

Abstract
Paraquat (PQ2+) is a prototypic toxin known to exert injurious effects through oxidative stress and bears a structural similarity to the Parkinson disease toxicant, 1-methyl-4-pheynlpyridinium. The cellular sources of PQ2+-induced reactive oxygen species (ROS) production, specifically in neuronal tissue, remain to be identified. The goal of this study was to determine the involvement of brain mitochondria in PQ2+-induced ROS production. Highly purified rat brain mitochondria were obtained using a Percoll density gradient method. PQ2+-induced hydrogen peroxide (H2O2) production was measured by fluorometric and polarographic methods. The production of H2O2 was evaluated in the presence of inhibitors and modulators of the mitochondrial respiratory chain. The results presented here suggest that in the rat brain, (a) mitochondria are a principal cellular site of PQ2+-induced H2O2 production, (b) PQ2+-induced H2O2 production requires the presence of respiratory substrates, (c) complex III of the electron transport chain is centrally involved in H2O2 production by PQ2+, and (d) the mechanism by which PQ2+ generates H2O2 depends on the mitochondrial inner transmembrane potential. These observations were further confirmed by measuring PQ2+-induced H2O2 production in primary neuronal cells derived from the midbrain. These findings shed light on the mechanism through which mitochondria may contribute to ROS production by other environmental and endogenous redox cycling agents implicated in Parkinson’s disease.
Parkinson disease (PD)2 is an age-related neurodegenerative disorder believed to originate in part via reactive oxygen species (ROS) overproduction, leading to oxidative stress and mitochondrial dysfunction (1). The disease is characterized pathologically by a progressive loss of dopaminergic neurons in the substantia nigra pars compacta. Both environmental and genetic factors are implicated in specific damage to the nigrostriatal system, contributing to the motor dysfunctions characteristic of PD (2, 3). However, the mechanisms by which such factors cause neurodegeneration remain relatively unknown.
Neurotoxins represent the most accepted tools used to produce experimental models of PD both in vivo and in vitro. The most commonly used agents include 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, 6-hydroxydopamine, rotenone, and paraquat (PQ2+) (4). PQ2+, a member of the widely used bipyridyl herbicides, is a prototypic compound known to exert its toxic effects via oxidative stress. A number of studies have exploited this action of PQ2+to test response and sensitivity to oxidative damage in various models (5–10). The possibility of PQ2+ as a neurotoxin has arisen from evidence that significant damage to the brain is observed in individuals exposed to lethal doses of the herbicide (11, 12). Furthermore, epidemiological studies have suggested an increased risk for developing PD following exposure to the herbicide (13). This has lead to an increased interest in the neurotoxic actions of PQ2+, along with the idea that PQ2+ and other herbicides may act as environmental triggers for PD (14). Therefore, in recent years, PQ2+ has become an increasingly popular model for studying the etiology of PD, alone and also in combination with other environmental toxins (15, 16). Systemic exposure of rodents to PQ2+ has been shown to reproduce many of the pathological features of PD, including selective degeneration of dopaminergic neurons in the substantia nigra and presence of intracellular α-synuclein deposits (15, 17–19). Although these studies show that agents such as PQ2+can produce selective neurodegeneration, the pathophysiological mechanisms underlying this damage remain unknown.
Interestingly, PQ2+ exhibits a remarkable structural similarity to MPP+, the toxic metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Although both of these compounds are believed to cause toxicity via oxidative stress, PQ2+ exerts its deleterious effects on dopamine neurons in a unique manner compared with MPP+ (20). PQ2+ belongs to a broad class of redox cycling compounds known to produce ROS. The proposed mechanism of redox cycling for PQ2+ involves the enzymatic reduction to form its cationic radical (Fig. 1, reaction 1), which then can reduce molecular oxygen (O2) to superoxide radical ( ) while also regenerating the parent compound (reaction 2). is subsequently converted to H2O2 either spontaneously (reaction 3) or through the actions of superoxide dismutase (reaction 4). The rate-limiting reaction in this process is the enzymatic reduction of PQ2+ depicted in reaction 1. The identities of enzymes capable of transferring electrons to PQ2+ in this reaction are not completely defined.
FIGURE 1.
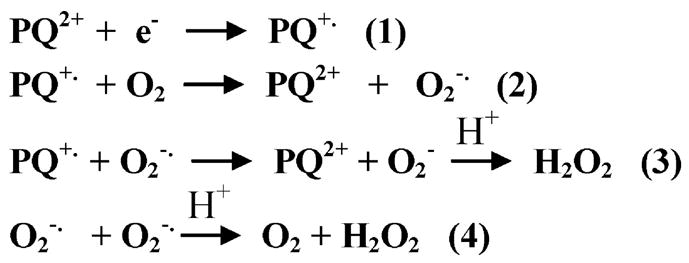
Chemical reactions proposed to participate in mechanisms of H2O2 production by PQ2+.
It is widely accepted that PQ2+-induced generation of ROS arises from a number of cellular sources. Enzymes capable of initiating the redox cycling process of PQ2+ have been identified in microsomal, plasma membrane, and cytosolic components through the actions of NADPH oxidase (21, 22), nitric-oxide synthase (23), and NADPH-cytochrome c reductase (24). However, the role of mitochondria in PQ2+ toxicity is remains unknown.
The purpose of this study was to examine the role that mitochondria play in PQ2+-induced ROS production in the brain. This was achieved by measuring rates of H2O2 production using polarographic and fluorometric methods. In particular, mitochondrial components capable of participating in the redox cycling-dependent ROS generation by PQ2+ were examined.
EXPERIMENTAL PROCEDURES
Isolation of Purified Rat Brain Mitochondria
Animal housing was conducted in compliance with University of Colorado at Denver Health Sciences Center procedures. Mitochondria were isolated from adult male Sprague-Dawley rats using Percoll density gradient centrifugation (25). Rat brain (excluding cerebellum) was homogenized in mitochondrial isolation buffer (70 mM sucrose, 210 mM mannitol, 5 mM Tris-HCl, 1 mM EDTA, pH 7.4) and then diluted 1:1 in 24% Percoll. Homogenates were centrifuged at 30,700 × g at 4 °C, for 10 min. The supernatant was saved as the cytosolic fraction, and the sediment was subjected to Percoll gradient (19% on 40%) centrifugation at 30,700 × g at 4 °C for 10 min. The material located at the interface of the lowest two layers was slowly diluted 1:4 with mitochondrial isolation buffer and centrifuged at 16,700 × g at 4 °C for 10 min. The pellets were resuspended in 5 ml of isolation buffer containing 1 mg/ml bovine serum albumin and centrifuged at 6700 × g at 4 °C for 10 min to obtain final pellets consisting of respiring mitochondria. Protein concentration was measured by using the Coomassie Plus™ protein assay reagent (Pierce).
Immunoblot Analysis of Mitochondrial Purity
Denatured proteins (20 μg) of cytosolic and mitochondrial fractions from Percoll isolation were separated by electrophoresis on 12% polyacrylamide gels (Bio-Rad) and transferred to polyvinylidene difluoride membrane (0.22 μM). Membrane blots were incubated with primary antibodies for anti-lactate dehydrogenase (1:100) (Rockland, Gilbertsville, PA) or anti-cytochrome oxidase subunit IV (1:1000) (Molecular Probes, Inc., Eugene, OR). For lactate dehydrogenase, membranes were incubated with horseradish peroxidase-conjugated anti-goat secondary antibody (Sigma), and for cytochrome oxidase subunit IV, membranes were incubated with horseradish peroxidase-conjugated anti-mouse secondary antibody (BD Biosciences). The membranes were developed using ECL Western blotting detection reagent (Amersham Biosciences). Cytochrome oxidase subunit IV was undetectable in cytosolic fractions and robustly expressed in mitochondrial fractions, whereas lactate dehydrogenase was only detectable in cytosolic fractions, indicating highly purified cellular fractions (data not shown).
Fluorometric Detection of H2O2 Production
H2O2 was measured using the horseradish peroxidase-linked Amplex Ultra Red™ fluorometric assay (Invitrogen). Cellular fractions (10 μg) were added to a 96-well plate containing 100 μl of reaction buffer containing 0.1 units/ml horseradish peroxidase, 50 μM Amplex UltraRed, and one of the following respiration substrates: 2.5 mM malate plus 5 mM pyruvate, 2.5 mM malate plus 10 mM glutamate, or 10 mM succinate. Additionally, carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 10 μM) was used as an uncoupler to determine the role of mitochondrial membrane potential. The reaction was started with the addition of 250 μM PQ2+ (final concentration). The following inhibitors were used in experiments: 10 μM rotenone (complex I), 10 μM 4,4,4-trifluoro-1-(2-thienyl)-1,3-butanedione (TTFA; complex II), 30 μg/ml antimycin A (complex III), 1 mM potassium cyanide (complex IV), and 10 μM FCCP. All inhibitors used in these studies were obtained from Sigma. Resorufin fluorescence was followed by a Gemini fluorescence microplate reader (Molecular Devices, Sunnyvale, CA). Superoxide dismutase (SOD) and catalase were added as controls at concentrations of 500 and 40 units/ml, respectively.
Polarographic Measurement of H2O2 Production
H2O2 production in cellular components was measured using an Apollo 4000 Free Radical Analyzer (WPI, Sarasota, FL) equipped with a 100-μm H2O2 sensor. The measurements were conducted in a water-jacketed open chamber at 30 °C with continuous stirring in a final reaction volume of 2 ml. Each measurement was started with the addition of reaction buffer (100 mM KCl, 75 mM mannitol, 25 mM sucrose, 10 mM Tris-Cl, 10 mM KH2PO4, pH 7.4) to the chamber. Once the output signal stabilized, the following were consecutively added: respiration substrate (2.5 mM malate plus 5 mM pyruvate, 2.5 mM malate plus 10 mM glutamate, or 10 mM succinate) and cellular fraction (200 μg of protein). The output signal was allowed to stabilize subsequent to each addition, followed by the addition of 250 μM PQ2+ to the chamber and the trace recorded. After 2–3 min of recording, vehicle (Me2SO) or inhibitor of the mitochondrial respiratory chain was added. Inhibitors of the respiratory chain were added at the concentrations described above.
Fluorometric Detection of PQ2+-induced H2O2 Production by Cell Cultures
Midbrain cell cultures were prepared from embryonic day 16 rats according to methods described previously for cortical cultures (26). For the assessment of PQ2+-induced H2O2 production, cells were plated in poly-D-lysine-coated 48-well plates at a density of 3.2 × 105 cells/well. All tissue culture reagents were obtained from Invitrogen. Primary midbrain cell cultures were maintained for 2 weeks before being tested for H2O2 production via fluorometry. Each well was incubated for 6 h at 37 °C in 250 μl of a medium containing 1.8 mM CaCl2, 5.4 mM KCl, 0.8 mM MgSO4, 116 mM NaCl, 14.7 mM NaHCO3, 1 mM NaH2PO4, 10 mM HEPES, 5.5 mM D-glucose, pH 7.4, also containing 0.1 units/ml horseradish peroxidase and 50 μM Amplex Red. PQ2+ and inhibitors were added at the same time at concentrations described previously. Resorufin fluorescence was followed using a Gemini fluorescence microplate reader (Molecular Devices, Sunnyvale, CA).
Measurement of Mitochondrial PQ2+ Uptake
Isolated brain mitochondria were incubated in reaction buffer (1 mg/ml protein) at 30 °C in the presence of 250 μM PQ2+, respiration substrates, and respiration inhibitors where indicated. After 5 min of incubation, the tubes were rapidly cooled at 4 °C and centrifuged for 8 min at 5400 ×g, and pellet volume was measured. The pellets were washed with 1 ml of reaction buffer containing respiration substrates and/or inhibitors but without PQ2+. The pellet was resuspended in 200 μl of distilled water and sonicated, followed by the addition of 1% perchloric acid, and centrifuged at 16,000 ×g for 10 min at 4 °C. The amount of PQ2+ in the supernatant was analyzed by high performance liquid chromatography using a published procedure (27) with minor modifications. Chromatographic separation was achieved on a YMC ODS-A S 3-μm 120-Å column (Waters, Milford, MA). The mobile phase consisted of sodium chloride with 4% acetonitrile, pH 2.8, pumped at 0.6 ml/min.
RESULTS
Mitochondria Are a Major Subcellular Source of PQ2+-induced ROS Generation
To address the question of what cellular components of the brain are involved in PQ2+-induced ROS production, brain homogenate, cytosolic, and mitochondrial fractions were analyzed for H2O2 production after the addition of PQ2+. Immunoblots for mitochondrial and cytosolic proteins were performed to confirm highly purified mitochondrial fractions void of any contamination from other cellular components (see “Experimental Procedures”). Fig. 2a shows the fluorometric determination of PQ2+-induced H2O2 production in rat brain homogenate, cytosolic, and mitochondrial fractions. Following the addition of PQ2+ in the presence of respiration substrates (malate + glutamate), mitochondria showed an immediate and robust production of H2O2, whereas rates were much lower in homogenate and cytosolic fractions. Concerns over the use of the Amplex UltraRed fluorescent assay to measure H2O2 have arisen from the possibility that endogenous reducing equivalents may interfere with the fluorescence (28). Therefore, in order to validate results from the high throughput fluorometric method to measure H2O2 production, a polarographic method was also used. Shown in Fig. 2b, H2O2 production using this polarographic method showed very similar rates in each cellular component following the addition of PQ2+. Rates of H2O2 production obtained from these traces under the conditions described are summarized in Table 1. All rates are compared with the PQ2+-dependent H2O2 production rate in mitochondria in the presence of malate and glutamate, which is expressed as 100%. In the absence of exogenous substrates, mitochondria generated H2O2 at a rate 5 times that of whole brain homogenates, whereas the cytosol generated the least amount of H2O2. The addition of substrates feeding the tricarboxylic acid cycle and electron transport chain (ETC) at the level of complex I (malate + glutamate, malate + pyruvate) or at the level of complex II (succinate) did not have any effect on rates of H2O2 production from the cytosolic fraction (Fig. 2). In the brain homogenate and mitochondrial fractions, the addition of respiration substrates significantly increased production in the range of 20–40- and 83–145-fold, respectively (Table 1). As expected, mitochondrial rates of H2O2 production with substrates were greatest (Fig. 2).
FIGURE 2. PQ2+-induced H2O2 production in cellular fractions from the brain.
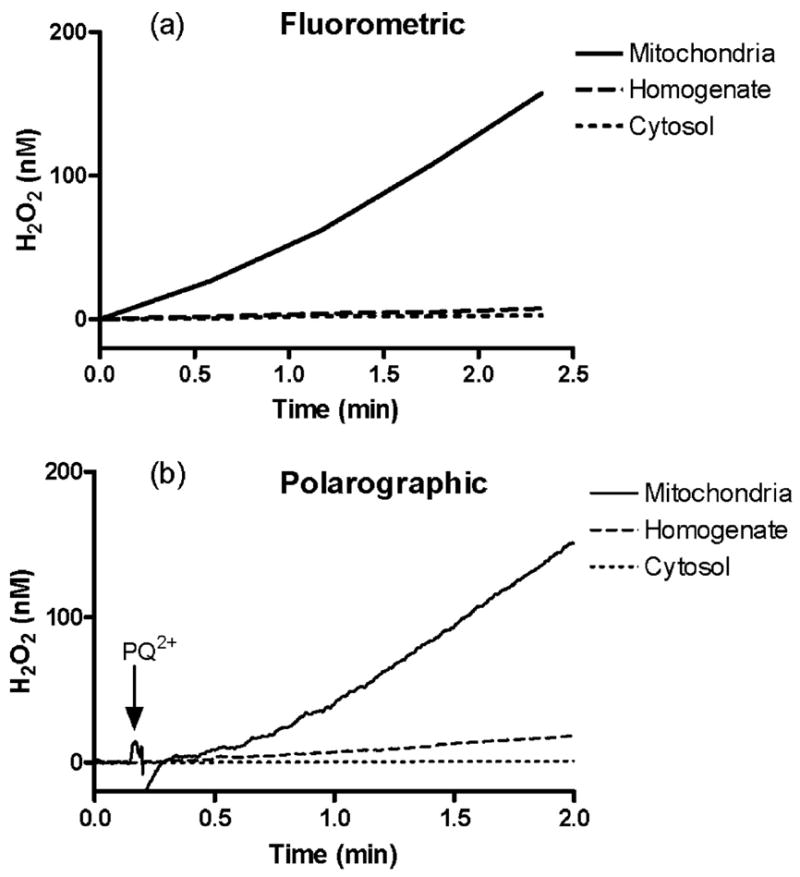
Fluorometric (a) and polarographic (b) assays were used to measure H2O2 in the presence of malate and glutmate following the addition of 250 μM PQ2+ to equal amounts of protein from each rat brain fraction: mitochondria (solid line), cytosol (dotted line), and homogenate (dashed line).
TABLE 1. Rates of PQ2+-induced H2O2 production in cellular fractions.
Equal protein amounts from each fraction were assayed for H2O2 production using a fluorometric method following the addition of 250 μM PQ2+. H2O2 production in mitochondrial fraction stimulated with malate plus glutamate as respiration substrates was considered 100%, and all values are expressed as a percentage of this (mean ± S.E., n = 3).
| Fraction | Substrate |
|||
|---|---|---|---|---|
| With endogenous substrate | With succinate | With Mal/Pyr | With Mal/Glu | |
| Homogenate | 0.21 ± 0.02a | 4.52 ± 0.19a | 7.99 ± 0.34a | 5.55 ± 0.17a |
| Cytosol | 0.02 ± 0.003 | 0.06 ± 0.01 | 0.03 ± 0.01 | 0.05 ± 0.02 |
| Mitochondria | 0.98 ± 0.07a,b | 81.48 ± 3.53a,b | 143.83 ± 6.11a,b | 100.00 ± 3.10a,b |
Significantly different from cytosolic fraction (p < 0.05, one-way analysis of variance) grouped by respiration substrate.
Significantly different from homogenate fraction (p < 0.05, one-way analysis of variance) grouped by respiration substrate.
Involvement of the Respiratory Chain in PQ2+-induced H2O2 Production in Brain Mitochondria
After establishing that mitochondria are a major subcellular source involved in PQ2+-induced ROS production, possible mitochondrial components implicated in this process were investigated. The generation of ROS via the redox cycling action of PQ2+requires reduction to its cationic radical PQ+· as an obligatory first step. The complexes of the ETC represent good candidates for this reduction, since they act to transport electrons and possess redox potentials in the range required for PQ2+. To test this hypothesis, PQ2+-dependent H2O2 production in brain mitochondria was assayed in the presence of inhibitors of the ETC. Fig. 3a shows rates of H2O2 production determined by a fluorometric assay. As expected, mitochondria stimulated by malate and glutamate in the presence of PQ2+ produced an immediate and robust increase in H2O2 production. Exogenous SOD had no effect on H2O2 production rates, whereas catalase almost completely attenuated this process (Fig. 3a). Antimycin A, a specific inhibitor of complex III, generated low rates of H2O2 by itself. However, co-administration with PQ2+ showed rates similar to those observed with antimycin A alone (Fig. 3a and Fig. 4). Using the polarographic method, antimycin A inhibition of PQ2+-induced H2O2 production was utilized to validate results obtained via fluorometry (Fig. 3b). Following the addition of PQ2+in the presence of malate and glutamate, H2O2 was measured at a rate termed “steady state 1.” Inhibitors of the ETC changed the rate of H2O2 production to the rate determined as “steady state 2.” The addition of antimycin A to inhibit complex III activity significantly decreased the rate of PQ2+-induced H2O2 production (dashed line). Catalase was added as a control (dotted line) at a concentration (12.5 units/ml) to provide a balance between H2O2 production and consumption in the system. Rotenone, an inhibitor of complex I, did not significantly alter the rate of H2O2 generation in the presence of malate and glutamate as respiration substrates (solid line).
FIGURE 3. Inhibition of PQ2+-induced H2O2 production in stimulated mitochondria.
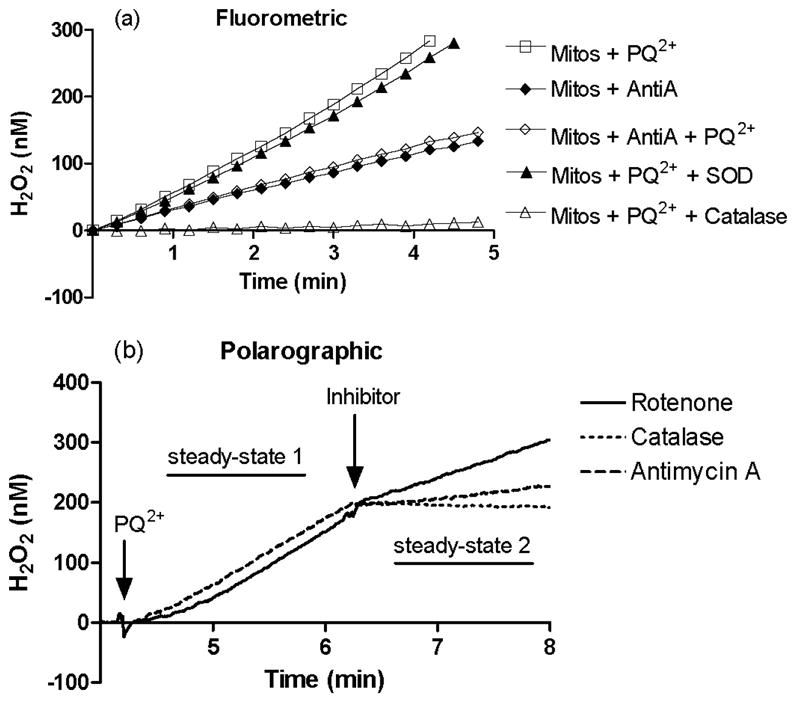
Fluorometric (a) measurement of H2O2 production from isolated mitochondria in the presence of succinate was measured in the presence of PQ2+(250 μM, □), antimycin A (◆), antimycin A and PQ2+(◇), PQ2+ and SOD (▲), and PQ2+and catalase (△). Polarographic (b) measurement was used to confirm results. Following the addition of 250 μM PQ2+, H2O2 production in mitochondria was monitored with inhibitors added as indicated: rote-none (solid line), catalase (dotted line), and antimycin A (dashed line). Rates of H2O2 production before and after the addition of inhibitor (steady state 1 and 2, respectively) were compared with obtain the percentage of H2O2 production under each condition (see Fig. 5).
FIGURE 4. Effect of ETC inhibitors on PQ2+-induced H2O2 production in isolated mitochondria.
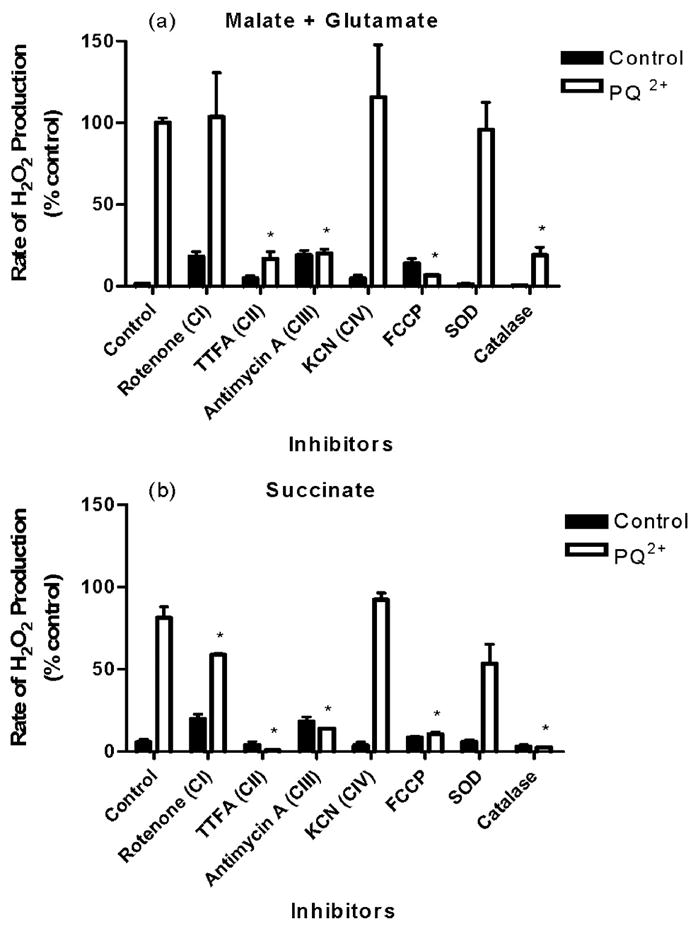
Mitochondria were stimulated with respiration substrates (malate plus glutamate (a) or succinate (b)), and H2O2 production was measured in the absence (closed bars) or presence (open bars) of 250 μM PQ2+. Inhibitors were added as described under “Experimental Procedures.” H2O2 production in mitochondrial fraction stimulated with malate plus glutamate as respiration substrates in the presence of PQ2+ was considered 100%, and all values are expressed as a percentage of this (mean ±S.E., n =3). *, p <0.05 compared with PQ2+-treated control mitochondria (one-way analysis of variance).
A more comprehensive screening was subsequently performed to determine effects on PQ2+-induced H2O2 production in the presence of inhibitors of all complexes of the ETC. H2O2 production rates are summarized in Fig. 4, in the presence of malate and glutamate (Fig. 4a) or succinate (Fig. 4b). These substrates act to feed electrons into the ETC at the level of complex I via NADH or directly to complex II, respectively. Inhibition of complexes I and III without the addition of PQ2+ produced a significant rate of H2O2 generation, but this production was never greater than 24% of the amount produced following PQ2+ administration in controls. Significant decreases in rates of H2O2 production were observed with both complex II and III inhibition by TTFA and antimycin A, respectively. This decrease was to an extent that is comparable with that obtained in the absence of PQ2+ (Fig. 4). Rotenone also significantly decreased H2O2 generation in mitochondria fed electrons via succinate, but to a lesser extent than antimycin A or TTFA. As shown in Fig. 3b, SOD had no effect on PQ2+-induced H2O2 production, whereas catalase was able to completely attenuate this effect.
Effect of Mitochondrial Membrane Potential on PQ2+-induced H2O2 Production
In respiring mitochondria, the transport of electrons through the ETC is accompanied by vectorial pumping of protons from the matrix to the intermembrane space, thereby creating an electrochemical gradient that establishes the inner mitochondrial transmembrane potential (ΔΨm). In intact mitochondria, ROS production is sharply regulated by the amplitude of ΔΨm (29). To evaluate the role of ΔΨm in PQ2+-induced ROS production, the effect of a mitochondrial uncoupler, FCCP, was examined. In the presence of substrates malate and glutamate (Fig. 4a) or succinate (Fig. 4b) to stimulate mitochondrial respiration, FCCP was able to completely abolish production of H2O2 by PQ2+.
Mitochondrial Uptake of PQ2+
The majority of the ETC components reside in the mitochondrial inner membrane and face either the lipid phase of the membrane, the intermembrane space, or the matrix. Therefore, for PQ2+ to interact with ETC complexes and generate ROS, it must be capable of traversing the outer mitochondrial membrane and entering the inter-membrane space and matrix. The ability of PQ2+to do this was assessed in the following manner. Mitochondria were incubated with PQ2+ under experimental conditions, extensively washed, and sonicated. PQ2+ was measured via HPLC with electrochemical detection as described under “Experimental Procedures.” Under nonrespiring conditions (i.e. in the absence of exogenous substrates) PQ2+ was effectively taken up into mitochondria at a rate of ~50% compared with the starting concentration (250 μM) in the incubation buffer (Table 2). Mitochondria sonicated prior to incubation showed very low levels of PQ2+ when measured by HPLC. Respiring mitochondria showed slightly increased uptake of PQ2+, but not significantly different from nonrespiring controls. Additionally, the addition of antimycin A or FCCP had no effect on mitochondrial PQ2+concentrations, indicating that these compounds do not prevent PQ2+ uptake (Table 2).
TABLE 2. PQ2+uptake by rat brain mitochondria.
Isolated mitochondria (Mito) were incubated in 250 μM PQ2+ in the presence of respiration substrates and inhibitors, and intramitochondrial PQ2+ concentrations were measured via HPLC. Values are presented as mean ± S.E., n = 3.
| Condition | Intramitochondrial PQ2+ |
|---|---|
| μM | |
| Mito + PQ2+ + buffer | 137.5 ± 50 |
| Mito + PQ2+ + buffer + sonication | 20 ± 12.5 |
| Mito + PQ2+ + buffer + succinate | 162.5 ± 75 |
| Mito + PQ2+ + buffer + succinate + antimycin A | 225 ± 50 |
| Mito + PQ2+ + buffer + succinate + FCCP | 200 ± 75 |
Confirmation of PQ2+-induced H2O2 Production in Cell Culture Model
To examine if mitochondrial mechanisms of PQ2+-dependent H2O2 production are involved in a more physiologically relevant model of whole cells, midbrain primary cultures obtained from embryonic rats were used. H2O2 production was determined exclusively via the horseradish peroxidase-linked Amplex UltraRed fluorometric assay due to methodological limitations of polarographic detection in cell culture. Following 6-h incubation of midbrain cultures with PQ2+ in the presence of ETC complex inhibitors, H2O2 production was measured and expressed as percentage of control (Fig. 5). In this in vitro model, H2O2 production was inhibited completely only in the presence of antimycin A compared with controls without PQ2+. Rotenone also significantly attenuated PQ2+-induced H2O2 production but to a much lesser extent than antimycin A. As observed in isolated mitochondria, FCCP abolished mitochondrial PQ2+-induced H2O2 production.
FIGURE 5. Effect of ETC inhibitors on PQ2+-induced H2O2 production in primary midbrain cultures.
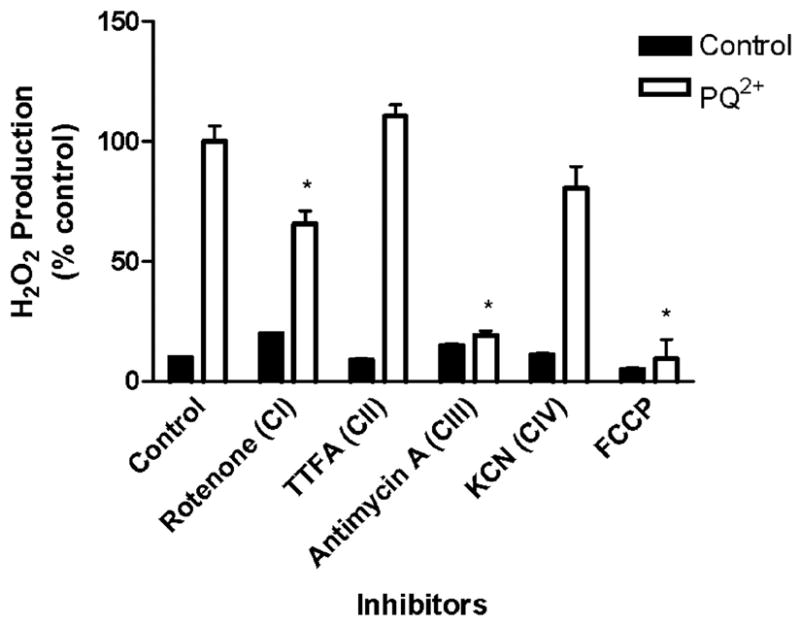
H2O2 production was measured in midbrain cultures incubated in buffer containing PQ2+ and inhibitors where indicated using a fluorometric method as described under “Experimental Procedures.” H2O2 production in the presence of PQ2+ was considered 100%, and all values are expressed as a percentage of this (mean ± S.E., n = 3). *, p < 0.05 compared with PQ2+-treated control mitochondria (one-way analysis of variance).
DISCUSSION
Several novel findings emerge from the present study regarding the mechanism of PQ2+-induced ROS production in brain tissue. First, mitochondria are a major cellular component involved in ROS generation induced by PQ2+ in the rat brain. PQ2+-induced H2O2 production by brain mitochondria is dependent on a constant electron flow provided by respiration substrates and a functional ETC. With the use of specific inhibitors of the ETC, complex III has been identified as a novel site of action for PQ2+ in the process of redox cycling to generate ROS. Last, the mechanism of PQ2+-induced ROS production is dependent on mitochondrial membrane potential.
Rat Brain Mitochondria Are a Major Cellular Component Involved in PQ2+-induced ROS Generation
The mechanism of ROS generation by redox cycling of PQ2+ has been widely studied in both in vivo and in vitro systems. These studies point out the involvement of several mitochondrial (30), microsomal (31, 32), and cytosolic (23, 33) enzymes in ROS production by PQ2+. Several studies have suggested the involvement of mitochondria in PQ2+-induced generation. In the yeast Saccharomyces cerevisiae, it was suggested that the mitochondrial ETC could be considered a potential site for PQ2+ reduction (34). Additionally, in Candida albicans, the inhibition of respiration almost completely suppressed PQ2+-induced generation of ROS (35). However, the relevance of mitochondria in mammalian systems has remained relatively unexplored. In this study, we clearly demonstrate that among the subcellular fractions of the rat brain, mitochondria are strongly implicated in PQ2+-induced ROS generation.
In the absence of exogenously added substrates, PQ2+-induced H2O2 production was highest in the mitochondrial fraction, followed by the homogenate and last the cystosol. This can be explained by the fact that mitochondria retain reducing equivalents in the matrix following isolation. Therefore, mitochondria are still able to undergo respiration at this time (36). The brain homogenate fraction also contains mitochondria, albeit at lesser amounts, which is why this component shows greater H2O2 production than cytosol in the absence of respiration substrates. This also explains why an increase in H2O2 production is observed in the homogenates following the addition of substrates. The addition of external respiration substrates produced significantly greater PQ2+-induced H2O2 in the mitochondria compared with other cell compartments. These data suggest that PQ2+-induced H2O2 production is highly dependent on substrates that stimulate mitochondrial respiration.
The issue regarding transport of PQ2+ into the brain and finally into the mitochondria remains an important aspect when examining its toxic actions as performed in this study. It has been demonstrated that PQ2+ can be taken up into the brain by neutral amino acid transporters in the blood-brain barrier and subsequently transported into cells in a sodium-dependent manner (37). It is expected that mitochondria may use similar transport systems for PQ2+. In this study, we provide evidence that mitochondria are able to efficiently transport PQ2+under both nonrespiring and respiring conditions. Additionally, antimycin A and FCCP had no effect on this uptake, confirming a transport system independent of ETC inhibition or transmembrane potential.
The use of SOD and catalase as controls in this study provide important insight regarding the nature of PQ2+-induced ROS production in isolated mitochondria. If PQ2+-induced ROS production occurred outside the mitochondria, exogenously added SOD would increase the rate of H2O2 production due to the rapidly catalyzed conversion of to H2O2 before other mechanisms could consume the oxygen radical. However, SOD failed to inhibit PQ2+-induced ROS production. This observation indicates that PQ2+ produces ROS in the mitochondria, which is then converted to H2O2 by mitochondrial systems, such as MnSOD, and/or by spontaneous dismutation. H2O2 is then capable of diffusing through the mitochondrial membrane into the extracellular medium, where it can be consumed enzymatically by exogenously added catalase or measured via polarographic and fluorescent methods shown here.
Involvement of the ETC and Role of Complex III in PQ2+-induced H2O2 Production
In this study, we demonstrate that respiration substrates are essential for mitochondria to generate significant amounts of H2O2 in the presence of PQ2+. Additionally, the inhibition of complex II and III in isolated mitochondria by TTFA and antimycin A, respectively, completely attenuated this effect. These data suggest that in mitochondria with a functional ETC, electrons can be transferred to PQ2+ at the level of these two complexes, thus initiating the redox cycling reaction and subsequent ROS production. However, in primary midbrain cultures, only complex III inhibition by antimycin A was able to block H2O2 production, whereas no effect was observed with complex II inhibition by TTFA. This somewhat contradictory effect may be explained as follows. First, the isolated mitochondria used in this study are maintained in a buffer that allows mitochondria to function under a high electrochemical potential. Under such conditions, complexes in the ETC exist in a highly reduced state more likely to exhibit electron leak and reverse electron transport that may lead in part to the observed generation of ROS. These electrons may also possess the capability of reducing PQ2+to initiate the redox cycling process involved in H2O2 production. However, in the whole cell model of primary midbrain cultures, mitochondria are functioning at a much lower electrochemical potential, more typical of what exists physiologically, where electron leak and reverse electron transport are not relevant (38). Therefore, complexes that may act to reduce PQ2+ via such mechanisms in isolated mitochondria may not be involved in this cell culture model.
Another possible explanation for the contrasting data regarding complexes II and III involves the metabolic processes being affected by the addition of substrates and inhibitors. Complex II of the ETC is also known as succinate dehydrogenase, an enzyme involved in the mitochondrial tricarboxylic acid cycle. In isolated mitochondria supplied with malate and glutamate or succinate, the use of TTFA inhibits both actions of the enzyme and may completely block electron flow into the ETC, since no other substrates are available to overcome this inhibition. In such an event, no electrons would be available to reduce PQ2+, and no ROS will subsequently be produced, as was observed in this study. Alternatively, in primary midbrain cultures, glucose is provided as the major energy source. In the presence of glucose, multiple metabolic pathways may also be acting as a means of producing reducing equivalents and substrates capable of feeding into the tricarboxylic acid cycle or ETC at alternate sites. For example, α-glycerol phosphate produced during glycolysis is able to feed electrons to ubiquinone in the ETC, downstream of complexes I and II. Therefore, in cultured cells where TTFA is added to inhibit complex II and block part of the tricarboxylic acid cycle, anaplerotic reactions may play a significant role in providing alternative substrates to overcome this inhibition in the ETC and tricarboxylic acid cycle.
Each of the above cases provides an explanation for why complex II inhibition does not attenuate PQ2+-induced H2O2 production in primary midbrain cultures. The data obtained from this in vitro model implicate a role for complex III of the ETC in the PQ2+ mechanism of toxic action.
Role of Mitochondrial Membrane Potential in PQ2+-induced ROS Production
The proton gradient between the mitochondrial matrix and intermembrane space establishes a transmembrane electrochemical potential of ΔΨm. The nonphosphorylating “state 4” conditions (39) used in this study favor a high membrane potential and high degree of reduction of the redox carriers in the complexes of the ETC. Based on the use of a mitochondrial uncoupler, FCCP, our results demonstrate that an established membrane potential is required for PQ2+-induced H2O2 production. In particular, a high reduction state of complex III may be a required step in the reduction of PQ2+ to its cationic radical capable of redox cycling. The dissipation of the membrane potential with the use of FCCP diminishes the reducing capacity of ETC complexes and, as a consequence, abolishes the ability to pass electrons to PQ2+ and lead to the generation of ROS.
Proposed Mechanisms Involved in Mitochondria-dependent PQ2+-induced ROS Production
The dependence of PQ2+-induced ROS generation on the presence of respiration substrates in mitochondria suggests the involvement of the ETC. The paired substrates malate and glutamate or malate and pyruvate are finally transformed to NADH in the tricarboxylic acid cycle and subsequently feed electrons into the ETC at the level of complex I, whereas succinate is an intermediate in the tricarboxylic acid cycle and is able to feed electrons directly at the level of complex II. The electrons from either of these two complexes then flow into the ubiquinone cycle and transfer their electrons to complex III. It is at this point in the flow of electrons through complex III that electrons are hypothesized to be transferred to PQ2+, thus initiating the process of redox cycling. This process is analogous to the production of from complex III that has been characterized (40). A critical role for complex III was revealed by Chen et al. (41) in mediating H2O2 production in heart subsarcolemmal mitochondria using substrates for complex I and complex II and a variety of ETC inhibitors. Based on their approach, we can speculate the following regarding PQ2+-induced H2O2 production in our studies. (a) The finding that rotenone inhibition produces minimal change in PQ2+-induced H2O2 production in mitochondria suggests that complex I may not be the major site of PQ2+-induced net H2O2 generation in mitochondria oxidizing complex I substrates. (b) Complex III inhibition produced a substantial reduction of PQ2+-induced H2O2 production in the presence of complex I or II substrates, suggesting that the site of ROS production is downstream in the respiratory chain, most likely at complex III. (c) Complex IV inhibition may be ruled out due to the lack of effect of cyanide on PQ2+-induced H2O2 production. (d) Inhibition of PQ2+-induced H2O2 production by complex II inhibitors in the presence of complex I or II substrates may be due to direct reduction of PQ2+ by the complex or indirectly by feeding electrons to complex III. However, our results using intact cells suggest that complex II has a minor role when compared with complex III as a primary site for net H2O2 production induced by PQ2+ in brain mitochondria.
It is difficult to speculate upon the precise mechanism responsible for PQ2+ reduction and ROS production at the level of the complex III. Assuming that antimycin A can block the electron transfers between cytochrome bL and bH and from cytochrome bL to iron-sulfur protein (42), a direct involvement of these cytochromes in PQ2+ reduction may be plausible. Since these cytochromes have deep membrane localization, it is difficult to predict the precise site of H2O2 generation after PQ2+ reduction. However, our studies show that a significant amount of H2O2 produced by PQ2+ is released from mitochondria, which would imply that reactions following reduction of PQ2+ that generate H2O2 occur in a site facing the intermembrane space, where mitochondria lack antioxidant defenses to remove H2O2.
Palmeira et al. (43) and more recently Gomez et al. (44) suggest that high concentrations of PQ2+ may exert nonselective direct oxidative damage toward all ETC complexes, leading to an impairment of their activity. This may be due to oxidative damage and compromised function of the ETC complexes as a consequence of their proximity to the site of mitochondrial ROS production by PQ2+, namely complex III. This mechanism is supported by the observation that mitochondrial ETC complexes are susceptible to inactivation by a wide range of oxidants (45). It has also been reported that microglial activation and consequent induction of NADPH oxidase plays a significant role in PQ2+-induced dopaminergic cell degeneration (9, 22). These studies suggest that NADPH oxidase may be one enzyme redox-cycling with PQ2+. However, the induction of NADPH oxidase is a delayed response following PQ2+ administration in vivo. Our results demonstrate a robust and instantaneous production of H2O2 by mitochondria after exposure to PQ2+. We propose that mitochondrial H2O2 production induced by PQ2+ in the brain is an early event that may later initiate other cellular events, such as nonspecific ETC inactivation, microgliosis, and NADPH oxidase activation (Fig. 6). Together, these results highlight the significance and potential consequences of PQ2+-induced mitochondrial ROS production.
FIGURE 6. Proposed involvement of ETC in PQ2+-dependent H2O2 generation in brain mitochondria.
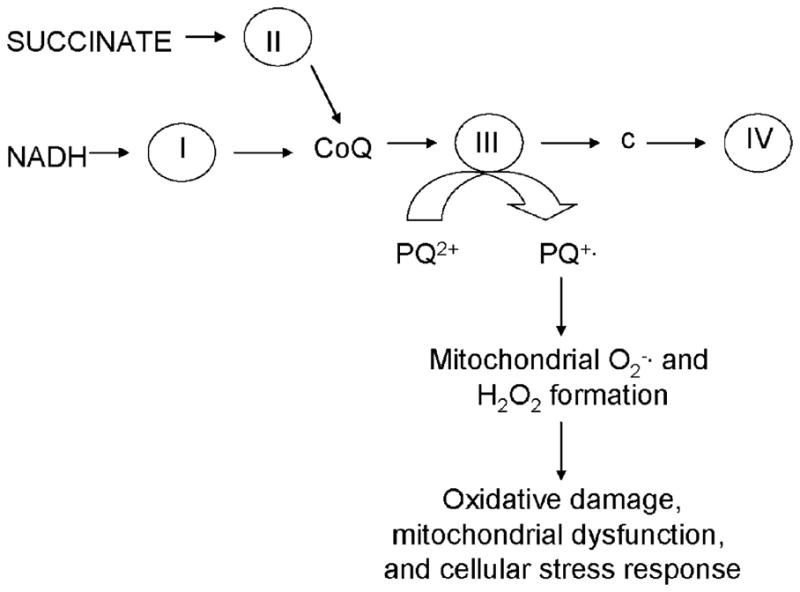
Electrons feed into the ETC at the level of complex I or complex II and are transferred to ubiquinone (CoQ), complex cytochrome c (c), and complex IV sequentially. Complex III has the ability transfer these electrons to PQ2+, thus forming the PQ+· radical, leading ROS formation and subsequent cell damage.
PQ2+ is an increasingly popular model of inducing systemic and cellular injury via the production of ROS (5–10). These findings may clarify possible mechanisms of the actions of PQ2+ in diverse models. Furthermore, they lead to the suggestion that similar mechanisms of mitochondrial ROS generation may be employed by other redox-cycling agents, both environmental and endogenous, that bear structural and functional resemblance to PQ2+.
Footnotes
The abbreviations used are: PD, Parkinson disease; ROS, reactive oxygen species; PQ2+, paraquat; FCCP, carbonylcyanide-p-trifluoromethoxyphenylhydrazone; TTFA, 4,4,4-trifluoro-1-(2-thienyl)-1,3-butanedione; ETC, electron transport chain; SOD, superoxide dismutase; HPLC, high pressure liquid chromatography.
This work was supported by National Institutes of Health Grant NS045748 (to M. P.) and an American Foundation for Pharmaceutical Education pre-doctoral fellowship (to D. A. D.).
References
- 1.Jenner P. Ann Neurol. 2003;53(Suppl 3):S26–S36. doi: 10.1002/ana.10483. [DOI] [PubMed] [Google Scholar]
- 2.Fahn S. Ann N Y Acad Sci. 2003;991:1–14. doi: 10.1111/j.1749-6632.2003.tb07458.x. [DOI] [PubMed] [Google Scholar]
- 3.Dauer W, Przedborski S. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 4.Bove J, Prou D, Perier C, Przedborski S. NeuroRx. 2005;2:484–494. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bus JS, Gibson JE. Environ Health Perspect. 1984;55:37–46. doi: 10.1289/ehp.845537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sato B, Tanaka A, Mori S, Yanabu N, Kitai T, Tokuka A, Inomoto T, Iwata S, Yamaoka Y, Chance B. Biochim Biophys Acta. 1995;1268:20–26. doi: 10.1016/0167-4889(95)00035-q. [DOI] [PubMed] [Google Scholar]
- 7.Bonilla E, Medina-Leendertz S, Villalobos V, Molero L, Bohorquez A. Neurochem Res. 2006;31:1425–1432. doi: 10.1007/s11064-006-9194-8. [DOI] [PubMed] [Google Scholar]
- 8.Arkblad EL, Tuck S, Pestov NB, Dmitriev RI, Kostina MB, Stenvall J, Tranberg M, Rydstrom J. Free Radic Biol Med. 2005;38:1518–1525. doi: 10.1016/j.freeradbiomed.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 9.Cicchetti F, Lapointe N, Roberge-Tremblay A, Saint-Pierre M, Jimenez L, Ficke BW, Gross RE. Neurobiol Dis. 2005;20:360–371. doi: 10.1016/j.nbd.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 10.Bishop AL, Rab FA, Sumner ER, Avery SV. Mol Microbiol. 2007;63:507–520. doi: 10.1111/j.1365-2958.2006.05504.x. [DOI] [PubMed] [Google Scholar]
- 11.Grant H, Lantos PL, Parkinson C. Histopathology. 1980;4:185–195. doi: 10.1111/j.1365-2559.1980.tb02911.x. [DOI] [PubMed] [Google Scholar]
- 12.Hughes JT. Neurotoxicology. 1988;9:243–248. [PubMed] [Google Scholar]
- 13.Liou HH, Tsai MC, Chen CJ, Jeng JS, Chang YC, Chen SY, Chen RC. Neurology. 1997;48:1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- 14.Di Monte DA. Lancet Neurol. 2003;2:531–538. doi: 10.1016/s1474-4422(03)00501-5. [DOI] [PubMed] [Google Scholar]
- 15.McCormack AL, Thiruchelvam M, Manning-Bog AB, Thiffault C, Langston JW, Cory-Slechta DA, Di Monte DA. Neurobiol Dis. 2002;10:119–127. doi: 10.1006/nbdi.2002.0507. [DOI] [PubMed] [Google Scholar]
- 16.Thiruchelvam M, Richfield EK, Baggs RB, Tank AW, Cory-Slechta DA. J Neurosci. 2000;20:9207–9214. doi: 10.1523/JNEUROSCI.20-24-09207.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manning-Bog AB, McCormack AL, Li J, Uversky VN, Fink AL, Di Monte DA. J Biol Chem. 2002;277:1641–1644. doi: 10.1074/jbc.C100560200. [DOI] [PubMed] [Google Scholar]
- 18.Thiruchelvam M, McCormack A, Richfield EK, Baggs RB, Tank AW, Di Monte DA, Cory-Slechta DA. Eur J Neurosci. 2003;18:589–600. doi: 10.1046/j.1460-9568.2003.02781.x. [DOI] [PubMed] [Google Scholar]
- 19.Peng J, Mao XO, Stevenson FF, Hsu M, Andersen JK. J Biol Chem. 2004;279:32626–32632. doi: 10.1074/jbc.M404596200. [DOI] [PubMed] [Google Scholar]
- 20.Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW. Toxicol Sci. 2005;88:193–201. doi: 10.1093/toxsci/kfi304. [DOI] [PubMed] [Google Scholar]
- 21.Wu XF, Block ML, Zhang W, Qin L, Wilson B, Zhang WQ, Veronesi B, Hong JS. Antioxid Redox Signal. 2005;7:654–661. doi: 10.1089/ars.2005.7.654. [DOI] [PubMed] [Google Scholar]
- 22.Purisai MG, McCormack AL, Cumine S, Li J, Isla MZ, Di Monte DA. Neurobiol Dis. 2007;25:327–331. doi: 10.1016/j.nbd.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS. Proc Natl Acad Sci U S A. 1999;96:12760–12765. doi: 10.1073/pnas.96.22.12760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mason RP. Environ Health Perspect. 1990;87:237–243. doi: 10.1289/ehp.9087237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson MF, Sims NR. Brain Res Protoc. 2000;5:95–101. doi: 10.1016/s1385-299x(99)00060-4. [DOI] [PubMed] [Google Scholar]
- 26.Patel M, Day BJ, Crapo JD, Fridovich I, McNamara JO. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 27.Taylor PJ, Salm P, Pillans PI. J Anal Toxicol. 2001;25:456–460. doi: 10.1093/jat/25.6.456. [DOI] [PubMed] [Google Scholar]
- 28.Votyakova TV, Reynolds IJ. Arch Biochem Biophys. 2004;431:138–144. doi: 10.1016/j.abb.2004.07.025. [DOI] [PubMed] [Google Scholar]
- 29.Andreyev AY, Kushnareva YE, Starkov AA. Biochemistry (Mosc) 2005;70:200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 30.Fukushima T, Tawara T, Isobe A, Hojo N, Shiwaku K, Yamane Y. J Neurosci Res. 1995;42:385–390. doi: 10.1002/jnr.490420313. [DOI] [PubMed] [Google Scholar]
- 31.Ilett KF, Stripp B, Menard RH, Reid WD, Gillette JR. Toxicol Appl Pharmacol. 1974;28:216–226. doi: 10.1016/0041-008x(74)90007-6. [DOI] [PubMed] [Google Scholar]
- 32.Montgomery MR. Toxicol Appl Pharmacol. 1976;36:543–554. doi: 10.1016/0041-008x(76)90233-7. [DOI] [PubMed] [Google Scholar]
- 33.Winterbourn CC, Sutton HC. Arch Biochem Biophys. 1984;235:116–126. doi: 10.1016/0003-9861(84)90260-1. [DOI] [PubMed] [Google Scholar]
- 34.Blaszczynski M, Litwinska J, Zaborowska D, Bilinski T. Acta Microbiol Pol. 1985;34:243–254. [PubMed] [Google Scholar]
- 35.Aoki S, Ito-Kuwa S, Nakamura K, Nakamura Y, Vidotto V, Takeo K. Med Mycol. 2002;40:13–19. doi: 10.1080/mmy.40.1.13.19. [DOI] [PubMed] [Google Scholar]
- 36.Veech R. In: Mitochondria in Health and Disease. Berdanier C, editor. CRC Press, Inc; Boca Raton, FL: 2005. pp. 65–94. [Google Scholar]
- 37.Shimizu K, Ohtaki K, Matsubara K, Aoyama K, Uezono T, Saito O, Suno M, Ogawa K, Hayase N, Kimura K, Shiono H. Brain Res. 2001;906:135–142. doi: 10.1016/s0006-8993(01)02577-x. [DOI] [PubMed] [Google Scholar]
- 38.Nicholls DG. Aging Cell. 2004;3:35–40. doi: 10.1111/j.1474-9728.2003.00079.x. [DOI] [PubMed] [Google Scholar]
- 39.Chance B, Williams GR. Adv Enzymol Relat Subj Biochem. 1956;17:65–134. doi: 10.1002/9780470122624.ch2. [DOI] [PubMed] [Google Scholar]
- 40.Zhang L, Yu L, Yu CA. J Biol Chem. 1998;273:33972–33976. doi: 10.1074/jbc.273.51.33972. [DOI] [PubMed] [Google Scholar]
- 41.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. J Biol Chem. 2003;278:36027–36031. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 42.Matsuno-Yagi A, Hatefi Y. J Biol Chem. 2001;276:19006–19011. doi: 10.1074/jbc.M101446200. [DOI] [PubMed] [Google Scholar]
- 43.Palmeira CM, Moreno AJ, Madeira VM. Biochim Biophys Acta. 1995;1229:187–192. doi: 10.1016/0005-2728(94)00202-g. [DOI] [PubMed] [Google Scholar]
- 44.Gomez C, Bandez MJ, Navarro A. Front Biosci. 2007;12:1079–1093. doi: 10.2741/2128. [DOI] [PubMed] [Google Scholar]
- 45.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. J Biol Chem. 1990;265:16330–16336. [PubMed] [Google Scholar]