2-O), 7.15–7.17 (m, 1H, Ar-H), 7.37–7.44 (brm, 3H, Ar-H), 9.96 (s, 1H, COH). 13C NMR (CDCl3) δ 14.56, 22.98, 28.71, 29.38, 68.86, 113.35, 122.50.123.82, 130.53, 138.34, 159.98, 192.75.
2-Benzyl-4-n-hexylbenzyl alcohol (22)
A 1.0 M solution of borane-THF complex (8 mL, 8 mmol) was added under N2 to a well stirred solution of 2-benzyl-4-n-hexylbenzoic acid (18d, 0.600 g, 2.00 mmol) at 0 °C. The solution was heated at reflux (5 h) and allowed to cool. HCl 6.0 M (5 mL) was added cautiously and the mixture was again heated at reflux (30 min). The solution was allowed to cool to room temperature, made basic with 15% NaOH (≈ 30 mL) and extracted with EtOAc (3 × 45 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide 22 (0.450 g, 79%) as a colorless oil. The product was used without further purification. 1H NMR (CDCl3) δ 0.85–0.89 (t, J=6 Hz, 2H, CH3), 1.28 (brs, 6H, CH2), 1.53–1.60 (m, 2H, CH2), 2.54–2.59 (t, J=8 Hz, 2H, CH3), 4.06 (s, 2H, CH2), 4.58 (s, 2H, CH2), 7.0–7.30 (brm, 8H, Ar-H). 13C NMR (CDCl3) δ 14.69, 23.20, 29.56, 32.03, 32.29, 36.19, 39.16, 63.72, 126.66, 127.37, 129.08, 129.17, 129.28, 131.48, 136.71, 138.52, 141.12, 143.42. IR (Film) 3334 cm−1.
2-(3-Methoxybenzyl)benzyl alcohol (23)
Compound 23 was prepared from 18e in a manner analogous to that of 22. The resulting oil was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 23 (0.540 g, 75%) as a colorless oil. 1H NMR (CDCl3) δ 3.76 (s, 3H, OCH3), 4.07 (s, 2H, Ar-CH2-OH), 4.65 (s, 2H, Ar-CH2-Ar), 6.7–6.76 (brm, 2H, Ar-H), 7.18–7.41 (brm, 6H, Ar-H). 13C NMR (CDCl3) 39.06, 55.70, 63.79, 111.87, 115.21, 121.66, 127.44, 128.57, 128.96, 130.08, 131.15, 138.94, 139.41, 142.77, 159.88. IR (Film) 3340 cm−1.
2-[3-(n-Pentyloxy)benzyl]benzyl alcohol (24)
Compound 24 was prepared from 18f in a manner analogous to that of 22. The resulting oil was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 24 (3.58 g, 100%) as a colorless oil. 1H NMR (CDCl3) δ 0.89–0.93 (t, J=7 Hz, 3H, CH3), 1.33–1.43 (m, 4H, CH2), 1.56 (brs, 1H, OH), 1.70–1.76 (m, 2H, CH2), 3.86–3.90 (t, J=7 Hz, 2H, CH2-O), 4.04 (s, 2H, Ar-CH2-Ar), 4.64 (s, 2H, CH2-OH), 6.68–6.71 (m, 2H, Ar-H), 6.74 (s, 1H, Ar-H), 7.14–7.28 (brm, 4H, Ar-H), 7.39–7.40 (m, 1H, Ar-H). 13C NMR (CDCl3) δ 14.59, 23.05, 28.77, 29.55, 39.11, 63.81, 68.42, 112.46, 115.76, 121.43, 127.41, 128.55, 128.95, 130.02, 131.17, 139.14, 139.24, 142.68, 159.51.
4-n-Hexyl-2-(3-methoxybenzyl)benzyl alcohol (25)
Compound 25 was prepared from 18h in a manner analogous to that of 22. The resulting oil was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 25 (2.21 g, 92%) as a colorless oil. 1H NMR (CDCl3) δ 0.85–0.89 (t, J=6 Hz, 3H, CH3), 1.26–1.29 (m, 6H, CH2), 1.53–1.60 (m, 2H, CH2), 2.54–2.59 (t, J=8 Hz, 2H, CH2-Ar), 3.76 (s, 3H, OCH3), 4.03 (s, 2H, Ar-CH2-Ar), 4.52 (s, 2H, CH2-OH), 6.68–6.74 (m, 3H, Ar-H), 7.00–7.30 (brm, 4H, Ar-H). 13C NMR (CDCl3) δ 14.62, 23.18, 29.54, 32.01, 32.28, 36.17, 39.16, 55.66, 63.75, 111.82, 115.10, 121.10, 127.41, 129.30, 130.02, 131.47, 136.70, 138.79, 143.03, 143.44. IR (Film) 3365 cm−1.
4-n-Hexyl-benzoylcyanide (26)
Trimethysilyl cyanide (0.62 mL, 4.67 mmol) was slowly added under N2 to a well stirred solution of 4-n-hexylbenzoyl chloride (1.0 g, 4.45 mmol) in anhydrous CH2Cl2 (10 mL). Tin(IV) chloride (0.10 mL, 0.850 mmol) was added over 30 min to the solution at room temperature and the mixture was allowed to stir for 2.5 h during which time the solution gradually turned from yellow to a dark brown. Ice cold water (30 mL) was added and the mixture was extracted with CH2Cl2 (3 × 30 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a brown oil. The oil was purified using medium pressure column chromatography (petroleum ether/acetone, 9.5:0.5) to provide 26 (0.820 g, 85%) as a pale yellow oil. 1H NMR (CDCl3) δ 0.86–0.88 (t, J= 8 Hz, 3H, CH3), 1.13 (s, 6H, CH2), 1.60–1.65 (m, 2H, CH2), 2.7–2.75 (t, J=7.8 Hz, 2H, Ar-CH2), 7.38–7.41 (d, J=8 Hz, 2H, Ar-H), 8.04–8.07 (d, J=8 Hz, 2H, Ar-H). 13C NMR (CDCl3) δ 14.59, 23.08, 29.43, 31.37, 32.14, 36.92, 130.17, 131.25, 154.24. IR (Film) 2221, 1687 cm−1.
2-(4-Bromophenyl)-2-phenylacetonitrile (27)
Bromine (0.82 g, 5.1 mmol) was slowly added over 1 h to 4-bromophenylacetonitrile (1.00 g, 5.10 mmol) at 110 °C (oil bath). The temperature was maintained between 105 °C and 110 °C for 30 min until the evolution of HBr had ceased. The solution was allowed to cool to room temperature and a steady stream of nitrogen was passed over the solution (30 min). The resulting yellow oil was dissolved in anhydrous benzene (1.20 g, 15.0 mmol) and AlCl3 (0.680 g, 5.10 mmol) was added. The solution was heated at reflux (3 h), cooled to room temperature and poured onto ice (25 g). The solution was made acidic to pH 2 with 5% HCl and extracted with Et2O (3 × 35 mL). The organic extracts were combined, washed with water (50 mL), sat. NaHCO3 (50 mL), brine (50 mL), dried (MgSO4) and concentrated under reduced pressure to provide a pale yellow semisolid. The semisolid was recrystallized from absolute EtOH to provide 27 (0.650 g, 46%) as pale yellow crystals; mp 80–82 °C. (Lit48 mp 79–81 °C). 1H NMR (CDCl3) δ 5.10 (s, 1H, CH), 7.21–7.53 (brm, 9H, Ar-H). IR (KBr) 2246 cm−1.
(4-n-Hexylphenyl)(phenyl)methanone (28)
AlCl3 (2.36 g, 17.8 mmol) was added slowly at 0 °C to 4-n-hexylbenzoyl chloride (2.00 g, 8.90 mmol) in anhydrous benzene (50 mL). The suspension was warmed to room temperature and heated at reflux (2 h). The solution was then cooled to room temperature and poured onto ice (30.0 g). The suspension was made acidic with 5% HCl (≈ 50 mL) and extracted with EtOAc (3 × 45 mL). The organic extracts were combined, washed with sat. NaHCO3 (50 mL), water (50 mL), brine (50 mL), dried (MgSO4) and concentrated under reduced pressure to provide 28 (2.00 g, 87%) as a pale yellow oil. The product was of sufficient purity to use in the next step. 1H NMR (CDCl3) δ 0.86–0.91 (t, J=7 Hz, 2H, CH3), 1.25 (s, 6H, CH2), 1.62–1.67 (m, 2H, CH2), 2.65–2.71 (t, J=8 Hz, 2H, CH2), 7.25–7.80 (brm, 9H, Ar-H). IR (Film) 1662 cm−1.
2-(4-n-Hexylphenyl)-2-hydroxy-2-phenylacetonitrile (29)
Trimethylsilyl cyanide (0.440 g, 4.51 mmol) was added to 28 (4-n-hexylphenyl)(phenyl)methanone (1.00 g, 3.76 mmol) and zinc iodide (cat. amount) in CH2Cl2 (5 mL). The suspension was heated at reflux (2 h), cooled to room temperature, and concentrated under reduced pressure to provide a pale yellow oil. The oil was dissolved in THF (5 mL) and 3 M HCl (3 mL) was added. The suspension was heated at reflux (1 h), cooled and water (100 mL) was added. The mixture was extracted with EtOAc (3 × 20 mL) and the organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a pale yellow oil that was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 29 (0.680 g, 62%) as a colorless oil. 1H NMR (CDCl3) δ 0.83–0.87 (t, J=8 Hz, 2H, CH3), 1.28 (s, 6H, CH2), 1.53–1.58 (m, 2H, CH2), 2.57–2.62 (t, J=8 Hz, 2H, CH2), 3.27 (brs, 1H, OH), 7.18–7.56 (brm, 9H, Ar-H). IR (Film) 3403 cm−1.
2-(4-n-Hexylphenyl)-2-phenylacetonitrile (30)
A NaBH4 pellet (1.00 g, 26.4 mmol) was added to a well stirred solution of 29 2-(4-n-hexylphenyl)-2-hydroxy-2-phenylacetonitrile (0.550 g, 1.88 mmol) in trifluoroacetic acid (15 mL). The viscous purple mixture was stirred at room temperature for 2 h and concentrated under reduced pressure. Water (40 mL) was added and the suspension was extracted with EtOAc (3 × 20 mL). The organic extracts were combined, dried (MgSO4) and concentrated under reduced pressure to provide a pale purple oil that was purified using medium pressure column chromatography (petroleum ether/acetone, 8:2) to provide 30 (0.350 g, 67%) as a colorless oil. 1H NMR (CDCl3) δ 0.85–0.89 (t, J=8 Hz, 2H, CH3), 1.28 (s, 6H, CH2), 1.52–1.59 (m, 2H, CH2), 2.55–2.60 (t, J=7.8 Hz, 2H, CH2), 5.20 (s, 1H, CH), 7.15–7.36 (brm, 9H, Ar-H). 13C NMR (CDCl3) δ 14.64, 23.14, 29.51, 31.86, 32.24, 36.10, 42.85, 128.15, 128.28, 128.70, 129.74, 129.75. IR (Film): 2246 cm−1.
Molecular Modeling
Molecular modeling investigations were conducted using the SYBYL molecular modeling package (version 7.1, 2005, Tripos Associates, St. Louis, MO) on MIPS R14K- and R16K-based IRIX 6.5 Silicon Graphics Fuel and Tezro workstations. Molecular mechanics-based energy minimizations were performed using the Tripos Force Field with Gasteiger-Hückel charges, a distance-dependent dielectric constant ε = 4 and a non-bonded interaction cutoff = 8 Å and were terminated at an energy gradient of 0.05 kcal/(mol·Å).
The primary sequences of the human dopamine D3 (P35462), human muscarinic cholinergic M1 (P11229), human vasopressin V1a (P37288), human adrenergic β2 (P07550), human δ-opioid (P41143), human 5-HT2A (P28223), human dopamine D2 (P14416) and bovine rhodopsin (P02699) receptors were retrieved from the ExPASy Proteomics Server (http://www.expasy.org/) at the Swiss Institute of Bioinformatics. An alignment profile consisting of the D3, M1, V1a, β2, δ-opioid and bovine rhodopsin receptors was created that reproduced the alignment of Bissantz, et al.79 An unambiguous (i.e. highly conserved residues previously identified80, 81 are aligned and no insertions or deletions in the helical regions) alignment of the 5-HT2A sequence with the aforementioned profile was performed using the ClustalX82 program. Within ClustalX, the slow-accurate alignment algorithm was used, the BLOSUM matrix series83 was employed and the gap opening penalty was increased from 10.0 to 15.0 to help maintain the continuity of the transmembrane helical segments. As in Bissantz, et al.,79 the alignment was carried out in two separate steps: the first alignment included all residues from the N-terminus to the i3 loop and the second alignment included all residues from the i3 loop to the C-terminus. Manual adjustment of the ClustalX alignment was also required to properly align the disulfide-forming cysteine residues in the e2 loop. The D2 sequence was subsequently aligned with that of 5-HT2A to identify loci within the binding sites of the two receptors where cognate amino acids differ and to identify those that may be responsible for 5-HT2A/D2 selectivity. The resulting alignment is presented in Figure 3.
The alignment described above was used as the basis for subsequent homology modeling of the 5-HT2A receptor. In light of the growing evidence35, 79, 84–91 that the binding of agonists versus antagonists may be more effectively modeled using at least two distinct static receptor models rather than a single static model, and our own observation that the agonist/antagonist properties of the 1-(2,5-dimethoxyphenyl)-2-aminopropane derivatives may be modulated by the nature of the substituent at the 4-position of the phenyl ring,15 separate agonist and antagonist 5-HT2A models were generated. The automodel routine in the MODELLER 8v1 software package92, 93 was thus used to generate an initial population of 100 5-HT2A receptor models. These models were constructed based on homology to bovine rhodopsin (chain A of PDB entry = 1U19), whose coordinates were downloaded from the RCSB Protein Data Bank (http://www.rcsb.org).
To maximize the variation in the sidechain conformations of the MODELLER-generated receptors, the residues falling within 12.0 Å of the retinal ligand in 1U19 were mutated to alanine. These residues are highlighted with a green background in Figure 3. This approach, which has been used to successfully model α2-adrenergic receptors,94 allows MODELLER to more fully explore sidechain conformational space, since the sidechains may then be placed onto the backbone without the added constraints imposed by the existing rhodopsin sidechains. This also ameliorates the problem of retinal leaving behind a “ghost site” in the newly-created 5-HT2A receptors.95
In addition to mutating the residues lining the 1U19 binding cavity to alanine, two other important aspects relating to the transfer of 3-D information from the 1U19 template to the 5-HT2A models (particularly in the less well-conserved regions) were specifically addressed. First, instead of attempting to accurately model the i3 loop of 5-HT2A (which is 40 residues longer than rhodopsin’s), the backbone of the i3 loop of rhodopsin was transferred to the 5-HT2A models and the residues therein mutated to glycine. These residues are highlighted with a purple background in Figure 3. This effectively created a tether that would keep the helices in place while having a minimal impact on the remaining portion of the receptor during minimizations and/or dynamics simulations. Pogozheva, Lomize and Mosberg96 have used a similar technique to build opioid receptors in which a contiguous primary amino acid sequence for the receptor was necessary. Second, since the N- and C-terminal domains of bovine rhodopsin and 5-HT2A are very dissimilar in terms of both sequence homology and length, and since these domains are not believed to be important for the binding of small molecules97, 98, these features were not included in the 5-HT2A models.
Each of the initial 100 5-HT2A models thus generated was energy-minimized as described above with a maximum of 100,000 iterations and with no constraints. The process of then selecting an agonist and an antagonist model from the receptor population was facilitated by the use of the automated docking program GOLD (version 3.0.1).99, 100 A high-potency agonist (DOB, 2b, Ki = 41 nM) and an antagonist (ketanserin, Ki = 0.4 nM) were selected as reference ligands and each was docked into all 100 receptors using the GOLD program. For DOB, separate docking runs were performed for each explicitly represented stereoisomer ((R)- and (S)- forms of 2b = 2 isomers) since each isomer shows low nanomolar binding affinity at the 5-HT2A receptor. Standard default settings were used (no speed-up), early termination was disabled, and 10 genetic algorithm (GA) runs were performed for each ligand. The ChemScore101 fitness function was used, and the binding site was defined to include all residues within a 15.0-Å radius of the D1553.32 Cγ carbon atom. A docking constraint was also enforced that biased the docking results toward solutions in which the ligand was hydrogen bonded to the conserved D1553.32 sidechain. When these initial docking runs were complete, the lists of ChemScores for each isomer were tabulated (each reference ligand isomer was docked into each of the 100 MODELLER-generated receptors) and sorted best to worst (highest to lowest for ChemScore in GOLD). The top-scoring complexes for each reference ligand are summarized in Supplemental Table 4.
The top-scoring receptor-ligand complexes for each of the reference compounds were then inspected visually to ensure that the receptor formed a chemically intuitive complex with the ligand and that the receptor-ligand complex could account for the observed point mutation data. High-scoring complexes that did not meet this requirement were discarded. Ligand-receptor complexes were discarded for a variety of reasons, including 1) the docked solution for DOB did not accept a hydrogen bond from the receptor at either the 2- or 5-position (rationale: methoxy groups are necessary for high affinity of the smaller 4-substituted phenylethylamines [Ki = 1770 nM, 3b; Ki = 41 nM, 2b]), 2) the docked solution for DOB did not interact with F340 (rationale: DOB most likely binds in the receptor site in the same manner as does DOI (Supplemental Figure 2), which loses all affinity for the F340A mutant (Table 2)), 3) the docked solution for ketanserin could not explain the effects of mutagenesis at F340, Y370 and/or W76 (Table 3), and 4) the e2 loop of the receptor was incorrectly modeled (rationale: MODELLER occasionally placed the e2 loop segment that joins the top of TM4 to the disulfide bond farther down in the receptor site than the segment joining the disulfide bond to the top of TM5 — the reverse of what is seen in the bovine rhodopsin crystal structure). A complete listing of the twelve top-scoring receptor-reference ligand complexes and comments on their binding modes is presented in Supplemental Table 4.
The ChemScores for the 100 top complexes covered a wide range (R(−)-DOB, 1.41 to 33.52; S(+)-DOB, 3.06 to 30.71; ketanserin, −31.84 to 35.21), a result of the receptor population containing members that exhibited both very high and very low degrees of complementarity to the docked ligands. The top-scoring receptors for a given ligand tended to have similar high scores (Supplemental Table 4), but the corresponding binding modes of the docked ligands were very different. It was thus necessary to use additional information from receptor mutagenesis and ligand SAR to eliminate from consideration those that were not consistent with this data and to select the most appropriate receptor models (vide supra). Of course, reconciliation of relevant experimentally-derived data with molecular models is a necessary part of any modeling study. Seven of the top twelve receptor models are common to both R(−)-DOB and S(+)-DOB. Interestingly, five of the top twelve receptor models for ketanserin are also found in the top receptor lists of either R(−)-DOB or S(+)-DOB. It should also be noted that for a given receptor-ligand model, the set of docked solutions from the ten GA runs tended to be qualitatively similar, differing only slightly in the ligands’ relative position and orientation within the binding site. For each of the reference ligands, then, the final receptor model chosen was the highest scoring chemically intuitive complex. For DOB, the same receptor model was coincidentally chosen for each of the two stereoisomers. In this selected agonist model, the DOB isomers docked in a similar fashion (Supplemental Figure 2).
The agonist and antagonist 5-HT2A models were subsequently analyzed using PROCHECK102 and the ProTable facility with in SYBYL to assess the geometric integrity of various structural elements (bond lengths, torsion angles, etc.) within each receptor. Unusual and unfavorable geometries were interactively corrected as necessary. There were two such regions in both the agonist and antagonist receptors. The first involved the region of TM7 proximal to position 7.43 where the retinal chromophore is covalently bound in rhodopsin. Backbone geometries in this region were visibly non-optimal (kinked), so the residues in the region V3647.37 to A3747.47 were assigned ideal alpha helix coordinates using SYBYL 7.1. A second visibly distorted region at location L2365.40 to V2415.45 was refined in a similar way. The modified receptors were then energy-minimized as described for the initial 5-HT2A receptors prior to docking. The final models are depicted together with the A chain of rhodopsin (1U19) in Supplemental Figure 1.
Ligand molecules were sketched manually using SYBYL 7.1 (Tripos, Inc., St. Louis, MO) and assigned three-dimensional coordinates using the CONCORD (v.6.1.0) facility within SYBYL. Since the synthesized and tested compounds are racemic mixtures in most if not all cases, R/S isomers were explicitly represented for each ligand where necessary. GOLD was used to dock the ligands into the agonist and antagonist 5-HT2A models in an automated fashion under the same conditions as described above for the receptor selection phase. Ligand preference for a particular receptor was determined by the difference in the ChemScore fitness function values for the agonist and antagonist receptor solutions (Supplemental Table 2). However, there was one exception: AMDA (1a), which was predicted to have a slight preference for the agonist receptor, was associated with the antagonist model on the strength of the F3406.52L mutation data, as described earlier. Final docked ligand-receptor complexes were energy-minimized as described above for the initial 5-HT2A models. The minimized complexes were further subjected to a short molecular dynamics simulation (Tripos Force Field, Gasteiger-Hückel charges, distance-dependent dielectric = 4.0, fixed aggregate = all residues > 8.0 Å from the GOLD-docked solution, 100 ps simulation time, 300K) to provide further evidence of the veracity of the docked solutions.
Affinity Determinations
Binding assays and data analysis were performed as previously described using [3H]ketanserin as the radioligand and stably transfected NIH3T3 cells expressing the 5-HT2A receptor (GF-62 cells).103 The F339L and F340L mutants were prepared and assayed as previously described.104
Supplementary Material
Acknowledgments
This work was supported by United States Public Health Service Grant MH57969 (RBW), R01MH61887, U19MH082441 (BLR) and the NIMH Psychoactive Drug Screening Program (BLR).
Footnotes
Abbreviations: AMDA, 9-aminomethyl-9,10-dihydroanthracene; DOB, 2,5-dimethoxy-4-bromoamphetamine; DOI, 2,5-dimethoxy-4-iodoamphetamine; 5-HT, serotonin; LSD, lysergic acid diethylamide; GPCR, G protein-coupled receptor; 5-HT2A, serotonin receptor subtype 2A; D2, dopamine receptor subtype 2; SERT, serotonin transporter; NET, norepinephrine transporter; TM, transmembrane; PDB, Protein Data Bank.
Supporting Information Available: Supplemental tables and figures are available free of charge via the internet at http://pubs.acs.org.
References
- 1.Roth BL. Multiple Serotonin Receptors: Clinical and Experimental Aspects. Ann Clin Psych. 1994;6:67–78. doi: 10.3109/10401239409148985. [DOI] [PubMed] [Google Scholar]
- 2.Kroeze WK, Kristiansen K, Roth BL. Molecular Biology of Serotonin Receptors — Structure and Function at the Molecaular Level. Curr Top Med Chem. 2002;2:507–528. doi: 10.2174/1568026023393796. [DOI] [PubMed] [Google Scholar]
- 3.Glennon RA, Dukat M, Westkaemper RB. Serotonin Receptors and Ligands. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: The Fourth Generation of Progress. Raven Press; New York: 1999. [Google Scholar]
- 4.Glennon RA, Dukat M. Novel Serotonergic Agents: 5-HT2 - Update 1997. Serotonin ID Resesarch Alert. 1997;2:107–113. [Google Scholar]
- 5.Runyon SP, Peddi S, Savage JE, Roth BL, Glennon RA, Westkaemper RB. Geometry-Affinity Relationships of the Selective Serotonin Receptor Ligand 9-(Aminomethyl)-9,10-dihydroanthracene. J Med Chem. 2002;45:1656–1664. doi: 10.1021/jm010354g. [DOI] [PubMed] [Google Scholar]
- 6.Runyon SP, Savage JE, Taroua M, Roth BL, Glennon RA, Westkaemper RB. Influence of Chain Length and N-Alkylation on the Selective Serotonin Receptor Ligand 9-(Aminomethyl)-9,10-dihydroanthracene. Bioorg Med Chem Lett. 2001;11:655–658. doi: 10.1016/s0960-894x(01)00023-3. [DOI] [PubMed] [Google Scholar]
- 7.Westkaemper RB, Glennon RA. Application of Ligand SAR, Receptor Modeling and Receptor Mutagenesis to the Discovery and Development of a New Class of 5-HT2A Ligands. Curr Topics Med Chem. 2002;2:575–598. doi: 10.2174/1568026023393741. [DOI] [PubMed] [Google Scholar]
- 8.Westkaemper RB, Runyon SP, Bondarev ML, Savage JE, Roth BL, Glennon RA. 9-(Aminomethyl)-9,10-dihydroanthracene is a Novel and Unlikely 5-HT2A Receptor Antagonist. Eur J Pharmacol. 1999;380:R5–R7. doi: 10.1016/s0014-2999(99)00525-7. [DOI] [PubMed] [Google Scholar]
- 9.Westkaemper RB, Runyon SP, Savage JE, Roth BL, Glennon RA. Exploring the Relationship Between Binding Modes of 9-(Aminomethyl)-9,10-dihydroanthracene and Cyproheptadine Analogues at the 5-HT2A Serotonin Receptor. Bioorg Med Chem Lett. 2001;11:563–566. doi: 10.1016/s0960-894x(01)00010-5. [DOI] [PubMed] [Google Scholar]
- 10.Rabideau P. The Conformational Analysis of 1,4-cyclohexadienes: 1,4-dihydrobenzenes, 1-4-dihydronaphthalenes and 9,10-dihydroanthracenes. Acc Chem Res. 1978;11:141–147. [Google Scholar]
- 11.Glennon RA, Liebowitz SM, Anderson GMI. Serotonin Receptor Affinities of Psychoactive Phenylalkylamine Derivatives. J Med Chem. 1980;23:294–299. doi: 10.1021/jm00177a017. [DOI] [PubMed] [Google Scholar]
- 12.Glennon RA, Young R, Benington F, Morin RD. Behavioral and Serotonin Receptor Properties of 4-Substituted Derivatives of the Hallucinogen 1-(2,5-Dimethoxy)-2-aminopropane. J Med Chem. 1982;25:1163–1168. doi: 10.1021/jm00352a013. [DOI] [PubMed] [Google Scholar]
- 13.Seggel MR, Yousif MY, Lyon RA, Titeler M, Roth BL, Suba EA, Glennon RA. A Structure-Affinity Study of the Binding of 4-Substituted Analogues of 1-(2,5-Dimethoxyphenyl)-2-aminopropane at 5-HT2 Serotonin Receptors. J Med Chem. 1990;33:1032–1036. doi: 10.1021/jm00165a023. [DOI] [PubMed] [Google Scholar]
- 14.Kornfeld EC. Raney Nickel Hydrogenolysis of Thioamides: A New Amine Synthesis. J Org Chem. 1951;16:131–138. [Google Scholar]
- 15.Dowd CS, Herrick-Davis K, Egan C, DuPre A, Smith C, Teitler M, Glennon RA. 1-[4-(3-Phenylalkyl)phenyl]-2-aminopropanes as 5-HT2A Partial Agonists. J Med Chem. 2000;43:3074–3084. doi: 10.1021/jm9906062. [DOI] [PubMed] [Google Scholar]
- 16.Pine SH, Shen GS, Hoang H. Ketone Methylenation Using the Tebbe and Wittig Reagents - A Comparison. Synthesis. 1991;1991:165–166. [Google Scholar]
- 17.Snyder SE, Avilez-Garay FA, Chakraborti R, Nichols DE, Watts VJ, Mailman RB. Synthesis and Evaluation of 6,7-Dihydroxy-2,3,4,8,9,13b-hexahydro-1H-benzo[6,7]cyclohepta[1,2,3-ef][3]benzazepine, 6,7-Dihydroxy-1,2,3,4,8,12b-hexahydroanthr[10,4a,4-cd]azepine, and 10-Aminomethyl-9,10-dihydro-1,2-dihydroxyanthracene as Conformationally Restricted Analogs of β-Phenyldopamine. J Med Chem. 1995;38:2395–2409. doi: 10.1021/jm00013a015. [DOI] [PubMed] [Google Scholar]
- 18.Ashwood MS, Bell LA, Houghton PG, Wright SHB. Synthesis of 1,1,-Diaryl-2,2-dimethoxyethanes. Synthesis. 1988;1988:379–381. [Google Scholar]
- 19.Bellamy FD, Ou K. Selective Reduction of Aromatic Nitro Compounds with Stannous Chloride in Non Acidic and Non Aqueous Medium. Tetrahedron Lett. 1984;25:839–842. [Google Scholar]
- 20.Meek JS, Dann JR, Poon BT. Diels-Alder Reactions of 9-Substituted Anthracenes. II. 9-Cyanoanthracene. J Am Chem Soc. 1956;78:5413–5416. [Google Scholar]
- 21.Miyaura N, Ishiyama T, Sasaki H, Ishikawa M, Sato M, Suzuki A. Palladium-Catalyzed Inter- and Intramolecular Cross-Coupling Reactions of B-Alkyl-9-borabycyclo[3.3.1]nonane Derivatives with 1-Halo-1-alkenes or Haloarenes. J Am Chem Soc. 1989;111:314–321. [Google Scholar]
- 22.Olah GA, Arvanaghi M, Surya Prakash GK. Tin(IV) Chloride-Catalyzed Preparation of Aroyl Cyanides from Aroyl Chlorides and Cyanotrimethylsilane. Synthesis. 1983;1983:636–637. [Google Scholar]
- 23.Kindler K, Hedemann B, Schärfe E. Studien über den Mechanismus Chemischer Reaktionen. X. Phenyl- and Cyclohexyl-alkylamine durch Hydrierung. Justus Liebigs Ann Chem. 1948;560:215–222. [Google Scholar]
- 24.Robb CM, Schultz EM. Diphenylacetonitrile. Org Syn Coll. 1955;3:347–349. [Google Scholar]
- 25.Gribble GW, Leese RM, Evans BE. Reactions of Sodium Borohydride in Acidic Media; IV. Reduction of Diarylmethanols and Triarylmethanols in Trifluoroacitic Acid. Synthesis. 1977;1977:172–176. [Google Scholar]
- 26.Jacoby E, Fauchère JL, Raimbaud E, Ollivier S, Michel A, Spedding M. A Three Binding Site Hypothesis for the Interaction of Ligands with Monoamine G Protein-coupled Receptors: Implications for Combinatorial Ligand Design. Quant Struct-Act Relat. 1999;18:561–572. [Google Scholar]
- 27.Surgand JS, Rodrigo J, Kellenberger E, Rognan D. A Chemogenomic Analysis of the Transmembrane Binding Cavity of Human G-Protein-Coupled Receptors. Proteins. 2006;62:509–538. doi: 10.1002/prot.20768. [DOI] [PubMed] [Google Scholar]
- 28.Glennon RA, Westkaemper RB. Serotonin Receptors, 5-HT Ligands and Receptor Modeling. In: Angeli P, Gulini U, Quaglia W, editors. Trends in Receptor Research. Elsevier; Amsterdam: 1992. pp. 185–207. [Google Scholar]
- 29.Westkaemper RB, Glennon RA. Molecular Graphics Models of the 5-HT2 Subfamily: 5-HT2A, 5-HT2B, 5-HT2C Receptors. Med Chem Res. 1993;3:317–334. [Google Scholar]
- 30.Westkaemper RB, Hyde EG, Choudhary MS, Khan N, Gelbar EI, Glennon RA, Roth BL. Engineering a Region of Bulk Tolerance in the 5-HT2A Receptor. Eur J Med Chem. 1999;34:441–447. [Google Scholar]
- 31.Kuipers W, van Wijngaarden I, Ijzerman AP. A Model of the Serotonin 5-HT1A Receptor: Agonist and Antagonist Binding Sites. Drug Des Discov. 1994;11:231–249. [PubMed] [Google Scholar]
- 32.Teodoro ML, Kavraki LE. Conformational Flexibility Models for the Receptor in Structure Based Drug Design. Curr Pharm Des. 2003;9:1635–1648. doi: 10.2174/1381612033454595. [DOI] [PubMed] [Google Scholar]
- 33.Unless specified otherwise, the receptor sequence number for a particular amino acid residue is listed first, followed by its Ballesteros-Weinstein110 residue identifier as a superscript. Following the convention of Xhaard et al.,94 the positions of the residues in the e2 (xl2) loop are given relative to the cysteine residue that forms the disulfide bond with position 3.25 of TM3, and is assigned the identifier xl2.50. Thus, this reference residue is referred to as C227xl2.50 in the 5-HT2A receptor.
- 34.Salom D, Lodowsky DT, Stenkamp RE, Le Trong I, Golczak M, Jastrzebska B, Harris T, Ballesteros JA, Palczewski K. Crystal Structure of a Photoactivated Deprotonated Intermediate of Rhodopsin. Proc Nat Acad Sci USA. 2006;103:16123–16128. doi: 10.1073/pnas.0608022103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. β2 Adrenergic Receptor Activation: Modulation of the Proline Kink in Transmembrane 6 by a Rotamer Toggle Switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- 36.Ariëns EJ. A General Introduction. In: Ariëns EJ, editor. Drug Design. Academic Press; New York: 1971. pp. 173–192. [Google Scholar]
- 37.Glennon RA, Westkaemper RB, Bartyzel P. Medicinal Chemistry of Serotonergic Agents. In: Peroutka SJ, editor. Serotonin Receptor Subtypes. Wiley-Liss; New York: 1991. pp. 19–64. [Google Scholar]
- 38.Stevenson GI, Smith AL, Lewis S, Michie SG, Neduvelil JG, Patel S, Marwood R, Patel S, Castro JL. 2-Aryl Tryptamines: Selective High-Affinity Antagonists for the h5-HT2A Receptor. Bioorg Med Chem Lett. 2000;10:2697–2699. doi: 10.1016/s0960-894x(00)00557-6. [DOI] [PubMed] [Google Scholar]
- 39.Bennett JP, Snyder SH. Stereospecific Binding of D-Lysergic Acid Diethylamide (LSD) to Brain Membranes: Relationship to Serotonin Receptors. Brain Res. 1975;94:523–544. doi: 10.1016/0006-8993(75)90234-6. [DOI] [PubMed] [Google Scholar]
- 40.Glennon RA, Dukat M, el-Burmawy M, Law H, de los Angeles J, Teitler M, King A, Herrick-Davis K. Influence of Amine Substituents on 5-HT2A Versus 5-HT2C Binding of Phenylalkyl- and Indolylalkylamines. J Med Chem. 1994;37:1929–1935. doi: 10.1021/jm00039a004. [DOI] [PubMed] [Google Scholar]
- 41.Roth BL, Berry SA, Kroeze WK, Willins DL, Kristiansen K. Serotonin 5-HT2A Receptors: Molecular Biology and Mechanisms of Regulation. Crit Rev Neurobiol. 1998;12:319–338. doi: 10.1615/critrevneurobiol.v12.i4.30. [DOI] [PubMed] [Google Scholar]
- 42.Parrish JC, Braden MR, Gundy E, Nichols DE. Differential Phospholipase C Activation by Phenylalkylamine Serotonin 5-HT2A Receptor Agonists. J Neurochem. 2005;95:1575–1584. doi: 10.1111/j.1471-4159.2005.03477.x. [DOI] [PubMed] [Google Scholar]
- 43.Süel G, Lockless SW, Wall MA, Ranganathan R. Evolutionarily Conserved Networks of Residues Mediate Allosteric Communication in Proteins. Nature Stuct Biol. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- 44.Roth BL, Shoham M, Choudhary MS, Khan N. Identification of Conserved Aromatic Residues Essential for Agonist Binding and Second Messenger Production at 5-Hydroxytryptamine2A Receptors. Mol Pharmacol. 1997;52:259–266. doi: 10.1124/mol.52.2.259. [DOI] [PubMed] [Google Scholar]
- 45.Almaula N, Ebersole BJ, Zhang D, Weinstein H, Sealfon SC. Mapping the Binding Site Pocket of the Serotonin 5-Hydroxytryptamine2A Receptor. Ser3.36(159) Provides a Second Interaction Site for the Protonated Amine of Serotonin but not of Lysergic Acid Diethylamide or Bufotenin. J Biol Chem. 1996;271:14672–14675. doi: 10.1074/jbc.271.25.14672. [DOI] [PubMed] [Google Scholar]
- 46.Johnson MP, Wainscott DB, Lucaites VL, Baez M, Nelson DL. Mutations of Transmembrane IV and V Serines Indicate that All Tryptamines Do Not Bind to the Rat 5-HT2A Receptor in the Same Manner. Mol Brain Res. 1997;49:1–6. doi: 10.1016/s0169-328x(97)00115-0. [DOI] [PubMed] [Google Scholar]
- 47.Shapiro DA, Kristiansen K, Kroeze WK, Roth BL. Differential Modes of Agonist Binding to 5-Hydroxytryptamine2A Serotonin Receptors Revealed by Mutation and Molecular Modeling of Conserved Residues in Transmembrane Region 5. Mol Pharmacol. 2000;58:877–886. doi: 10.1124/mol.58.5.877. [DOI] [PubMed] [Google Scholar]
- 48.Grånäs C, Nordvall G, Larhammar D. Site-directed Mutagenesis of the Human 5-HT1B Receptor. Eur J Med Chem. 1998;349:367–375. doi: 10.1016/s0014-2999(98)00213-1. [DOI] [PubMed] [Google Scholar]
- 49.Hwa J, Graham RM, Perez DM. Chimeras of α1-Adrenergic Receptor Subtypes Identify Critical Residues that Modulate Active State Isomerization. J Biol Chem. 1996;271:7956–7964. doi: 10.1074/jbc.271.14.7956. [DOI] [PubMed] [Google Scholar]
- 50.Lundstrom K, Turpin MP, Large C, Robertson G, Thomas P, Lewell XQ. Mapping of Dopamine D3 Receptor Binding Site by Pharmacological Characterization of Mutants Expressed in CHO Cells with the Semliki Forest Virus System. J Recept Signal Transduct Res. 1998;18:133–150. doi: 10.3109/10799899809047741. [DOI] [PubMed] [Google Scholar]
- 51.Weiland K, ter Laak AM, Smit MJ, Kühne R, Timmerman H, Leurs R. Mutational Analysis of the Antagonist-binding Site of the Histamine H1 Receptor. J Biol Chem. 1999;274:29994–30000. doi: 10.1074/jbc.274.42.29994. [DOI] [PubMed] [Google Scholar]
- 52.Weiland K, Zuurmond HM, Krasel C, Ijzerman AP, Lohse MJ. Involvement of Asn-293 in Stereospecific Agonist Recognition and in Activation of the β2-Adrenergic Receptor. Proc Nat Acad Sci USA. 1996;93:9276–9281. doi: 10.1073/pnas.93.17.9276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zuurmond HM, Hessling J, Blüml K, Lohse MJ, Ijzerman AP. Study of Interaction between Agonists and Asn293 in Helix VI of Human β2-Adrenergic Receptor. Mol Pharmacol. 1999;56:909–916. doi: 10.1124/mol.56.5.909. [DOI] [PubMed] [Google Scholar]
- 54.Javitch JA, Ballesteros JA, Weinstein H, Chen J. A Cluster of Aromatic Residues in the Sixth Membrane-Spanning Segment of the Dopamine D2 Receptor is Accessible in the Binding-Site Crevice. Biochemistry. 1998;37:998–1006. doi: 10.1021/bi972241y. [DOI] [PubMed] [Google Scholar]
- 55.Yao X, Parnot C, Deupi X, Ratnala VRP, Swaminath G, Farrens D, Kobilka B. Coupling Ligand Structure to Specific Confromational Switches in the β2-Adrenoceptor. Nature Chem Biol. 2006;2:417–422. doi: 10.1038/nchembio801. [DOI] [PubMed] [Google Scholar]
- 56.Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SGF, Shi L, Gether U, Javitch JA. Activation of the β2-Adrenergic Receptor Involves Disruption of an Ionic Lock between the Cytoplasmic Ends of Transmembrane Segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- 57.Perola E, Charifson PS. Conformational Analysis of Drug-Like Molecules Bound to Proteins: An Extensive Study of Ligand Reorganization upon Binding. J Med Chem. 2004;47:2499–2510. doi: 10.1021/jm030563w. [DOI] [PubMed] [Google Scholar]
- 58.Stockwell GR, Thornton JM. Conformational Diversity of Ligands Bound to Proteins. J Mol Biol. 2006;356:928–944. doi: 10.1016/j.jmb.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 59.Glennon RA, Metwally K, Dukat M, Ismael AM, De Los Angeles J, Herndon J, Teitler M, Khorana N. Ketanserin and Spiperone as Templates for Novel Serotonin 5-HT2A Antagonists. Curr Topics Med Chem. 2002;2:539–558. doi: 10.2174/1568026023393787. [DOI] [PubMed] [Google Scholar]
- 60.Squillacote M, SSR, Chapman OL, Anet FAL. Spectroscopic Detection of the Twist-Boat Comformation of Cyclohexane. A Direct Measurement of the Free Energy Difference between the Chair and the Twist-Boat. J Am Chem Soc. 1975;97:3244–3246. [Google Scholar]
- 61.Bostrom J, Norrby PO, Liljefors T. Conformational Energy Penalties of Protein-Bound Ligands. J Comp-Aided Mol Des. 1998;12:383–396. doi: 10.1023/a:1008007507641. [DOI] [PubMed] [Google Scholar]
- 62.Nicklaus MC, Wang S, Driscoll JS, Milne GW. Conformational Changes of Small Molecules Binding to Proteins. Bioorg Med Chem. 1995;3:411–428. doi: 10.1016/0968-0896(95)00031-b. [DOI] [PubMed] [Google Scholar]
- 63.Dezi C, Brea J, Alvarado M, Raviña E, Masaguer CF, Loza MI, Sanz F, Pastor M. Multistructure 3D-QSAR Studies on a Series of Conformationally Constrained Butyrophenones Docked into a New Homolgy Model of the 5-HT2A Receptor. J Med Chem. 2007;50:3242–3255. doi: 10.1021/jm070277a. [DOI] [PubMed] [Google Scholar]
- 64.Schetz JA, Benjamin PS, Sibley DR. Nonconserved Residues in the Second Transmembrane-spanning Domain of the D4 Dopamine Receptor Are Molcular Determinants of D4-Selective Pharmacology. Mol Pharmacol. 2000;57:144–152. [PubMed] [Google Scholar]
- 65.Simpson MM, Ballesteros JA, Chiappa V, Chen J, Suehiro M, Hartman DS, Godel T, Snyder LA, Sakmar TP, Javitch JA. Dopamine D4/D2 Receptor Selectivity is Determined by a Divergent Aromatic Microdomain Contained Within the Second, Third, and Sevcenth Membrane-spanning Segments. Mol Pharmacol. 1999;56:1116–1126. doi: 10.1124/mol.56.6.1116. [DOI] [PubMed] [Google Scholar]
- 66.Boess FG, Monsma FJJ, Sleight AJ. Identification of Residues in Transmembrane Regions III and VI that Contribute to the Ligand Binding Site of the Serotonin 5-HT6 Receptor. J Neurochem. 1998;71:2169–2177. doi: 10.1046/j.1471-4159.1998.71052169.x. [DOI] [PubMed] [Google Scholar]
- 67.Lu ZL, Hulme EC. The Funcional Topography of Transmembrane Domain 3 of the M1 Muscarinic Acetylcholine Receptor, Revealed by Scaning Mutagenesis. J Biol Chem. 1999;274:7309–7315. doi: 10.1074/jbc.274.11.7309. [DOI] [PubMed] [Google Scholar]
- 68.Matsui H, Lazareno S, Birdsall NJM. Probing the Location of the Allosteric Site on M1 Muscarinic Receptors by Site-directed Mutagenesis. Mol Pharmacol. 1995;47:88–98. [PubMed] [Google Scholar]
- 69.Daniell SJ, Strange PG, Naylor LH. Site-directed Mutagenesis of Tyr417 in the Rat D2 Dopamine Receptor. Biochem Soc Trans. 1994;22:144S. doi: 10.1042/bst022144s. [DOI] [PubMed] [Google Scholar]
- 70.Fu D, Ballesteros JA, Weinstein H, Chen J, Javitch JA. Residues in the Seventh Membrane-spanning Segment of the Dopamine D2 Receptor Accessible in the Binding-site Crevice. Biochemistry. 1996;35:11278–11285. doi: 10.1021/bi960928x. [DOI] [PubMed] [Google Scholar]
- 71.Rezmann-Vitti LA, Nero TL, Jackman GP, Machida CA, Duke BJ, Louis WJ, Louis SNS. Role of Tyr356(7.43) and Ser190(4.57) in Antagonist Binding in the Rat β1-Adrenergic Receptor. J Med Chem. 2006;49:3467–3577. doi: 10.1021/jm050624l. [DOI] [PubMed] [Google Scholar]
- 72.Adham N, Tamm JA, Salon JA, Vaysse PJJ, Weinshank RL, Branchek TA. A Single Point Mutation Increases the Affinity of Serotonin 5-HT1Dα, 5-HT1Dβ, 5-HT1E and 5-HT1F Receptors for β-adrenergic Antagonists. Neuropharmacology. 1994;33:387–391. doi: 10.1016/0028-3908(94)90068-x. [DOI] [PubMed] [Google Scholar]
- 73.Glennon RA, Dukat M, Westkaemper RB, Ismaiel AM, Izzarelli DG, Parker EM. The Binding of Propanolol at 5-Hydroxytryptamine1Dβ T355N Mutant Receptors May Involve Formation of Two Hydrogen Bonds to Asparagine. Mol Pharmacol. 1996;49:198–206. [PubMed] [Google Scholar]
- 74.Guan XM, Peroutka SJ, Koblika BK. Identification of a Single Amino Acid Residue Responsible for the Binding of a Class of β-Adrenergic Receptor Antagonists to 5-Hydroxytryptamine1A Receptors. Mol Pharmacol. 1992;41:695–698. [PubMed] [Google Scholar]
- 75.Kuipers W, Link R, Standaar PJ, Stoit AR, van Wijngaarden I, Leurs R, Ijzerman AP. Study of the Interaction Between Aryloxypropanolamines and Asn386 in Helix VII of the Human 5-Hydroxytryptamine1A Receptor. Mol Pharmacol. 1997;51:889–896. doi: 10.1124/mol.51.5.889. [DOI] [PubMed] [Google Scholar]
- 76.Oksenberg D, Marsters SA, O’Dowd BF, Jin H, Havlik S, Peroutka SJ, Ashkenazi A. A Single Amino Acid Difference Confers Major Pharmacological Variation Between Human and Rodent 5-HT1B Receptors. Nature. 1992;360:161–163. doi: 10.1038/360161a0. [DOI] [PubMed] [Google Scholar]
- 77.Suryanarayana S, Daunt DA, von Zastrow M, Koblika BK. A Point Mutation in the Seventh Hydrophobic Domain of the α2 Adrenergic Receptor Increases Its Affinity for a Family of β Receptor Antagonists. J Biol Chem. 1991;266:15488–15492. [PubMed] [Google Scholar]
- 78.Suryanarayana S, Koblika BK. Amino Acid Substitutions at Position 312 in the Seventh Hydrophobic Segment of the β2-Adrenergic Receptor Modify Ligand-binding Specificity. Mol Pharmacol. 1993;44:111–114. [PubMed] [Google Scholar]
- 79.Bissantz C, Bernard P, Hilbert M, Rognan D. Protein-Based Virtual Screening of Chemical Databases. II. Are Homology Models of G-Protein Coupled Receptors Suitable Targets? Proteins. 2003;50:5–25. doi: 10.1002/prot.10237. [DOI] [PubMed] [Google Scholar]
- 80.Baldwin JM. The Probable Arrangement of the Helices in G Protein-Coupled Receptors. EMBO J. 1993;12:1693–1703. doi: 10.1002/j.1460-2075.1993.tb05814.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baldwin JM, Schertler GFX, Unger VM. An Alpha-Carbon Template for the Transmembrane Helices in the Rhodopsin Family of G-Protein-Coupled Receptors. J Mol Biol. 1997;272:144–164. doi: 10.1006/jmbi.1997.1240. [DOI] [PubMed] [Google Scholar]
- 82.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Multiple Sequence Alignment with the Clustal Series of Programs. Nuc Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Henikoff S, Henikoff JG. Amino Acid Substitution Matrices from Protein Blocks. Proc Nat Acad Sci USA. 1992;89:10915–10919. doi: 10.1073/pnas.89.22.10915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bissantz C. Conformational Changes of G Protein-Coupled Recetpros During their Activation by Agonist Binding. J Recept Signal Transduct. 2003;23:123–152. doi: 10.1081/rrs-120025192. [DOI] [PubMed] [Google Scholar]
- 85.Crocker E, Eilers M, Ahuja S, Hornak V, Hirshfeld A, Sheves M, Smith SO. Location of Trp265 in Metarhodopsin II: Implications for the Activation Mechanism of the Visual Receptor Rhodopsin. J Mol Biol. 2006;357:163–172. doi: 10.1016/j.jmb.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 86.Hallmen C, Wiese M. Molecular Dynamics Simulation of the Human Adenosine A3 Receptor: Agonist Induced Conformational Changes of Trp243. J Comp-Aided Mol Des. 2006;20:673–684. doi: 10.1007/s10822-006-9088-5. [DOI] [PubMed] [Google Scholar]
- 87.Niv MY, Skrabanek L, Filizola M, Weinstein H. Modeling Activated States of CPCRs: The Rhodopsin Template. J Comp-Aided Mol Des. 2006;20:437–448. doi: 10.1007/s10822-006-9061-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Patel AB, Crocker E, Eilers M, Hirshfeld A, Sheves M, Smith SO. Coupling of Retinal Isomerization to the Activation of Rhodopsin. Proc Nat Acad Sci USA. 2004;101:10048–10053. doi: 10.1073/pnas.0402848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh R, Hurst DP, Barnett-Norris J, Lynch DL, Reggio PH, Guarnieri F. Activation of the Cannabinoid CB1 Receptor may Involve a W6.48/F3.36 Rotamer Toggle Switch. J Peptide Res. 2002;60:357–370. doi: 10.1034/j.1399-3011.2002.21065.x. [DOI] [PubMed] [Google Scholar]
- 90.Spooner PJR, Sharples JM, Goodall SC, Bovee-Geurts PHM, Verhoeven MA, Lugtenburg J, Pistorius AMA, DeGrip WJ, Watts A. The Ring of the Rhodopsin Chromophore in a Hydrophobic Activation Switch Within the Binding Pocket. J Mol Biol. 2004;343:719–730. doi: 10.1016/j.jmb.2004.08.049. [DOI] [PubMed] [Google Scholar]
- 91.Swaminath G, Deupi X, Lee TW, Zhu W, Thain FS, Kobilka TS, Koblika BK. Probing the β2 Adrenoceptor Binding Site with Catechol Reveals Differences in Binding and Activation by Agonists and Partial Agonists. J Biol Chem. 2005;280:22165–22171. doi: 10.1074/jbc.M502352200. [DOI] [PubMed] [Google Scholar]
- 92.Fiser A, Šali A. MODELLER: Generation and Refinement of Homology-based Protein Structure Models. In: Carter CWJ, Sweet RM, editors. Methods in Enzymology: Macromolecular Crystallography: Part D. Vol. 374. 2003. pp. 461–491. [DOI] [PubMed] [Google Scholar]
- 93.Šali A, Blundell TL. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 94.Xhaard H, Nyrönen T, Rantanen VV, Ruuskanen JO, Laurila J, Salminen T, Scheinin M, Johnson MS. Model Structures of α-2 Adrenoreceptors in Complex with Automatically Docked Antagonist Ligands Raise the Possibility of Interactions Dissimilar from Agonist Ligands. J Struct Biol. 2005;150:126–143. doi: 10.1016/j.jsb.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 95.Westkaemper RB, Roth BL. Structure and Function Reveals Insights in the Pharmacology of 5-HT Receptor Subtypes. In: Roth BL, editor. The Serotonin Receptors: From Molecular Pharmacology to Human Therapeutics. Humana Press; Totowa, NJ: 2006. [Google Scholar]
- 96.Pogozheva ID, Lomize AL, Mosberg HI. Opioid Receptor Three-Dimensional Structures from Distance Geometry Calculations with Hydrogen Bonding Constraints. Biophys J. 1998;75:612–634. doi: 10.1016/S0006-3495(98)77552-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Meng F, Hoversten MT, Thompson RC, Taylor L, Watson SJ, Akil H. A Chimeric Study of the Molecular Basis of Affinity and Selectivity of the κ and δ Opioid Receptors. Potential Role of Extracellular Domains. J Biol Chem. 1995;270:12730–12736. doi: 10.1074/jbc.270.21.12730. [DOI] [PubMed] [Google Scholar]
- 98.Watson B, Meng F, Akil H. A Chimeric Analysis of the Opioid Receptor Domains Critical for the Binding Selectivity of μ Opioid Receptors. Neurobiol Dis. 1996;3:87–96. doi: 10.1006/nbdi.1996.0009. [DOI] [PubMed] [Google Scholar]
- 99.Jones G, Willett P, Glen RC. Molecular Recognition of Receptor Sites using a Genetic Algorithm with a Description of Desolvation. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 100.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 101.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. Empirical Scoring Functions: I. The Development of a Fast Empirical Scoring Function to Estimate the Binding Affinity of Ligands in Receptor Complexes. J Comp-Aided Mol Des. 1997;11:425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 102.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A Program to Check the Stereochemical Quality of Protein Structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- 103.Roth BL, Palvimaki EP, Berry S, Khan N, Sachs N, Uluer A, Choudhary MS. 5-Hydroxytryptamine2A (5-HT2A) Receptor Desensitization Can Occur Without Down-regulation. J Pharmacol Exp Ther. 1995;275:1638–1646. [PubMed] [Google Scholar]
- 104.Choudhary MS, Sachs N, Uluer A, Glennon RA, Westkaemper RB, Roth BL. Differential Ergopeptine Binding to 5-Hydroxytryptamine2A Receptors: Ergolines Require an Aromatic Residue at Position 340 for High Affinity Binding. Mol Pharmacol. 1995;47:450–457. [PubMed] [Google Scholar]
- 105.Wang CD, Gallaher TK, Shih JC. Site-directed Mutagenesis of the Serotonin 5-Hydroxytryptamine2 Receptor: Identification of Amino Acids Necessary for Ligand Binding and Receptor Activation. Mol Pharmacol. 1993;43:931–40. [PubMed] [Google Scholar]
- 106.Choudhary MS, Craigo S, Roth BL. A Single Point Mutation (Phe340 → Leu340) of a Conserved Phenylalanine Abolishes 1-[125I]Iodo-(2,5-dimethoxy)phenylisopropylamine and [3H]Mesulergine but not [3H]Ketanserin Binding to 5-Hydroxytryptamine2 Receptors. Mol Pharmacol. 1993;43:755–761. [PubMed] [Google Scholar]
- 107.Roth BL, Choudhary MS, Craigo S. Mutagenesis of 5-HT2 Serotonin Receptors: What Does an Analysis of Many Mutant ReceptorsTell Us? Med Chem Res. 1993;3:297–305. [Google Scholar]
- 108.Kristiansen K, Kroeze WK, Willins DL, Gelber EI, Savage JE. A Highly Conserved Aspartic Acid (Asp 155) Anchors the Terminal Amine Moiety of Tryptamines and is Involved in Membrane Targeting of the 5-HT2A Serotonin Receptor but Does Not Participate in Activation via a “Salt-bridge Disruption” Mechanism. J Pharmacol Exp Ther. 2000;293:735–746. [PubMed] [Google Scholar]
- 109.Almaula N, Ebersole BJ, Ballesteros JA, Weinstein H, Sealfon SC. Contribution of a Helix 5 Locus to Selectivity of Hallucinogenic and Nonhallucinogenic Ligands for the Human 5-Hydroxytryptamine2A and 5-Hydroxytryptamine2C Receptors: Direct and Indirect Effects on Ligand Affinity Mediated by the Same Locus. Mol Pharmacol. 1996;50:34–42. [PubMed] [Google Scholar]
- 110.Ballesteros JA, Weinstein H. Integrated Methods for the Construction of Three Dimensional Models and Computational Probing of Structure-function Relationships in G-protein Coupled Receptors. Methods Neurosci. 1995;25:366–428. [Google Scholar]
- 111.Barton GJ. ALSCRIPT: A Tool to Format Multiple Sequence Alignments. Protein Eng. 1993;6:37–40. doi: 10.1093/protein/6.1.37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.