

Abstract
Zn2+ plays essential roles in biology, and cells have adopted exquisite mechanisms for regulating steady-state Zn2+ levels. Although much is known about total Zn2+ in cells, very little is known about its subcellular distribution. Yet defining the location of Zn2+ and how it changes with signaling events is essential for elucidating how cells regulate this essential ion. Here we create fluorescent sensors genetically targeted to the endoplasmic reticulum (ER) and Golgi to monitor steady-state Zn2+ levels as well as flux of Zn2+ into and out of these organelles. These studies reveal that ER and Golgi contain a concentration of free Zn2+ that is 100 times lower than the cytosol. Both organelles take up Zn2+ when cytosolic levels are elevated, suggesting that the ER and Golgi can sequester elevated cytosolic Zn2+ and thus have the potential to play a role in influencing Zn2+ toxicity. ER Zn2+ homeostasis is perturbed by small molecule antagonists of Ca2+ homeostasis and ER Zn2+ is released upon elevation of cytosolic Ca2+ pointing to potential exchange of these two ions across the ER. This study provides direct evidence that Ca2+ signaling can influence Zn2+ homeostasis and vice versa, that Zn2+ dynamics may modulate Ca2+ signaling.
Keywords: zinc homeostasis, calcium signaling, zinc sensor, FRET
Zinc is the second most abundant transition metal in biological organisms, with mammalian cells containing an estimated 100–500 μM total Zn2+ (1, 2). The majority of this Zn2+ is bound to proteins and enzymes and indeed it has been estimated that approximately 10% of the human proteome or 3,200 proteins require zinc for their structure and function (3). It is now well established that mammalian cells contain a small but measurable pool of free or labile Zn2+ in the cytosol that is buffered in the picomolar range (4–6). Fluctuations in cytosolic Zn2+ have been shown to influence signaling cascades such as the mitogen activated protein kinase pathway (7), as well as cellular processes such as mitochondrial function (8, 9), apoptosis (10, 11), and dendritic cell maturation (12). Although many studies have focused on cytosolic Zn2+, the overall distribution of zinc at the subcellular level is not well defined. In particular, it is not clear whether organelles contain labile Zn2+, if so how these Zn2+ pools influence organelle function, and whether organelles modulate fluctuations in cytosolic Zn2+. Yet defining where Zn2+ is located, whether it translocates from one place to another in response to stimuli, and how Zn2+ flux influences cellular processes is a fundamental part of elucidating how cells regulate and are influenced by this essential metal ion.
Mammalian cells contain an elaborate network of Zn2+ transporters, including 10 ZnT/cation diffusion facilitator (SLC30) family members and 14 Zrt-, Irt-like protein (SLC39) family members (13). These transporters help mediate zinc flux into and out of the cell and intracellular organelles. It is well established that Zn2+ can be concentrated into secretory vesicles (14), and recent studies have demonstrated a labile pool of Zn2+ in mitochondria (9, 15, 16), yet little is known about other intracellular organelles such as the endoplasmic reticulum (ER) and Golgi. Numerous proteins found within the secretory pathway require Zn2+ for their function, including resident ER chaperones such as calnexin and calreticulin, as well as secreted proteins such as metalloproteases and alkaline phosphatases (2). Moreover, deletion of an ER-localized ZnT in both yeast and higher eukaryotic cells led to activation of the unfolded protein response and general ER dysfunction (17, 18), suggesting Zn2+ plays an essential role in normal ER function.
In this work, we develop high-affinity genetically encoded Zn2+ sensors targeted to organelles to allow direct monitoring of ER and Golgi Zn2+ levels. These sensors reveal free Zn2+ in both the ER and Golgi at a concentration just under 1 pM. We demonstrate that the ER and Golgi sequester excess cytosolic Zn2+ indicating that organelles have the potential to play a role in modulating bioavailability of cytosolic Zn2+. Moreover, using a combination of Ca2+ and Zn2+ sensors along with small molecule antagonists of calcium homeostasis, we discover a connection between ER Ca2+ stores and ER Zn2+ dynamics, providing evidence of interplay between Zn2+ homeostasis and Ca2+ signaling.
Results
Generation of High-Affinity Zn2+ Sensors Comprising Zap1, Cyan, and Yellow Fluorescent Proteins (ZapCY1 and ZapCY2).
In this work, we optimized a sensor developed by Qiao et al. (19) which is comprised of the first and second zinc fingers of Saccharomyces cerevisiae Zap1 sandwiched between two fluorescent proteins [enhanced cyan fluorescent protein (CFP) and enhanced yellow fluorescent protein (EYFP)]. Zn2+ binding induces a conformational change in the pair of zinc fingers leading to an increase in FRET from CFP to YFP (Fig. 1A). In yeast suspensions of the original sensor, Zn2+ led to a 1.3-fold increase in the FRET ratio (R), defined as the FRET emission intensity divided by the CFP emission intensity (19). To optimize the sensor, we truncated CFP, replaced EYFP with the more pH stable citrine (20), and changed the amino acids in the linker regions to those found in Ca2+ sensors. Such changes have previously been shown to optimize the FRET response of genetically encoded calcium sensors (21). To characterize the sensor, which we named ZapCY1, we measured the apparent dissociation constant ( ) of purified sensor protein in vitro. The Zn2+ binding curve was fit to a one-site saturation model yielding a
) of purified sensor protein in vitro. The Zn2+ binding curve was fit to a one-site saturation model yielding a 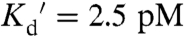 (Fig. 1B). Titration with a series of metal ions revealed that ZapCY1 was selective for Zn2+ over other metal ions (Ca2+, Mg2+, Cu1+, Cu2+, Mn2+, Co2+, Ni2+, Fe2+) (Fig. 1C and SI Appendix, Fig. S1A). Only Zn2+ gave rise to a FRET ratio change when ZapCY1 protein was treated with excess metal ions. Because Ca2+ and Mg2+ are present at high concentrations in cells, we verified that micromolar concentrations of these ions do not perturb the sensor in vitro (SI Appendix, Fig. S1C) or in HeLa cells (SI Appendix, Fig. S2E). Treatment with neocuproine, a Cu1+ chelator, and 2,2-bipyridine, an Fe2+ chelator, had no effect on the sensor in HeLa cells, indicating that physiological Cu1+ and Fe2+ do not interfere with the sensor in cells under resting conditions (SI Appendix, Fig. S2). Fig. 1D demonstrates that ZapCY1 exhibits a large decrease in the FRET ratio when treated with a membrane permeable metal chelator N,N,N′,N′-tetrakis-(2-pyridylmethyl)-ethylenediamine (TPEN), followed by an increase upon saturation with Zn2+, yielding the minimum and maximum ratio (Rmin and Rmax), respectively. In cells, ZapCY1 exhibits a 4.15-fold dynamic range (Rmax/Rmin) (Fig. 1D).
(Fig. 1B). Titration with a series of metal ions revealed that ZapCY1 was selective for Zn2+ over other metal ions (Ca2+, Mg2+, Cu1+, Cu2+, Mn2+, Co2+, Ni2+, Fe2+) (Fig. 1C and SI Appendix, Fig. S1A). Only Zn2+ gave rise to a FRET ratio change when ZapCY1 protein was treated with excess metal ions. Because Ca2+ and Mg2+ are present at high concentrations in cells, we verified that micromolar concentrations of these ions do not perturb the sensor in vitro (SI Appendix, Fig. S1C) or in HeLa cells (SI Appendix, Fig. S2E). Treatment with neocuproine, a Cu1+ chelator, and 2,2-bipyridine, an Fe2+ chelator, had no effect on the sensor in HeLa cells, indicating that physiological Cu1+ and Fe2+ do not interfere with the sensor in cells under resting conditions (SI Appendix, Fig. S2). Fig. 1D demonstrates that ZapCY1 exhibits a large decrease in the FRET ratio when treated with a membrane permeable metal chelator N,N,N′,N′-tetrakis-(2-pyridylmethyl)-ethylenediamine (TPEN), followed by an increase upon saturation with Zn2+, yielding the minimum and maximum ratio (Rmin and Rmax), respectively. In cells, ZapCY1 exhibits a 4.15-fold dynamic range (Rmax/Rmin) (Fig. 1D).
Fig. 1.

Generation of high-affinity Zn2+ sensor ZapCY. (A) Schematic of the ZapCY sensor. (B) In vitro titration of ZapCY1 (●, Kd = 2.53 pM) and ZapCY2 (▪, Kd = 811 pM, n = 0.44) sensor. (C) Metal selectivity of ZapCY1 and ZapCY2 sensor. Two micromolar ZapCY1 solutions were combined with 15 μM EDTA and 20 μM indicated metals, to yield 2∶5 molar ratio of protein:metal. For ZapCY2, 1 μM metal was added to 2 μM sensor solutions to yield 2∶1 solutions. Data represent the FRET ratio upon addition of a given metal minus the Rmin. (D) Representative experiment (n = 5 cells) demonstrating changes in the FRET ratio of ZapCY1 in the cytosol of HeLa cells. (E) Representative experiment (n = 6 cells) demonstrating changes in the FRET ratio of ZapCY2 in the cytosol of HeLa cells. For D and E, FRET ratios differ from in vitro ratios due to different instrumentation and data processing. Microscope filter combinations for FRET and CFP: 430/24 excitation filter, 455 dichroic, 535/25, and 470/24 emission filters, respectively.
Mutation of two cysteines in the binding domain of ZapCY1 to histidine lowered the 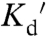 , generating another sensor ZapCY2. In vitro Zn2+ titration data fit equally well to an empirical Hill expression (
, generating another sensor ZapCY2. In vitro Zn2+ titration data fit equally well to an empirical Hill expression ( of 811 pM, n = 0.44, Fig. 1B) and a two-site saturation model (SI Appendix, Methods). This sensor yielded a 1.4-fold dynamic range in HeLa cells (Fig. 1E). Pseudocolored FRET ratio images of a representative titration are presented in SI Appendix, Fig. S3. ZapCY2 was less selective than ZapCY1 at a 2∶5 molar ratio of protein:metal (SI Appendix, Fig. S1D), but was reasonably selective for Zn2+ at a 2∶1 molar ratio (Fig. 1C). However, ZapCY2 was perturbed by Cu1+, even at very low molar excesses in vitro (SI Appendix, Fig. S1A). Therefore, we tested whether Cu1+ was capable of influencing measurements of resting cytosolic Zn2+; neocuproine had no effect on the resting FRET ratio, indicating that physiological Cu1+ does not perturb the sensor under resting conditions (SI Appendix, Fig. S2D). In addition, ZapCY2 was not perturbed by endogenous Fe2+ or micromolar levels of Ca2+ and Mg2+ in cells (SI Appendix, Figs. S1 and S2).
of 811 pM, n = 0.44, Fig. 1B) and a two-site saturation model (SI Appendix, Methods). This sensor yielded a 1.4-fold dynamic range in HeLa cells (Fig. 1E). Pseudocolored FRET ratio images of a representative titration are presented in SI Appendix, Fig. S3. ZapCY2 was less selective than ZapCY1 at a 2∶5 molar ratio of protein:metal (SI Appendix, Fig. S1D), but was reasonably selective for Zn2+ at a 2∶1 molar ratio (Fig. 1C). However, ZapCY2 was perturbed by Cu1+, even at very low molar excesses in vitro (SI Appendix, Fig. S1A). Therefore, we tested whether Cu1+ was capable of influencing measurements of resting cytosolic Zn2+; neocuproine had no effect on the resting FRET ratio, indicating that physiological Cu1+ does not perturb the sensor under resting conditions (SI Appendix, Fig. S2D). In addition, ZapCY2 was not perturbed by endogenous Fe2+ or micromolar levels of Ca2+ and Mg2+ in cells (SI Appendix, Figs. S1 and S2).
Fig. 1 demonstrates that ZapCY1 was fully saturated in the cytosol under resting conditions (i.e., Rresting = Rmax), whereas ZapCY2 was only partially saturated. Using ZapCY2, we measured Rresting, Rmin, and Rmax in 16 individual cells and used these parameters to estimate the concentration of free Zn2+ in the cytosol to be 80 ± 7 pM (SI Appendix, Table S1). This estimate is consistent with previous reports that resting cytosolic concentrations in human colon adenocarcinoma HT-29, HEK 293, and rat insulinoma INS-1(832/13) cells are in the hundreds of picomolar range (5, 22).
Measurement of Zn2+ in the ER and Golgi.
Because we were unsure whether ER and Golgi Zn2+ levels would be higher or lower than the cytosol, the high-affinity ZapCY1 and a low-affinity Zn2+ sensor (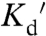 1 μM), ZifCY1, previously developed in our lab (15) were targeted to lumen of the ER and the inner surface of the Golgi membrane (Fig. 2 A and D). Colocalization with established ER and Golgi markers confirmed targeting to the desired organelles (SI Appendix, Figs. S4 and S5). Treatment with TPEN and Zn2+ revealed that both sensors were functional in each of the targeted locations (Fig. 2). TPEN led to a decrease in the FRET ratio for both ER-ZapCY1 and Golgi-ZapCY1 (Fig. 2 B and E), indicating the presence of free Zn2+ in the ER and Golgi. Under the same conditions, no decrease in the FRET ratio was observed for the low-affinity ER-ZifCY1 and Golgi-ZifCY1, likely because the level of Zn2+ in ER and Golgi was below the detection limit. Under resting conditions, ZapCY1 was 100% saturated in cytosol, but only 26% saturated in ER and 18% in Golgi, revealing that the labile Zn2+ level in ER and Golgi was lower than in cytosol (SI Appendix, Fig. S6A). To calculate resting Zn2+ concentrations in the ER and Golgi, HeLa cells were treated with TPEN to obtain Rmin and perfused with Zn2+ and pyrithione to saturate the sensor and obtain Rmax (Fig. 2 B and E). For the ER, thapsigargin was used in combination with TPEN because this aided depletion of the ER. Pseudocolored FRET ratio images of a representative titration are presented in SI Appendix, Fig. S3. The free Zn2+ in ER ([Zn2+]ER) was estimated to be 0.9 ± 0.1 pM and 0.6 ± 0.1 pM in Golgi ([Zn2+]Golgi) (SI Appendix, Table S1). The dynamic range of the targeted sensors were comparable [ER-ZapCY1, 2.16 ± 0.06 (n = 13 cells, three independent experiments); Golgi-ZapCY1, 2.09 ± 0.07 (n = 13 cells, three independent experiments)], but were lower than that observed in the cytosol. These data demonstrate that the ER and Golgi contain free Zn2+ that is buffered at a concentration significantly lower than the cytosol.
1 μM), ZifCY1, previously developed in our lab (15) were targeted to lumen of the ER and the inner surface of the Golgi membrane (Fig. 2 A and D). Colocalization with established ER and Golgi markers confirmed targeting to the desired organelles (SI Appendix, Figs. S4 and S5). Treatment with TPEN and Zn2+ revealed that both sensors were functional in each of the targeted locations (Fig. 2). TPEN led to a decrease in the FRET ratio for both ER-ZapCY1 and Golgi-ZapCY1 (Fig. 2 B and E), indicating the presence of free Zn2+ in the ER and Golgi. Under the same conditions, no decrease in the FRET ratio was observed for the low-affinity ER-ZifCY1 and Golgi-ZifCY1, likely because the level of Zn2+ in ER and Golgi was below the detection limit. Under resting conditions, ZapCY1 was 100% saturated in cytosol, but only 26% saturated in ER and 18% in Golgi, revealing that the labile Zn2+ level in ER and Golgi was lower than in cytosol (SI Appendix, Fig. S6A). To calculate resting Zn2+ concentrations in the ER and Golgi, HeLa cells were treated with TPEN to obtain Rmin and perfused with Zn2+ and pyrithione to saturate the sensor and obtain Rmax (Fig. 2 B and E). For the ER, thapsigargin was used in combination with TPEN because this aided depletion of the ER. Pseudocolored FRET ratio images of a representative titration are presented in SI Appendix, Fig. S3. The free Zn2+ in ER ([Zn2+]ER) was estimated to be 0.9 ± 0.1 pM and 0.6 ± 0.1 pM in Golgi ([Zn2+]Golgi) (SI Appendix, Table S1). The dynamic range of the targeted sensors were comparable [ER-ZapCY1, 2.16 ± 0.06 (n = 13 cells, three independent experiments); Golgi-ZapCY1, 2.09 ± 0.07 (n = 13 cells, three independent experiments)], but were lower than that observed in the cytosol. These data demonstrate that the ER and Golgi contain free Zn2+ that is buffered at a concentration significantly lower than the cytosol.
Fig. 2.

Targeting Zn2+ sensors to ER and Golgi in HeLa cells. (A) Fluorescence image of ER-ZapCY1. (B) Representative traces (n = 6 cells) for ER-ZapCY1. The FRET ratio decreased with 150 μM TPEN/10 μM thapsigargin and increased with addition of 5 μM pyrithione and 10 nM Zn2+. (C) Representative traces (n = 6 cells) for ER-ZifCY1. The FRET ratio did not change with TPEN but increased with 25 μM pyrithione, 0.001 mg/mL saponin, and 500 μM Zn2+. (D) Fluorescence image of Golgi-ZapCY1. (E) Representative traces (n = 4 cells) for Golgi-ZapCY1. The FRET ratio decreased with 150 μM TPEN and increased with addition of 5 μM pyrithione and 10 nM Zn2+. (F) Representative traces for Golgi-ZifCY2 (n = 7 cells). The FRET ratio did not change with TPEN but increased with addition of 25 μM pyrithione, 0.001 mg/mL saponin, and 500 μM Zn2+.
Zn2+ Sequestration into ER and Golgi.
Intracellular Zn2+ levels are tightly regulated by transporters and buffering by cytosolic proteins such as metallothionein (2, 22, 23). A fundamental unanswered question is whether organelles sequester zinc, particularly given that there is an excess of high-affinity ligands in the cytosol (22). To address this question the plasma membrane was permeabilized with saponin and buffered Zn2+ solutions were added to cells. Zn2+ uptake was observed with both the high-affinity ER-ZapCY1 and low-affinity ER-ZifCY1, demonstrating that the ER can concentrate high levels of Zn2+ (Fig. 3 A and B). In the Golgi, only Golgi-ZapCY1 detected Zn2+ influx, suggesting that the Golgi can concentrate cytosolic Zn2+ but to a lower level than the ER (Fig. 3). Next, we explored whether changes in the extracellular environment could affect cytosolic Zn2+ levels and organelle sequestration. Fig. 3C demonstrates that when 100 μM Zn2+ is added to cell media, in the absence of permeabilizing agents, Zn2+ is transported into the cytosol, reaching levels of approximately 5.5 nM (SI Appendix, Fig. S7). In this experiment, excess Zn2+ that accumulates in the cytosol is sequestered into both the ER and Golgi, providing direct evidence that Zn2+ is transported into organelles when cytosolic levels reach the nanomolar range. SI Appendix, Fig. S11 presents pseudocolor ratio images of the changes in [Zn2+]ER over the time course presented in Fig. 3C.
Fig. 3.

Comparison of Zn2+ uptake in ER and Golgi. (A) HeLa cells expressing ZifCY1 targeted to ER and Golgi were permeabilized with saponin for 30 min and then incubated with ZnCl2 (ER: black square, 100 μM, n = 10 cells; red circle, 12 μM, n = 4 cells; green diamond, 0.58 μM, n = 4 cells; violet triangle, 0.044 μM, n = 7 cells; and blue square, Golgi, 100 μM, n = 9 cells). (B) HeLa cells expressing ER-ZapCY1 were permeabilized with saponin for 30 min and then incubated with 193 pM (violet triangle, n = 4 cells), 2 nM (green circle, n = 7 cells), and 44 nM (red square, n = 4 cells) Zn2+. Uptake in ER was recorded as low as 2 nM Zn2+. (C) HeLa cells expressing high-affinity Zn2+ sensor ER-ZapCY1 (n = 6 cells), Golgi-ZapCY1 (n = 4 cells), and ZapCY2 were incubated with 100 μM ZnCl2. All Zn2+ concentrations below 100 μM were buffered solutions. The left axis represents the cytosolic concentrations whereas the right axis represents the FRET changes in the ER and Golgi.
Regulation of ER Zn2+ Uptake.
To examine the mechanism of Zn2+ uptake into the ER, we explored whether the sarco/endoplasmic-reticulum-Ca2+-ATPase (SERCA) was responsible for Zn2+ uptake. SERCA is the primary means by which Ca2+ is pumped into the ER and Zn2+ has been shown to penetrate many Ca2+ channels (24), leading us to question whether SERCA could also pass Zn2+. Surprisingly, treatment with a SERCA inhibitor (100 nM thapsigargin) did not inhibit Zn2+ uptake, but instead enhanced uptake (Fig. 4A). The ER is the main storehouse of Ca2+ within the cell and ER Ca2+ levels are regulated by SERCA, as well as Ca2+ release channels such as the inositol-1,4,5-trisphosphate receptor (IP3R) and ryanodine receptor (RyR). Inhibition of SERCA causes Ca2+ to slowly leak out of the ER, leading to ER Ca2+ depletion, elevation of cytosolic Ca2+, and activation of store-operated Ca2+ channels to promote influx of Ca2+ across the plasma membrane. Thus we next explored whether other agents that perturb ER Ca2+ impact ER Zn2+ uptake. Cells were treated with histamine which leads to ER Ca2+ release through the IP3R, 2-aminoethoxydiphenyl borate (2-APB) an IP3R inhibitor, and diltiazem, an L-type Ca2+ antagonist and inhibitor of RyR. All reagents were used at standard concentrations known to perturb Ca2+ homeostasis (25–27). Both agents (thapsigargin and histamine), which induce release of ER Ca2+ into the cytosol, led to enhanced Zn2+ uptake, whereas 2-APB which inhibits Ca2+ release through the IP3R did not have a statistically significant effect on Zn2+ uptake (Fig. 4B). Diltiazem, a RyR inhibitor (27), also gave rise to enhanced Zn2+ uptake. At present, we do not have a cohesive explanation for why pharmacological agents with widely different direct impacts on ER Ca2+ (two agents promote release, whereas one inhibits release) all enhance Zn2+ uptake. However, it is clear that maintenance of ER Ca2+ involves a delicately balanced network of processes and perturbation of one component often results in multiple changes in ER Ca2+ handling. The pharmacological perturbations presented in this paper do suggest that disruption of the ER Ca2+ regulatory network influences Zn2+ uptake.
Fig. 4.

Regulation of Zn2+ uptake in ER. (A) Thapsigargin enhanced Zn2+ uptake in ER. HeLa cells expressing ER-ZapCY1 sensor were treated with DMSO (n = 17 cells, three experiments) or 100 nM thapsigargin (n = 17 cells, three experiments) for 5 min (R0), and then incubated with 100 μM ZnCl2 (R). The relative ratio (R/R0) indicates the changes of Zn2+ in ER. (B) Effect of different drugs on Zn2+ uptake in ER. HeLa cells expressing ER-ZapCY1 sensors were treated with DMSO (n = 17 cells from three experiments), 100 nM thapsigargin (n = 17 cells, three experiments), 50 μM 2-APB (n = 23 cells, three experiments), 100 μM histamine (n = 18 cells from three experiments), or 50 μM diltizem (n = 17 cells, three experiments) for 5 min followed by addition of 100 μM ZnCl2 to activate ER Zn2+ uptake for 30 min. Compared with DMSO, thapsigargin, histamine, and diltiazem induced greater Zn2+ uptake (**p < 0.01, ANOVA, Tukey honestly significant difference post hoc test).
To further characterize the connection between Ca2+ dynamics and ER Zn2+ homeostasis, we explored whether [Ca2+]cyto impacts [Zn2+]ER. For these experiments, genetically targeted Ca2+ sensors D1ER (28) and D3cpv (29) were used to measure [Ca2+]ER and [Ca2+]cyto, respectively. Treatment of HeLa cells with thapsigargin in either the presence or absence of extracellular Ca2+ led to Ca2+ efflux from ER where reduction of [Ca2+]ER was independent of the extracellular media employed in the experiment (Fig. 5A). Calcium efflux from the ER led to rapid elevation of [Ca2+]cyto and a decrease in [Zn2+]ER (Fig. 5 B and C). ER Ca2+ depletion upon thapsigargin treatment activates store-opened Ca2+ channels on plasma membrane, which, in the presence of extracellular Ca2+, led to sustained elevation of [Ca2+]cyto and continued decrease in [Zn2+]ER (Fig. 5B). On the other hand, in the absence of extracellular Ca2+, [Ca2+]cyto returned back to resting levels and coincident with this [Zn2+]ER increased, possibly as a result of influx from the cytosol (Fig. 5C). These perturbations suggest that Ca2+ levels in the cytosol influence whether Zn2+ is taken up into or released from the ER.
Fig. 5.

Ca2+ dynamics affect Zn2+ homeostasis in the ER. (A) Effect of thapsigargin on [Ca2+]ER. Treatment with 100 nM thapsigargin, caused [Ca2+]ER to decrease in the presence (n = 6 cells) and absence of (n = 6 cells) extracellular Ca2+. (B) Increasing [Ca2+]cyto reduced [Zn2+]ER. Treatment with 100 nM thapsigargin caused [Ca2+]cyto (n = 5 cells) to increase; [Zn2+]ER decreased (n = 6 cells) concomitantly. (C) Thapsigargin increased ER Zn2+ when extracellular calcium was absent. [Ca2+]cyto increased (n = 4 cells) and then decreased to resting level upon treatment with 100 nM thapsigargin in calcium free Hepes-buffered Hanks Balanced Salt Solution. [Zn2+]ER slightly decreased (n = 5 cells) when [Ca2+]cyto increased and then increased when [Ca2+]cyto returned to the resting level. (D) Increasing [Ca2+]cyto by activating TrpA1 reduced [Zn2+]ER. HeLa cells expressing TrpA1 were treated with AITC, causing [Ca2+]cyto (n = 4 cells) to increase and [Zn2+]ER (n = 4 cells) to decrease. (E) Effects of extracellular Ca2+ on [Zn2+]ER (n = 6 cells). HeLa cells were preincubated with 100 μM ZnCl2 for 30 min, then increasing extracellular Ca2+ reduced [Zn2+]ER. (F) Effects of extracellular Zn2+ on [Ca2+]ER (n = 5 cells). HeLa cells were treated with thapsigargin for 30 min to block the SERCA pump. Increasing extracellular Zn2+ reduced [Ca2+]ER.
The above experiments all utilized thapsigargin which raises cytosolic Ca2+ by inducing release from the ER. To elevate cytosolic Ca2+ by an independent means, cells were transfected with the transient receptor potential channel TrpA1. TrpA1 is a cation permeable channel that can be activated by phytochemicals such as mustard oil and cinnemaldehyde (30), and has been shown to transport both Ca2+ and Zn2+ (31). Treatment of cells expressing TrpA1 with the active ingredient in mustard oil, allyl-isothiocyanate (AITC), led to Ca2+ influx across the plasma membrane and concomitant decrease in [Zn2+]ER in accord with the rising [Ca2+]cyto (Fig. 5D). Zinc release required the presence of extracellular Ca2+ because it did not occur when extracellular Ca2+ was absent (SI Appendix, Fig. S8). Because Zn2+ also acts as an activator for TrpA1 (31), addition of 100 μM extracellular Zn2+ in the presence of extracellular Ca2+ to HeLa cells expressing TrpA1 also increased [Ca2+]cyto and decreased [Zn2+]ER (SI Appendix, Fig. S8). These experiments demonstrate that cytosolic Ca2+ fluctuations that arise from ER release or influx across the plasma membrane directly impact ER Zn2+ homeostasis.
The connection between Ca2+ and Zn2+ was further explored by changing extracellular media conditions. HeLa cells were pretreated with 100 μM Zn2+ for 30 min to increase the [Zn2+]ER level, and subsequent addition of increasing extracellular Ca2+ (10, 50, and 100 mM) caused [Zn2+]ER to decrease in a stepwise fashion (Fig. 5E), further demonstrating that cytosolic Ca2+ alters [Zn2+]ER. To explore whether the cytosolic Zn2+ could in turn impact [Ca2+]ER, we examined ER Ca2+ upon elevation of cytosolic Zn2+. HeLa cells were treated with thapsigargin for 30 min to lower ER Ca2+ and prevent refilling of the organelle. Addition of 100 μM ZnCl2 to the extracellular media results in an increase in cytosolic Zn2+ to nanomolar levels (Fig. 3C), and this stimulus induced release of Ca2+ from the ER (Fig. 5F). These experiments indicate that elevated cytosolic Ca2+ can induce ER Zn2+ release and, conversely, elevated cytosolic Zn2+ can induce ER Ca2+ release.
Discussion
With the growing recognition that labile Zn2+ plays important roles in influencing signaling pathways and cellular functions, there has been extensive effort over the past few years to develop fluorescent sensors for monitoring Zn2+ fluxes in cells (5, 15, 32, 33). Previously we developed two zinc sensors that utilized a single zinc finger from Zif268 as the sensing module (ZifCY1  , ZifCY2
, ZifCY2  ) (15). A complementary design based on the copper binding proteins Atox1 and WD4 has also been developed (called eCALWY1-6) with
) (15). A complementary design based on the copper binding proteins Atox1 and WD4 has also been developed (called eCALWY1-6) with 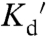 ranging from 2 pM to 2.9 nM (5). Although the high-affinity sensor (
ranging from 2 pM to 2.9 nM (5). Although the high-affinity sensor (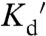 2 pM) was sensitive to Cu1+, lower-affinity versions were specific for Zn2+. In this work, we develop ZapCY1, which exhibits high-affinity (
2 pM) was sensitive to Cu1+, lower-affinity versions were specific for Zn2+. In this work, we develop ZapCY1, which exhibits high-affinity ( ) and selectivity (Fig. 1C), and a 4.15-fold dynamic range in the cytosol (approximately twofold in the ER and Golgi); but the dissociation kinetics are slow (Fig. 1D). On the other hand, ZapCY2 exhibits faster dissociation kinetics (Fig. 1E), a lower Zn2+ affinity (
) and selectivity (Fig. 1C), and a 4.15-fold dynamic range in the cytosol (approximately twofold in the ER and Golgi); but the dissociation kinetics are slow (Fig. 1D). On the other hand, ZapCY2 exhibits faster dissociation kinetics (Fig. 1E), a lower Zn2+ affinity (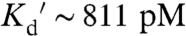 ), reduced selectivity, and a smaller dynamic range (approximately 1.4-fold). The fact that these sensors differ by only two amino acids highlights how difficult it is to predict how sensor properties will be altered by changes in the primary sequence.
), reduced selectivity, and a smaller dynamic range (approximately 1.4-fold). The fact that these sensors differ by only two amino acids highlights how difficult it is to predict how sensor properties will be altered by changes in the primary sequence.
Numerous studies have reported measurement of cytosolic free Zn2+ in a variety of cell types, including red blood cells (34), leukemic cells (35), splenocytes and thymocytes (36), PC-12 (6), HT-29 (22), HEK 293 (5), INS-1 (832/13) (5), hepatocytes (37), and neurons (38), yielding estimates for resting free Zn2+ concentrations from 5 to 1,000 pM. In this paper, we report estimates of free Zn2+ in ER and Golgi, which are 0.9 and 0.6 pM, respectively, at least 100 times lower than our estimate of the cytosolic Zn2+ concentration (80 pM) in HeLa cells (SI Appendix, Table S1).
As suggested by the wide range of reported Zn2+ levels, estimates of absolute Zn2+ concentration should be regarded with caution. Some discrepancies in Zn2+ measurements may reflect true variation in Zn2+ levels in different cell types and/or cell state. Indeed in HT29 cells it has been shown that Zn2+ levels change with cell state (614 pM in resting vs. 1.25 nM in differentiated cells) (22). However, it is also likely that both the method and nature of the probe influence the absolute estimate. Important considerations include the concentration of the sensor in the cell, potential buffering of endogenous Zn2+, reliability of in situ calibration, and the accuracy of intensity-based vs. ratiometric measurements. For example, the concentration of some small molecule Zn2+ sensors has been shown to perturb the sensitivity of the sensor, thus influencing calculation of Zn2+ concentrations (39). The most reliable estimates will likely result from measurements in which the sensor minimally perturbs the cellular environment, sensor concentration is minimized, and the 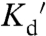 of the sensor is close to the free Zn2+ concentration or, better yet, multiple probes with slightly different
of the sensor is close to the free Zn2+ concentration or, better yet, multiple probes with slightly different 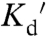 values are utilized to constrain the measurements.
values are utilized to constrain the measurements.
In the present study, we explored whether sensor expression level influenced estimates of cytosolic free Zn2+ levels by examining the calculated Zn2+ concentration as a function of YFP fluorescence intensity. The direct YFP signal does not depend on the presence of Zn2+ and hence is an indicator of protein concentration. Over the intensity range examined, we did not see a correlation suggesting that, at the expression levels used in this study, the sensor concentration did not significantly impact our estimates of free Zn2+ (SI Appendix, Fig. S6). Still, we acknowledge that each sensor has attributes that limit the accuracy of absolute estimates of Zn2+. However, our data clearly provide a reasonable estimate of the relative levels of Zn2+ in the cytosol, ER, and Golgi. This relative difference is evident using the same sensor (ZapCY1) targeted to different locations where the sensor is 100% saturated in the cytosol but only partially occupied in the ER and Golgi.
In this work, we sought to characterize the role of the ER and Golgi in transporting cytosolic Zn2+. Zn2+ uptake into the ER is particularly intriguing because ER function has been shown to depend on Zn2+ (17, 18), and the ER plays a central role in integrating cellular signaling events, serving as the hub of Ca2+ signaling. Surprisingly we found that Ca2+ dynamics influenced ER Zn2+ homeostasis and, conversely, that Zn2+ could influence ER Ca2+ homeostasis. These data suggest a critical link between Ca2+ signaling and metal homeostasis. Elevation of cytosolic Ca2+ led to ER Zn2+ release, whereas elevation of cytosolic Zn2+ led to ER Ca2+ release, suggesting possible exchange of these two ions across the ER membrane. Given that Ca2+ dynamics can be initiated by numerous stimuli and affect a wide range of downstream cellular processes, it will be intriguing to explore whether different Ca2+ signaling pathways result in concomitant changes in Zn2+ homeostasis.
The electrochemical driving force for movement of ions across the ER membrane can be estimated by considering the difference between the ER membrane potential (VER) and the equilibrium potential for a given ion (e.g., EZn2+). The equilibrium potential exactly balances the chemical driving force caused by a concentration gradient and can be calculated using the Nernst equation:
 |
[1] |
With our values of [Zn2+]cyto = ∼ 80 pM and [Zn2+]ER ∼ 0.9 pM, the EZn2+ for movement of Zn2+ from the ER to the cytosol would correspond to EZn2+ = 58 mV, whereas movement from the cytosol to the ER would correspond to -58 mV. Currently, there is a dearth of direct experimental evidence for VER. However, some studies have used theoretical arguments combined with experimental data on Ca2+ fluxes from the ER into the cytosol to estimate VER of neurons (-95 mV) and pancreatic acinar cells (-74 mV) (40). If the VER of HeLa cells is in a similar range, movement of Zn2+ ions from the cytosol into the ER would yield VER - EZn2+ = +74 mV - (-58 mV) = 132 mV. Because the difference in the membrane and equilibrium potential yields a positive value, movement of Zn2+ into the ER would be an energetically downhill process (ΔG < 0). However, even if ion movement is energetically favorable, membrane permeability is controlled by channels and pumps, which may need to be activated by a cellular signal (such as increased cytosolic Zn2+). Indeed, our experiments demonstrate that elevation of cytosolic Zn2+ above 1 nM induces transport of Zn2+ into the ER. Treatment with thapsigargin in the absence of extracellular Ca2+ led to more rapid Zn2+ uptake, perhaps due to alteration of VER or enhanced membrane permeability to Zn2+(activation of a Zn2+ channel). Future studies will be aimed at using the genetically encoded sensors developed in this study to identify the protein(s) responsible for this uptake.
Unexpectedly, elevation of cytosolic Ca2+ led to release of Zn2+ from the ER. Using estimates similar to above, Zn2+ release from the ER into the cytosol would correspond to VER - EZn2+ = -74 mV - (58 mV) = -132 mV, an energetically uphill process. Therefore, it is possible this transport is energy dependent. Although, the membrane potential of the ER under this experimental paradigm is likely to be complicated by Ca2+ and counterion flux across the ER membrane and is likely changing over the course of the experiment (40, 41).
In conclusion, we have generated two high-affinity genetically encoded sensors for Zn2+ and demonstrated that these sensors enable measurement of steady-state Zn2+ levels within the ER and Golgi as well as flux of Zn2+ into and out of these organelles. Our study reveals a surprising correlation between Zn2+ and Ca2+ regulation in the ER that suggests potential exchange of these ions across the ER membrane. We suspect that, as the tools for monitoring cellular metals continue to grow, they will help uncover unique connections between metal ions and cellular signaling pathways.
Methods
SI Appendix, Methods includes the design and in vitro characterization of sensors, colocalization protocol, comparison of in vitro and cellular FRET ratios, and methodology for conversion of FRET ratios into Zn2+ concentrations.
Cellular Imaging.
Sensor constructs were transiently transfected into HeLa cells and imaged in phosphate-free Hepes-buffered Hanks Balanced Salt Solution, pH 7.4, 48 h after transfection. Imaging experiments were carried out on an Axiovert 200 M inverted fluorescence microscope (Zeiss) with a Cascade 512B CCD camera (Roper Scientific), equipped with a Xenon arc lamp (XBO75), and data were collected using Metafluor software (Universal Imaging). Details regarding data collection and processing are presented in SI Appendix, Methods.
Statistical Analysis.
Statistical analysis was performed using ANOVA with a post hoc test in KaleidaGraph program. Error bars indicate SEM.
Supplementary Material
Acknowledgments.
We thank Prof. David Eide (University of Wisconin, Madison, WI) for the gift of pFRET-ZnF1/2 and Dr. Ardem Patapoutian (The Scripps Research Institute, La Jolla, CA) for the gift of TrpA1. This work was supported by a Molecular Biophysics Training Grant (National Institutes of Health T32 GM-065103 to P.J.D.), National Institutes of Health Grant (GM084027 to A.E.P.), and The Alfred P. Sloan Foundation (A.E.P.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database, http://www.ncbi.nlm.nih.gov/genbank/ (accession nos. JF261177, JF261178, JF261179, and JF261180).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1015686108/-/DCSupplemental.
References
- 1.Takeda A. Movement of zinc and its functional significance in the brain. Brain Res Brain Res Rev. 2000;34:137–148. doi: 10.1016/s0165-0173(00)00044-8. [DOI] [PubMed] [Google Scholar]
- 2.Eide DJ. Zinc transporters and the cellular trafficking of zinc. Biochim Biophys Acta. 2006;1763:711–722. doi: 10.1016/j.bbamcr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 3.Andreini C, Banci L, Bertini I, Rosato A. Counting the zinc-proteins encoded in the human genome. J Proteome Res. 2006;5:196–201. doi: 10.1021/pr050361j. [DOI] [PubMed] [Google Scholar]
- 4.Maret W, Krezel A. Cellular zinc and redox buffering capacity of metallothionein/thionein in health and disease. Mol Med. 2007;13:371–375. doi: 10.2119/2007-00036.Maret. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinkenborg JL, et al. Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat Methods. 2009;6:737–740. doi: 10.1038/nmeth.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bozym RA, Thompson RB, Stoddard AK, Fierke CA. Measuring picomolar intracellular exchangeable zinc in PC-12 cells using a ratiometric fluorescence biosensor. ACS Chem Biol. 2006;1:103–111. doi: 10.1021/cb500043a. [DOI] [PubMed] [Google Scholar]
- 7.Du S, McLaughlin B, Pal S, Aizenman E. In vitro neurotoxicity of methylisothiazolinone, a commonly used industrial and household biocide, proceeds via a zinc and extracellular signal-regulated kinase mitogen-activated protein kinase-dependent pathway. J Neurosci. 2002;22:7408–7416. doi: 10.1523/JNEUROSCI.22-17-07408.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maret W. Metallothionein/disulfide interactions, oxidative stress, and the mobilization of cellular zinc. Neurochem Int. 1995;27:111–117. doi: 10.1016/0197-0186(94)00173-r. [DOI] [PubMed] [Google Scholar]
- 9.Sensi SL, et al. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc Natl Acad Sci USA. 2003;100:6157–6162. doi: 10.1073/pnas.1031598100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Truong-Tran AQ, Ruffin RE, Zalewski PD. Visualization of labile zinc and its role in apoptosis of primary airway epithelial cells and cell lines. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1172–1183. doi: 10.1152/ajplung.2000.279.6.L1172. [DOI] [PubMed] [Google Scholar]
- 11.Ho LH, Ratnaike RN, Zalewski PD. Involvement of intracellular labile zinc in suppression of DEVD-caspase activity in human neuroblastoma cells. Biochem Biophys Res Commun. 2000;268:148–154. doi: 10.1006/bbrc.2000.2090. [DOI] [PubMed] [Google Scholar]
- 12.Kitamura H, et al. Toll-like receptor-mediated regulation of zinc homeostasis influences dendritic cell function. Nat Immunol. 2006;7:971–977. doi: 10.1038/ni1373. [DOI] [PubMed] [Google Scholar]
- 13.Lichten LA, Cousins RJ. Mammalian zinc transporters: Nutritional and physiologic regulation. Annu Rev Nutr. 2009;29:153–176. doi: 10.1146/annurev-nutr-033009-083312. [DOI] [PubMed] [Google Scholar]
- 14.Frederickson CJ, Suh SW, Silva D, Thompson RB. Importance of zinc in the central nervous system: The zinc-containing neuron. J Nutr. 2000;130(Suppl 5):1471S–1483S. doi: 10.1093/jn/130.5.1471S. [DOI] [PubMed] [Google Scholar]
- 15.Dittmer PJ, Miranda JG, Gorski JA, Palmer AE. Genetically encoded sensors to elucidate spatial distribution of cellular zinc. J Biol Chem. 2009;284:16289–16297. doi: 10.1074/jbc.M900501200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atkinson A, et al. Mzm1 influences a labile pool of mitochondrial zinc important for respiratory function. J Biol Chem. 2010;285:19450–19459. doi: 10.1074/jbc.M110.109793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ellis CD, et al. Zinc and the Msc2 zinc transporter protein are required for endoplasmic reticulum function. J Cell Biol. 2004;166:325–335. doi: 10.1083/jcb.200401157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ellis CD, Macdiarmid CW, Eide DJ. Heteromeric protein complexes mediate zinc transport into the secretory pathway of eukaryotic cells. J Biol Chem. 2005;280:28811–28818. doi: 10.1074/jbc.M505500200. [DOI] [PubMed] [Google Scholar]
- 19.Qiao W, Mooney M, Bird AJ, Winge DR, Eide DJ. Zinc binding to a regulatory zinc-sensing domain monitored in vivo by using FRET. Proc Natl Acad Sci USA. 2006;103:8674–8679. doi: 10.1073/pnas.0600928103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miyawaki A, Griesbeck O, Heim R, Tsien RY. Dynamic and quantitative Ca2+ measurements using improved cameleons. Proc Natl Acad Sci USA. 1999;96:2135–2140. doi: 10.1073/pnas.96.5.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 22.Krezel A, Maret W. Zinc-buffering capacity of a eukaryotic cell at physiological pZn. J Biol Inorg Chem. 2006;11:1049–1062. doi: 10.1007/s00775-006-0150-5. [DOI] [PubMed] [Google Scholar]
- 23.Maret W, Larsen KS, Vallee BL. Coordination dynamics of biological zinc “clusters” in metallothioneins and in the DNA-binding domain of the transcription factor Gal4. Proc Natl Acad Sci USA. 1997;94:2233–2237. doi: 10.1073/pnas.94.6.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camello C, Lomax R, Petersen OH, Tepikin AV. Calcium leak from intracellular stores—the enigma of calcium signalling. Cell Calcium. 2002;32:355–361. doi: 10.1016/s0143416002001926. [DOI] [PubMed] [Google Scholar]
- 25.Perocchi F, et al. MICU1 encodes a mitochondrial EF hand protein required for Ca(2+) uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Landowski TH, Megli CJ, Nullmeyer KD, Lynch RM, Dorr RT. Mitochondrial-mediated disregulation of Ca2+ is a critical determinant of Velcade (PS-341/bortezomib) cytotoxicity in myeloma cell lines. Cancer Res. 2005;65:3828–3836. doi: 10.1158/0008-5472.CAN-04-3684. [DOI] [PubMed] [Google Scholar]
- 27.Ong DS, Mu TW, Palmer AE, Kelly JW. Endoplasmic reticulum Ca2+ increases enhance mutant glucocerebrosidase proteostasis. Nat Chem Biol. 2010;6:424–432. doi: 10.1038/nchembio.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Palmer AE, Jin C, Reed JC, Tsien RY. Bcl-2-mediated alterations in endoplasmic reticulum Ca2+ analyzed with an improved genetically encoded fluorescent sensor. Proc Natl Acad Sci USA. 2004;101:17404–17409. doi: 10.1073/pnas.0408030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer AE, et al. Ca2+ indicators based on computationally redesigned calmodulin-peptide pairs. Chem Biol. 2006;13:521–530. doi: 10.1016/j.chembiol.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Jordt SE, et al. Mustard oils and cannabinoids excite sensory nerve fibres through the TRP channel ANKTM1. Nature. 2004;427:260–265. doi: 10.1038/nature02282. [DOI] [PubMed] [Google Scholar]
- 31.Hu H, Bandell M, Petrus MJ, Zhu MX, Patapoutian A. Zinc activates damage-sensing TRPA1 ion channels. Nat Chem Biol. 2009;5:183–190. doi: 10.1038/nchembio.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Que EL, Domaille DW, Chang CJ. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem Rev. 2008;108:1517–1549. doi: 10.1021/cr078203u. [DOI] [PubMed] [Google Scholar]
- 33.Tomat E, Lippard SJ. Imaging mobile zinc in biology. Curr Opin Chem Biol. 2010;14:225–230. doi: 10.1016/j.cbpa.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simons TJ. Intracellular free zinc and zinc buffering in human red blood cells. J Membr Biol. 1991;123:63–71. doi: 10.1007/BF01993964. [DOI] [PubMed] [Google Scholar]
- 35.Adebodun F, Post JF. Role of intracellular free Ca(II) and Zn(II) in dexamethasone-induced apoptosis and dexamethasone resistance in human leukemic CEM cell lines. J Cell Physiol. 1995;163:80–86. doi: 10.1002/jcp.1041630109. [DOI] [PubMed] [Google Scholar]
- 36.Zalewski PD, Forbes IJ, Betts WH. Correlation of apoptosis with change in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II) Biochem J. 1993;296:403–408. doi: 10.1042/bj2960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brand IA, Kleineke J. Intracellular zinc movement and its effect on the carbohydrate metabolism of isolated rat hepatocytes. J Biol Chem. 1996;271:1941–1949. doi: 10.1074/jbc.271.4.1941. [DOI] [PubMed] [Google Scholar]
- 38.Sensi SL, et al. Measurement of intracellular free zinc in living cortical neurons: Routes of entry. J Neurosci. 1997;17:9554–9564. doi: 10.1523/JNEUROSCI.17-24-09554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dineley KE, Malaiyandi LM, Reynolds IJ. A reevaluation of neuronal zinc measurements: Artifacts associated with high intracellular dye concentration. Mol Pharmacol. 2002;62:618–627. doi: 10.1124/mol.62.3.618. [DOI] [PubMed] [Google Scholar]
- 40.Burdakov D, Petersen OH, Verkhratsky A. Intraluminal calcium as a primary regulator of endoplasmic reticulum function. Cell Calcium. 2005;38:303–310. doi: 10.1016/j.ceca.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 41.Jafri MS, Gillo B. A membrane potential model with counterions for cytosolic calcium oscillations. Cell Calcium. 1994;16:9–19. doi: 10.1016/s0143-4160(05)80003-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.