

Abstract
Pore-forming toxins (PFTs) are potent cytolytic agents secreted by pathogenic bacteria that protect microbes against the cell-mediated immune system (by targeting phagocytic cells), disrupt epithelial barriers, and liberate materials necessary to sustain growth and colonization. Produced by gram-positive and gram-negative bacteria alike, PFTs are released as water-soluble monomeric or dimeric species, bind specifically to target membranes, and assemble transmembrane channels leading to cell damage and/or lysis. Structural and biophysical analyses of individual steps in the assembly pathway are essential to fully understanding the dynamic process of channel formation. To work toward this goal, we solved by X-ray diffraction the 2.9-Å structure of the 450-kDa heptameric Vibrio cholerae cytolysin (VCC) toxin purified and crystallized in the presence of detergent. This structure, together with our previously determined 2.3-Å structure of the VCC water-soluble monomer, reveals in detail the architectural changes that occur within the channel region and accessory lectin domains during pore formation including substantial rearrangements of hydrogen-bonding networks in the pore-forming amphipathic loops. Interestingly, a ring of tryptophan residues forms the narrowest constriction in the transmembrane channel reminiscent of the phenylalanine clamp identified in anthrax protective antigen [Krantz BA, et al. (2005) Science 309:777–781]. Our work provides an example of a β-barrel PFT (β-PFT) for which soluble and assembled structures are available at high-resolution, providing a template for investigating intermediate steps in assembly.
Keywords: hemolysin, membrane protein, X-ray crystallography, virulence factor
The devastating human pathogen Vibrio cholerae, which is endemic in many parts of the globe and responsible for thousands of deaths annually, produces a cell-damaging toxin, Vibrio cholerae cytolysin (VCC), that permeabilizes human intestinal and immune cells (1–3) and significantly facilitates intestinal colonization in mouse models (4). Although VCC’s involvement as a virulence factor in the human cholera pandemic remains unclear, this toxin belongs to a much larger class of pore-forming toxins (PFTs) secreted by a wide variety of human pathogens (5). PFTs are released by bacteria as water-soluble species and can attack cells at a distance, especially if they enter into the bloodstream. This poses an interesting physical problem in that the toxin must shield the hydrophobic residues that anchor the lytic channel in the membrane from the aqueous phase during transit to the intended cellular target. As a consequence, the channel-forming region comprises only a portion of the entire secreted toxin, and formation of a complete pore requires oligomerization of multiple subunits. The rest of the secreted polypeptide contains regions that target receptors, facilitate oligomerization, and regulate activity of the toxin.
Individual PFTs are generally classified into two primary groups depending on whether the final transmembrane channel contains an arrangement of α-helical or β-sheet motifs. VCC is classified as a β-PFT, a diverse family unified by amphipathic hairpin loops that insert to form β-barrel structures. Similar to VCC are the staphylococcal PFTs, which display weak sequence identity (approximately 15%) but strong structural similarity with the VCC core domain (6). Structures exist for the water-soluble bicomponent LukF (7, 8) and LukS (9) toxins as well as the homomeric α-hemolysin (α-HL) heptamer (10), which represents the only fully assembled β-PFT crystal structure determined until now. Distinct in sequence and structure from VCC (and each other), but with a similar heptameric stoichiometry, are the extensively studied aerolysin (11) and anthrax toxins (12) produced by Aeromonas hydrophila and Bacillus anthracis, respectively. Anthrax toxin, which consists of three proteins, is unique in that the heptameric pore [formed by protective antigen (PA)] facilitates the translocation of two toxic enzymes (lethal factor and edema factor) into the cytosol following clathrin-mediated endocytosis of the proteolytically activated and receptor-bound toxin complex. A third important class of β-PFTs is the cholesterol-dependent cytolysin (CDC) family, which are unrelated in sequence to VCC and include perfringolysin O from Clostridium perfringens and intermedilysin from Streptococcus intermedius (13). CDCs form large channels consisting of approximately 50 subunits, contribute two amphipathic hairpins per subunit (14), and, similar to VCC (15, 16), prefer cholesterol-rich membranes for lysis.
The VCC protoxin (17) consists of a core cytolytic domain containing the single prestem (pore-forming) loop, an amino-terminal 15-kDa prodomain, and two carboxyl-terminal domains with lectin-like folds that may aid in binding to carbohydrate receptors (18). Functional studies indicate that proteolytic activation must first occur within a consecutive string of protease sites that connect the prodomain to the toxin core (19). Extensive experimental evidence supports a model for β-PFT assembly in which individual toxin monomers bind to membranes, oligomerize into nonlytic prepore intermediates, and insert their stem domains into the membrane in a concerted fashion (20–23).
A wealth of structural and biophysical studies has outlined many key features relevant to the targeting and assembly of β-PFTs, but no high-resolution structures of identical toxins in water-soluble and membrane-inserted states exist. For VCC, our recent low-resolution structure determined by single-particle electron cryomicroscopy (24) confirms a heptameric endpoint for assembly (25) and suggests that substantial rearrangements of structural domains occur during the transition to the membrane-embedded pore. Although this structure provides an image of the gross domain organization of the heptamer, the resolution is insufficient to determine details of the transmembrane channel and to fully utilize the high-resolution monomeric crystal structure in understanding the molecular transitions that occur during assembly. With these limitations in mind, we solved the crystal structure of the VCC heptamer purified in detergent micelles. Our structure reveals an unexpected aromatic ring of residues within the channel lumen and a pore rich in charged amino acids. Furthermore, a comparison with the VCC monomer structure suggests that assembly entails a substantial rearrangement of secondary structure within the stem domain demonstrating why, at least for some β-PFTs, the prepore to pore transition is the rate-limiting step in assembly (21, 23).
Results and Discussion
Crystallization and X-Ray Diffraction.
To produce assembled VCC for structural studies, we activated monomeric protoxin with trypsin and added milligram quantities to asolectin liposomes containing 20% wt/wt cholesterol. Following solubilization in the nonionic detergent hexaethylene glycol monodecyl ether (C10E6), we purified heptameric material by size exclusion chromatography. Several additional detergents were successful in stabilizing monodisperse heptamer in solution, but C10E6 produced the best diffracting crystals for X-ray experiments. These crystals belonged to a P212121 space group with approximate unit cell dimensions of a = 172 Å, b = 182 Å, and c = 430 Å. This large unit cell, containing two heptamers per asymmetric unit (67% solvent content), provided a challenging data collection and phasing problem, and exhaustive attempts at using molecular replacement techniques proved unsuccessful. Phases were ultimately derived from a three-wavelength multiwavelength anomalous diffraction (MAD) experiment (26) using crystals soaked in 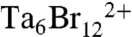 clusters (Fig. S1). Extension of low-resolution (4 Å) phases to the final native 2.9-Å data was greatly facilitated by the 14-fold noncrystallographic symmetry (NCS) and the high-resolution water-soluble monomer structure (2.3 Å) yielding excellent electron density maps. The final model refined against the 2.9-Å native dataset contained two VCC heptamers (residues 136–716), 650 water molecules, and had Rwork and Rfree factors of 21.8 and 24.9%, respectively (Table S1). Weak density for 14 detergent molecules surrounding the channel and fragments of the disordered amino terminus (residues 124–135) were discernable, but the density was too discontinuous to include in the model.
clusters (Fig. S1). Extension of low-resolution (4 Å) phases to the final native 2.9-Å data was greatly facilitated by the 14-fold noncrystallographic symmetry (NCS) and the high-resolution water-soluble monomer structure (2.3 Å) yielding excellent electron density maps. The final model refined against the 2.9-Å native dataset contained two VCC heptamers (residues 136–716), 650 water molecules, and had Rwork and Rfree factors of 21.8 and 24.9%, respectively (Table S1). Weak density for 14 detergent molecules surrounding the channel and fragments of the disordered amino terminus (residues 124–135) were discernable, but the density was too discontinuous to include in the model.
VCC Heptamer Structure.
The VCC heptamer forms a ring-like structure approximately 140 Å high perpendicular to the membrane plane with a widest outer diameter of 135 Å (Fig. 1 A and B). The channel pore, which runs along the 7-fold symmetry axis of the heptamer, is separated into an upper large vestibule, formed by β-prism and cytolysin domains, and a 14-strand transmembrane β-barrel formed by the stem domains. The β-barrel has a backbone-to-backbone diameter of approximately 25 Å and is ringed by a single layer of aromatic residues (composed of F288 and Y313) commonly observed in transmembrane β-barrel proteins near the lipid/solvent interface (27). However, VCC is missing a second ring on the opposite side of the barrel predicted to occur in many β-PFTs [primarily phenylalanines (28)]. At its narrowest point, the β-barrel has an approximately 8-Å-wide constriction [calculated by the program HOLE (29)] formed by a heptad of tryptophan residues (W318) conserved throughout many Vibrio species (except Vibrio vulnificus) but not Aeromonas species (which also lack the β-prism lectin). The upper vestibule surface is primarily acidic, whereas the β-barrel region contains alternating bands of basic and acidic amino acids with two rings of lysine residues near the intracellular opening of the stem (K304 and K306; Fig. 1 C and D). This is in contrast to staph α-HL, which is more neutral in character and wider within the channel (approximately 10-Å constriction formed by M113 and K147; Fig. 1E and Fig. S2). Consistent with these observations, electrophysiological measurements of single channels in planar lipid-bilayers indicate that VCC is moderately anion-selective and has a 4-fold lower conductivity than α-HL (30). Previous modeling studies of the VCC pore predicted an excess of positive charge near the intracellular opening (31), which may also help to explain the low permeability of Ca2+ ions through the channel (1). The VCC outer vestibule is more open than in α-HL, but repulsion of ions due to charges within the barrel could underlie the lower conductivity of VCC, similar to the 10-fold drop in conductance seen between ScrY and LamB, two glycoporins with nearly identical pore geometries but different electrostatic profiles (32).
Fig. 1.

Structure of the VCC heptamer. (A) Ribbon representation of the assembled heptamer. Core cytolysin domain (including rim region), blue; β-trefoil lectin, purple; β-prism lectin, gold; β-barrel stem, green. Side chains of aromatic residues near the putative membrane-solvent interface are shown in red. The approximate outline of the membrane is in gray. (B) Top view of the heptamer. (C) Surface representation of the heptamer sliced in half along the sevenfold axis and colored by electrostatic potential. Figure generated using APBS (55) and Chimera (56). (D) Outline of the central vestibule/channel of the VCC heptamer generated using HOLE (29). The sevenfold axis is shown as a yellow bar. (E) Graph showing the inner pore diameter along the sevenfold symmetry axis for VCC (purple solid line) and α-HL (dotted black line).
The ring of β-trefoil lectin domains sits atop the cytolysin domains and heightens the upper vestibule by 30 Å. An extended loop connects the β-trefoil lectin to the second β-prism lectin in a VCC protomer. This lectin domain interacts with the outer surface of the cytolysin domain, burying approximately 600 Å2 of accessible surface area. The cytolytic core region of the VCC protomer shares an overall topology with α-HL (RMSD 2.9 Å for 1453 of 2051 Cα residues, 11.9% sequence identity), with important differences discussed below.
Structural Rearrangements During Assembly.
Superimposing the cytolytic domains (residues 136–278 and 325–459) of the protoxin monomer and a protomer from the VCC oligomer reveals five major rearrangements that occur along the pathway between the protoxin and assembled states (Fig. 2 A and B and Movies S1 and S2). Firstly, the amino-terminal prodomain (amino acids 1–105) is absent from the heptameric assembly after liberation by proteolytic cleavage. Secondly, the β-prism lectin domain swings around the cytolytic domain to a location 180° opposite its starting place. This new position on the exterior of the ring (forming the “spikes” seen in the low-resolution EM structure) partially overlaps the previous location of the prodomain in the water-soluble protoxin and involves a different surface of the β-prism domain than utilized in interactions with the prestem. Our water-soluble monomer structure exhibits electron density for a bound glucoside moiety consistent with reports that this domain interacts with carbohydrate receptors on cell membranes. It appears that the β-prism rearrangement could occur while still bound to a carbohydrate receptor, with the final location of the site facing downward toward the cell membrane (as modeled in Fig. 2B). The third transition involves a 35° rigid-body rotation of the β-trefoil lectin domain around the loop connecting it to the cytolytic domain. The short helical turn that precedes the β-trefoil linker is anchored within the cytolysin domain by a phenylalanine residue (F455) that is necessary for oligomerization in the related Vibrio vulnificus hemolysin (33). The fourth transition is a movement of the loop that cradles the prestem in the water-soluble structure (residues 191–203) through hydrophobic side-chain interactions (notably L192, Y194, L307, and A309) and backbone hydrogen bonds involving G291 (Fig. S3). Superposition of the water-soluble monomer on top of the heptameric structure indicates this loop would sterically bump with each neighboring protomer if a rearrangement did not occur.
Fig. 2.

Comparison of the VCC protoxin structure and a protomer from the VCC heptamer. (A) The VCC water-soluble monomer structure with bound glucoside (PDB ID code 1XEZ) (17). Domains are colored as in Fig. 1 with the prodomain in red and W318 shown as green spheres. (B) In the assembled form, the stem domain is completely unfurled and the β-prism lectin domain moves to the opposite side of the cytolysin domain. The cradle loop has rearranged, contacting the neighboring protomer. The sugar headgroup seen in A is modeled into the β-prism lectin-binding site. (C) Schematic of the putative backbone hydrogen-bonding pattern in the prestem. Hydrogen bonds (using a 3.2-Å cutoff) are shown as black dashed lines. (D) The shifted hydrogen-bonding pattern of the assembled stem loop. Amino-acid side chains facing the membrane are marked with black dots, and the aromatic residues near the membrane/solvent interface are marked with gold asterisks.
Reordering of the cradle loop may destabilize the interactions holding the stem in the water-soluble position and initiate unfolding of the stem loop, which constitutes the fifth major transition. Rearrangement of the prestem loop from the water-soluble to assembled state requires a significant breaking and reforming of numerous polar and nonpolar interactions (Fig. 2 C and D). The prestem in the water-soluble monomer consists of a 10-residue-long antiparallel β-sheet with an intervening 18-residue loop with 310 helical characteristics. Analysis of hydrogen-bonding patterns identifies approximately 18 bonds between backbone atoms within the prestem that are broken during pore assembly. Upon formation of the β-barrel stem, each of the seven stem loops transforms into 19-residue-long antiparallel β-sheets held together by 20 newly formed backbone hydrogen bonds. Each protomer loop additionally forms 21 main chain hydrogen bonds with each of its two neighboring protomer stem loops. The net gain in hydrogen-bonding interactions explains the irreversibility of oligomer assembly, and the rearrangement of bonds likely constitutes an energy barrier that must be overcome to initiate stem unfolding. This transition represents the rate-limiting step in α-HL assembly, with a measured t1/2 of 8 min on rabbit erythrocyte membranes (21). In VCC, the rate-limiting step for pore formation is highly dependent on the membrane cholesterol content, an additional requirement necessary for the insertion of the stem (34). Hydrophobic residues that contact the inner leaflet of the target membrane in the assembled stem are mostly packed against hydrophobic residues on the cytolytic core in the water-soluble monomer, shielding them from water.
Most of the interactions between protomers in the heptamer are localized within the interface between cytolysin domains and stem loops and, with the exception of stem hydrogen bonds, are distinct from interactions that bridge α-HL protomers. The VCC cradle loop, which is absent in α-HL, forms multiple interactions between the cytolytic and lectin domains of neighboring protomers (Fig. 3A) and is in a position to coordinate assembly-related conformational movements between domains. This loop may play a functionally analogous role to the amino latch in the staph toxins, which prevents premature oligomerization of monomers in solution and may form cooperative interactions with the prestem (35). In α-HL, a key histidine residue (H35) forms important interprotomer contacts, and mutations to this residue arrest assembly at the prepore state (36). In VCC, the H35 position is replaced by a unique loop structure containing three consecutive aspartate residues that form salt bridges within and between protomers (Fig. 3B). Together, interactions between each pair of protomers bury 2,854 Å2 of accessible surface area with a total of 19,978 Å2 in the entire heptamer.
Fig. 3.

Key residues at the interprotomer interface (two protomers shown). (A) The cradle loop (yellow) knits together multiple domains through hydrogen bonding (dashed lines) and van der Waals (dotted surface) interactions (see Fig. S3 for more details). (B) An insertion containing three consecutive aspartate residues not present in staph toxins (red) participates in multiple salt-bridge interactions. Additional putative hydrogen bonds involving R330 form links between the protomers.
VCC Membrane Interactions.
The functional role of cholesterol in the assembly of VCC and other β-PFTs is still an area of intense investigation and may vary between different toxins. For some proteins such as aerolysin and anthrax toxin, cholesterol may serve to cluster membrane receptors and facilitate productive oligomerization of bound monomers (37, 38). In contrast, cholesterol is a receptor for the CDC perfringolysin O, which contains a two-residue motif (T490–L491 in YTTL sequence) within a membrane-interacting loop responsible for cholesterol recognition (39). Inspection of the membrane-proximal rim domain of VCC reveals an identical motif (T237–L238 in TTLY sequence) within a loop facing the membrane surface, which could similarly mediate interactions with cholesterol (the L238 side chain is disordered in our maps). A second possible site (A360–L361) is located in a comparable orientation on an adjacent loop (Fig. S2). We do not observe any bound lipid or cholesterol moieties in our crystal structure even though the toxin was solubilized from membranes containing 20% cholesterol. It is possible that such interactions are weak, disrupted by detergent, or nonspecific in nature; or that lipids and cholesterol only play a role in earlier stages of assembly. Additionally, the lipid-headgroup binding pocket observed within the LukF (7) and α-HL (40) rim domains is absent in VCC (neither staph toxin rim contains a Thr–Leu motif). A superposition of the VCC and α-HL β-barrel stem domains indicates that the hydrophobic and presumably membrane buried region of the VCC stem is approximately 3–5 Å longer than the α-HL stem, possibly due to complementarity with thicker membranes, such as found in cholesterol-rich “lipid-raft” subdomains (41). It remains to be seen whether a longer stem is a consequence of the toxin evolving toward increased stability within thicker regions of the membrane and to what extent cholesterol or lipid-binding motifs are responsible for membrane specificity.
Another distinction between the VCC heptamer and α-HL is the significantly longer loops within the VCC membrane-proximal rim domain (Fig. S2B). These loops are 10–15 Å longer in VCC, adopt a conformation nearly identical to the VCC water-soluble monomeric state, and would presumably extend much deeper into the membrane bilayer than the loops in α-HL. Aside from two valine residues (V422 and V423) on the tip of the longest loop and the two previously mentioned leucine residues (L238 and L361), the amino-acid composition within the rim domain loops is richly aromatic and contains a balance of tyrosine, tryptophan, and histidine residues: side chains less likely to penetrate deep into the bilayer. We note that our VCC EM structure (24) is consistent with the loop conformation seen in the crystal structure, which is not surprising because both were determined from solubilized heptamer in micelles rather than in a lipid bilayer environment. It is possible that these loops adopt a splayed conformation when the heptamer is sitting on the membrane and that detergent solubilization of the complex may have allowed them to relax to their preferred water-soluble conformation. Without significant reorientation, it is also possible that the loops distort the local lipid bilayer structure in a way that might facilitate insertion of the stem domain.
The VCC Pore.
Analysis of alignments of the predicted membrane-spanning loops of β-PFTs reveals common features within the family of β-barrel membrane proteins. Many channels are lined with charged amino acids and have an approximately 5-residue loop (or turn) containing hydrophobic and/or aromatic residues at the trans (opposite the side of entry) end of the pore. A “rivet” model for membrane insertion has been proposed, where this hydrophobic loop at the end of the stem hairpin folds back into the membrane and anchors the channel (28), a motif also observed in some bacterial outer membrane proteins. The α-HL toxin is an exception, with a neutral pore surface and two charged aspartate residues in the trans-loop (with sequence DDTGK). VCC combines features from both groups, having a highly charged lumen and a loop containing a single aspartate residue (with sequence SGDG). Instead of fluting outward on the trans side as α-HL does, the tip of the VCC channel backbone curves slightly inward (see Fig. S2B).
Another feature linking VCC with a nonhomologous β-PFT is the W318 aromatic ring positioned at the narrowest aperture of the pore. Within the anthrax PA channel lies a “φ-clamp,” a heptameric ring of phenylalanine residues (F427) that may also outline a pore constriction (42, 43). This hydrophobic belt forms a seal around translocating polypeptides and facilitates unidirectional transport through the pore (42). Mutations at position F427 in PA disrupt polypeptide translocation and in some cases also inhibit the prepore to pore transition (44). An interprotomer salt bridge lying directly above the φ-clamp (formed by K397 and D426) may help position F427 and neutralize charges near the constriction that interact strongly with a translocating protein (45). Quite remarkably, two charged residues in VCC reside directly above the W318 ring (K283 and E281) within close enough proximity to form a salt bridge (less than 4 Å), and VCC has an acidic central vestibule similar to anthrax toxin. At this time, we are unaware of any evidence that VCC serves as a translocation channel, although electrophysiological experiments indicate that synthetic peptides can fit within the α-HL pore (46) and presumably VCC as well. Certainly these intriguing similarities between VCC and anthrax toxin require further inquiry to elucidate whether analogous features exist in other β-PFTs and to determine to what extent they participate in the assembly mechanism.
Conclusions
The VCC heptamer structure provides a deeper understanding of the specific rearrangements that occur during assembly of a β-PFT as well as structural insight into the properties of the VCC channel. It supports an assembly model where interactions between large interprotomer surfaces drive loop rearrangements that lead to the unfolding of the channel stem (Fig. S4). The stem rearrangement requires the breaking and reforming of virtually all of the backbone hydrogen bonds within the stem loop as well as numerous van der Waals surface interactions. Our structure reveals intriguing similarities with structurally unrelated β-PFTs including an aromatic ring similar to anthrax toxin and putative cholesterol-binding sites shared with CDCs. This information will provide a launching point for new experiments aimed at understanding the specificity of VCC and related PFTs against target cell membranes and the involvement of individual membrane components and structural motifs in the assembly process.
Methods
Assembly of the VCC Oligomer.
Monomeric VCC was expressed and purified as described previously (17). Trypsin-cleaved toxin was added to freshly prepared soybean asolectin liposomes (1∶2 protein∶lipid ratio) containing 20% cholesterol and incubated for 30 min at room temperature. Following centrifugation for 30 min at 40 K rpm in a Beckman TL-100 rotor tabletop ultracentrifuge, oligomeric toxin was solubilized in a buffer containing 20 mM Tris pH 7.6, 150 mM NaCl, 1 mM EDTA, and 40 mM C10E6. After a second centrifugation step, the solubilized toxin was purified over a Superose 6 column in a buffer containing 20 mM Tris pH 7.6, 150 mM NaCl, 1 mM EDTA, and 1 mM C10E6. Peak fractions were concentrated to 10 mg/mL before crystallization using a 100-kDa cutoff membrane (Millipore Ultrafree).
Crystallization and Data Collection.
Oligomeric toxin was crystallized by the hanging drop method. One microliter of concentrated oligomer was added to an equal volume of buffer containing 20 mM HEPES pH 7.6, 10% PEG 2000, and 20 mM Co(NH3)6Cl3. Crystals were cryoprotected in 20% glycerol before freezing in liquid nitrogen. For heavy atom derivativatization, crystals were soaked overnight in a buffer containing 20 mM HEPES pH 7.6, 10% PEG 2000, 10% glycerol, and 1 mM 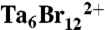 clusters (Jena Bioscience). All native and heavy atom data were collected at beamline 12-2 at the Stanford Synchrotron Radiation Laboratory (SSRL), and X-ray data were processed by HKL2000 (47). MAD data were collected using the inverse-beam method at the peak, infection point, and high remote energies based on tantalum fluorescence scans.
clusters (Jena Bioscience). All native and heavy atom data were collected at beamline 12-2 at the Stanford Synchrotron Radiation Laboratory (SSRL), and X-ray data were processed by HKL2000 (47). MAD data were collected using the inverse-beam method at the peak, infection point, and high remote energies based on tantalum fluorescence scans.
Structure Solving and Refinement.
The program SHELX (48) located cluster sites and provided preliminary phases (using data to 6.5 Å) resulting in electron density maps with a clear molecular outline for two VCC heptamer molecules in the asymmetric unit. Phases were extended to 4.0 Å by solvent flattening and 14-fold NCS averaging using the program PARROT (49). Real-space 14-fold NCS averaging by COOT (50) allowed manual docking of the three main domains from the 2.3-Å VCC monomer structure [Protein Data Bank (PDB) ID code 1XEZ]. The resulting 42 domains were subjected to rigid-body refinement [REFMAC (51)], first in the 4-Å data and then in a native dataset diffracting to 2.9 Å (resulting in an R factor of 44%). Missing regions, including the transmembrane channel region and several connecting loops, were built into 14-fold NCS averaged and B-factor sharpened 2Fo - Fc electron density maps. Alternating refinement and model-rebuilding steps were carried out using the PHENIX suite (52) and COOT. NCS restraints (coordinate sigma = 0.05 and B-factor weight = 10) were used in refinement, with residues 589–716 restrained in a second NCS group for chains A and L (due to crystal-contact induced loop rearrangements). Model validation was performed using Molprobity (53). Movies were created using the Gerstein morph script (52, 53) and CNS (54).
Supplementary Material
Acknowledgments.
We thank Dr. Eric Gouaux and Dr. Pamela Bjorkman for advice and financial support throughout this project. Thanks to Dr. Michelle Montoya for help in editing this manuscript. Dr. Jens Kaiser assisted with data collection at the Molecular Observatory (beamline 12-2 at SSRL), a gift from the Gordon and Betty Moore Foundation. A Beckman Institute Fellowship at California Institute of Technology (R.O.) also supported this work. We thank Wesleyan University for computer time, supported by the National Science Foundation under Grant CNS-0619508.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: Coordinates and structure factors for the VCC heptamer have been deposited in the Protein Data Bank, www.pdb.org (PDB ID code 3O44).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1017442108/-/DCSupplemental.
References
- 1.Zitzer A, Wassenaar TM, Walev I, Bhakdi S. Potent membrane-permeabilizing and cytocidal action of Vibrio cholerae cytolysin on human intestinal cells. Infect Immun. 1997;65:1293–1298. doi: 10.1128/iai.65.4.1293-1298.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valeva A, et al. Pro-inflammatory feedback activation cycle evoked by attack of Vibrio cholerae cytolysin on human neutrophil granulocytes. Med Microbiol Immunol. 2008;197:285–293. doi: 10.1007/s00430-007-0061-2. [DOI] [PubMed] [Google Scholar]
- 3.Debellis L, et al. The Vibrio cholerae cytolysin promotes chloride secretion from intact human intestinal mucosa. PLoS One. 2009;4:e5074. doi: 10.1371/journal.pone.0005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olivier V, Queen J, Satchell KJ. Successful small intestine colonization of adult mice by Vibrio cholerae requires ketamine anesthesia and accessory toxins. PLoS One. 2009;4:e7352. doi: 10.1371/journal.pone.0007352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iacovache I, van der Goot FG, Pernot L. Pore formation: An ancient yet complex form of attack. Biochim Biophys Acta. 2008;1778:1611–1623. doi: 10.1016/j.bbamem.2008.01.026. [DOI] [PubMed] [Google Scholar]
- 6.Olson R, Gouaux E. Vibrio cholerae cytolysin is composed of an alpha-hemolysin-like core. Protein Sci. 2003;12:379–383. doi: 10.1110/ps.0231703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson R, Nariya H, Yokota K, Kamio Y, Gouaux E. Crystal structure of staphylococcal LukF delineates conformational changes accompanying formation of a transmembrane channel. Nat Struct Biol. 1999;6:134–140. doi: 10.1038/5821. [DOI] [PubMed] [Google Scholar]
- 8.Pedelacq JD, et al. The structure of a Staphylococcus aureus leucocidin component (LukF-PV) reveals the fold of the water-soluble species of a family of transmembrane pore-forming toxins. Structure. 1999;7:277–287. doi: 10.1016/s0969-2126(99)80038-0. [DOI] [PubMed] [Google Scholar]
- 9.Guillet V, et al. Crystal structure of leucotoxin S component: New insight into the staphylococcal beta-barrel pore-forming toxins. J Biol Chem. 2004;279:41028–41037. doi: 10.1074/jbc.M406904200. [DOI] [PubMed] [Google Scholar]
- 10.Song L, et al. Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 11.Fivaz M, Abrami L, Tsitrin Y, van der Goot FG. Not as simple as just punching a hole. Toxicon. 2001;39:1637–1645. doi: 10.1016/s0041-0101(01)00151-9. [DOI] [PubMed] [Google Scholar]
- 12.Young JA, Collier RJ. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 13.Tweten RK. Cholesterol-dependent cytolysins, a family of versatile pore-forming toxins. Infect Immun. 2005;73:6199–6209. doi: 10.1128/IAI.73.10.6199-6209.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shatursky O, et al. The mechanism of membrane insertion for a cholesterol-dependent cytolysin: A novel paradigm for pore-forming toxins. Cell. 1999;99:293–299. doi: 10.1016/s0092-8674(00)81660-8. [DOI] [PubMed] [Google Scholar]
- 15.Ikigai H, Akatsuka A, Tsujiyama H, Nakae T, Shimamura T. Mechanism of membrane damage by El Tor hemolysin of Vibrio cholerae O1. Infect Immun. 1996;64:2968–2973. doi: 10.1128/iai.64.8.2968-2973.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zitzer A, Zitzer O, Bhakdi S, Palmer M. Oligomerization of Vibrio cholerae cytolysin yields a pentameric pore and has a dual specificity for cholesterol and sphingolipids in the target membrane. J Biol Chem. 1999;274:1375–1380. doi: 10.1074/jbc.274.3.1375. [DOI] [PubMed] [Google Scholar]
- 17.Olson R, Gouaux E. Crystal structure of the Vibrio cholerae cytolysin (VCC) pro-toxin and its assembly into a heptameric transmembrane pore. J Mol Biol. 2005;350:997–1016. doi: 10.1016/j.jmb.2005.05.045. [DOI] [PubMed] [Google Scholar]
- 18.Saha N, Banerjee KK. Carbohydrate-mediated regulation of interaction of Vibrio cholerae hemolysin with erythrocyte and phospholipid vesicle. J Biol Chem. 1997;272:162–167. doi: 10.1074/jbc.272.1.162. [DOI] [PubMed] [Google Scholar]
- 19.Nagamune K, et al. In vitro proteolytic processing and activation of the recombinant precursor of El Tor cytolysin/hemolysin (pro-HlyA) of Vibrio cholerae by soluble hemagglutinin/protease of V. cholerae, trypsin, and other proteases. Infect Immun. 1996;64:4655–4658. doi: 10.1128/iai.64.11.4655-4658.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walker B, Krishnasastry M, Zorn L, Bayley H. Assembly of the oligomeric membrane pore formed by staphylococcal alpha-hemolysin examined by truncation mutagenesis. J Biol Chem. 1992;267:21782–21786. [PubMed] [Google Scholar]
- 21.Walker B, Braha O, Cheley S, Bayley H. An intermediate in the assembly of a pore-forming protein trapped with a genetically-engineered switch. Chem Biol. 1995;2:99–105. doi: 10.1016/1074-5521(95)90282-1. [DOI] [PubMed] [Google Scholar]
- 22.Valeva A, Palmer M, Bhakdi S. Staphylococcal alpha-toxin: Formation of the heptameric pore is partially cooperative and proceeds through multiple intermediate stages. Biochemistry. 1997;36:13298–13304. doi: 10.1021/bi971075r. [DOI] [PubMed] [Google Scholar]
- 23.Bayley H, Jayasinghe L, Wallace M. Prepore for a breakthrough. Nat Struct Mol Biol. 2005;12:385–386. doi: 10.1038/nsmb0505-385. [DOI] [PubMed] [Google Scholar]
- 24.He Y, Olson R. Three-dimensional structure of the detergent-solubilized Vibrio cholerae cytolysin (VCC) heptamer by electron cryomicroscopy. J Struct Biol. 2010;169:6–13. doi: 10.1016/j.jsb.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 25.Harris JR, et al. Interaction of the Vibrio cholerae cytolysin (VCC) with cholesterol, some cholesterol esters, and cholesterol derivatives: A TEM study. J Struct Biol. 2002;139:122–135. doi: 10.1016/s1047-8477(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 26.Hendrickson WA, Horton JR, LeMaster DM. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 1990;9:1665–1672. doi: 10.1002/j.1460-2075.1990.tb08287.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gouaux E. Roll out the barrel. Nat Struct Biol. 1998;5:931–932. doi: 10.1038/2804. [DOI] [PubMed] [Google Scholar]
- 28.Iacovache I, et al. A rivet model for channel formation by aerolysin-like pore-forming toxins. EMBO J. 2006;25:457–466. doi: 10.1038/sj.emboj.7600959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smart OS, Goodfellow JM, Wallace BA. The pore dimensions of gramicidin A. Biophys J. 1993;65:2455–2460. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menzl K, Maier E, Chakraborty T, Benz R. HlyA hemolysin of Vibrio cholerae O1 biotype E1 Tor. Identification of the hemolytic complex and evidence for the formation of anion-selective ion-permeable channels. Eur J Biochem. 1996;240:646–654. doi: 10.1111/j.1432-1033.1996.0646h.x. [DOI] [PubMed] [Google Scholar]
- 31.Pantano S, Montecucco C. A molecular model of the Vibrio cholerae cytolysin transmembrane pore. Toxicon. 2006;47:35–40. doi: 10.1016/j.toxicon.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Ranatunga KM, Adcock C, Kerr ID, Smith GR, Sansom MS. Ion channels of biological membranes: Prediction of single channel conductance. Theor Chem Acc. 1999;101:97–102. [Google Scholar]
- 33.Kashimoto T, et al. The aromatic ring of phenylalanine 334 is essential for oligomerization of Vibrio vulnificus hemolysin. J Bacteriol. 2009;192:568–574. doi: 10.1128/JB.01049-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krasilnikov OV, et al. Pore formation by Vibrio cholerae cytolysin requires cholesterol in both monolayers of the target membrane. Biochimie. 2007;89:271–277. doi: 10.1016/j.biochi.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 35.Jayasinghe L, Miles G, Bayley H. Role of the amino latch of staphylococcal alpha-hemolysin in pore formation: A co-operative interaction between the N terminus and position 217. J Biol Chem. 2006;281:2195–2204. doi: 10.1074/jbc.M510841200. [DOI] [PubMed] [Google Scholar]
- 36.Panchal RG, Bayley H. Interactions between residues in staphylococcal alpha-hemolysin revealed by reversion mutagenesis. J Biol Chem. 1995;270:23072–23076. doi: 10.1074/jbc.270.39.23072. [DOI] [PubMed] [Google Scholar]
- 37.Abrami L, van Der Goot FG. Plasma membrane microdomains act as concentration platforms to facilitate intoxication by aerolysin. J Cell Biol. 1999;147:175–184. doi: 10.1083/jcb.147.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrand AJ, LaChapelle S, Hotze EM, Johnson AE, Tweten RK. Only two amino acids are essential for cytolytic toxin recognition of cholesterol at the membrane surface. Proc Natl Acad Sci USA. 2010;107:4341–4346. doi: 10.1073/pnas.0911581107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galdiero S, Gouaux E. High resolution crystallographic studies of alpha-hemolysin-phospholipid complexes define heptamer-lipid head group interactions: Implication for understanding protein-lipid interactions. Protein Sci. 2004;13:1503–1511. doi: 10.1110/ps.03561104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 42.Krantz BA, et al. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science. 2005;309:777–781. doi: 10.1126/science.1113380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katayama H, et al. Three-dimensional structure of the anthrax toxin pore inserted into lipid nanodiscs and lipid vesicles. Proc Natl Acad Sci USA. 2010;107:3453–3457. doi: 10.1073/pnas.1000100107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun J, Lang AE, Aktories K, Collier RJ. Phenylalanine-427 of anthrax protective antigen functions in both pore formation and protein translocation. Proc Natl Acad Sci USA. 2008;105:4346–4351. doi: 10.1073/pnas.0800701105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Melnyk RA, Collier RJ. A loop network within the anthrax toxin pore positions the phenylalanine clamp in an active conformation. Proc Natl Acad Sci USA. 2006;103:9802–9807. doi: 10.1073/pnas.0604000103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Movileanu L, Schmittschmitt JP, Scholtz JM, Bayley H. Interactions of peptides with a protein pore. Biophys J. 2005;89:1030–1045. doi: 10.1529/biophysj.104.057406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 48.Sheldrick GM. A short history of SHELX. Acta Crystallogr A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 49.Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 50.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 51.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 52.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 53.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brünger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 55.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pettersen EF, et al. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.