

Abstract
In the development of new nanoparticle-based technologies for therapeutic and diagnostic purposes, understanding the fate of nanoparticles in the body is crucial. We recently developed a multistage vector delivery system comprising biodegradable and biocompatible nanoporous silicon particles (first-stage microparticles [S1MPs]) able to host, protect, and deliver second-stage therapeutic and diagnostic nanoparticles (S2NPs) on intravenous injection. This delivery system aims at sequentially overcoming the biologic barriers en route to the target delivery site by separating and assigning tasks to the coordinated logic-embedded vectors constituting it. In this work, by conjugating a near-infrared dye on the surface of the S1MP without compromising the porous structure and potential loading of S2NPs, we were able to monitor the in vivo distribution of S1MPs in healthy mice using an optical imaging system. It was observed that particles predominantly accumulated in the liver and spleen at the end of 24 hours. Further quantification of S1MPs in the major organs of the animals by elemental analysis of silicon using inductively coupled plasma-atomic electron spectroscopy verified the accuracy of in vivo near-infrared imaging as a tool for evaluation of nanovector biodistribution.
The growing use of nanoparticles as theranostic agents requires new methodologies to study their fate on systemic injection. Optical imaging allows noninvasive longitudinal analysis based on fluorescent and bioluminescent reporters to provide real-time, in vivo access to critical information at the molecular scale. Deep tissue imaging exploits the near-infrared (NIR) window (650–900 nm) where hemoglobin and water are highly transparent1 to discern the function, localization, affinity, and fate of nanoparticles either through innate infrared (IR) fluorescence or by conjugation of fluorescent molecules.2,3 The photonic properties of metal nanoparticles (quantum dots, Au nanoshells, nanoparticles, and nanorods) originating from quantum confinement and tunable with particle size offer a direct ability to assess their interaction within biologic systems and provide diagnostic capability.2,4 However, metal nanoparticles are not biodegradable; thus, their tissue accumulation poses concerns of toxicity.5 Porous silicon (pSi) emerged as a promising drug delivery material when its ability to load and deliver therapeutic agents was established.6 Since then, pSi has been shown to load drugs with markedly different characteristics and modulate their solubility,7–9 as well as proteins,10 diagnostic agents, and nanoparticles.11,12 pSi biocompatibility and bioresorption in biologic environments have been established in vivo,13–16 the by-products of degradation are known to be benign,15,17,18 and the degradation rates can be engineered by tailoring pSi's porosity and surface chemistry.12,18,19 pSi quantum sponge structure20 provides tunable photonic properties.21
The IR photoluminescence (PL)12 of pSi vectors has been exploited to assess their fate on systemic administration14; however, efficient IR PL is obtained only through imposing severe constraints on the physical characteristics of the porous structure that limit the vector's versatility as a delivery system.14,22,23 Pore size and porosity control the pore wall thickness of pSi structures that determines their PL spectrum. Thus, the porous structure must be specifically engineered to obtain efficient IR PL at the expense of versatility in degradation kinetics and loading capacity for therapeutic and diagnostic nanoparticles. Recently, we introduced a multistage vector (MSV) as a versatile delivery platform for bioactive materials. The MSV comprises biodegradable and biocompatible pSi particles (first-stage microparticles or nanoparticles [S1MPs]) able to host, protect, and deliver second-stage theranostic nanoparticles (S2NPs) on intravenous injection. The scope of the MSV is to overcome the biologic barriers in a sequential manner on its way to the target delivery site. Such scope is achieved by separating and assigning tasks to the coordinated logic-embedded vectors that constitute the MSV.12,24–26 The versatility of the manufacturing processes allowed for the optimization of the porous structure (porosity and pore size) and of size and shape.27 Similarly, a number of postfabrication chemical functionalizations of the pSi surface enable the control of the surface charge and the conjugation of fluorescent dyes and targeting agents. Given that the ability to tailor the porosity and pore size of S1MPs is crucial to attain optimal loading, protection, and release of the S2NPs, the innate IR PL of pSi cannot be relied on to assess the biodistribution of the S1MP. Thus, alternative techniques for in vivo assessment of the fate of MSVs should be sought. In this article, we present the conjugation of an NIR dye to the S1MP surface, the biodegradation and biocompatibility of the S1MP, and the ability to monitor their biodistribution based on in vivo imaging supported by the quantitative analysis of silicon in various tissues by inductively coupled plasma-atomic emission spectroscopy (ICP-AES).
Materials and Methods
Fabrication of S1MPs
S1MPs were fabricated by semiconductor processing and electrochemical etching in the Microelectronics Research Center at The University of Texas at Austin based on the methods previously described.12,27 Particles with 20 to 50 nm pores were formed by selective electrochemical etch of a silicon-rich silicon nitride (SiN) masked array of 2 μm cylindrical trenches in the silicon in a mixture of hydrofluoric acid (49% HF) and ethanol (3:7 v/v). The etch was performed initially by applying a current density of 100 mA cm−2 for 40 seconds, followed by a high-porosity layer applying a current density of 380 mA cm−2 for 6 seconds. After removing the SiN layer by HF, particles were released by ultrasound in isopropyl alcohol (IPA) for 1 minute and preserved in a sealed centrifuge tube at 4°C in controlled humidity. The process is schematically summarized in Figure 1.
Figure 1.

Stage 1 porous silicon microparticles. A–D, Schematics of the microfabrication process: (A) array patterning on photoresist; (B) pattern transfer into the SiN and Si by reactive ion etch; (C) selective porosification of trenches to form particles by electro-chemical etch; (D) release of particles from substrate by sonication in iso-propanol. E, Scanning electron micrograph of a collection of porous silicon microparticles. F, Scanning electron micrograph of the porous silicon microparticles' pore openings.
NIR Dye Conjugation to S1MPs and Physicochemical Characterization
Surface Modifications and Conjugation of N-Hydroxysuccinimide Dye to S1MP Surface
To enable suspension in aqueous media and to impart NIR absorption properties, pSi S1MP underwent a series of surface modifications, schematically summarized in Figure 2A. First, to introduce the hydroxyl groups on the surface, the particles were oxidized in piranha solution (1:2 H2O2:H2SO4 [v/v], Sigma Aldrich, St. Louis, MO) at 100 to 110°C for 2 hours, with intermittent sonication to disperse aggregates. We obtained scanning and transmission electron microscopy indication that neither the porous structure nor the particle geometry was altered by the oxidation treatment (Figure S1 [online version only]).27,28 The particles were washed with deionized water and either used immediately or stored at 4°C until further use (for a maximum of 7 days). Positive charges on the particle surface to which the fluorophores could be conjugated were introduced through silane chemistry using 3-aminopropyltriethoxysilane (APTES; Sigma). The particles were suspended in IPA containing APTES (2% v/v) for 2 to 8 hours at room temperature, washed five times with IPA, and stored in IPA at 4°C. APTES-modified S1MPs were reacted for 1 hour with N-hydroxysuccinimide (NHS) esters: DyLight 488 or DyLight 750 (Pierce, Pierce Protein Research Products, Rockford, IL). The succinimidyl ester on the probes reacted with amino groups exposed on the surface of the APTES particles to provide a stable chemical linkage of the probe to the particles. The particles were then washed (by centrifugation) in deionized water four times to remove any unreacted molecules. In the initial conjugation set to establish the optimal probe conjugation conditions that enable maximum particle fluorescence, the particles were reacted with various concentrations of DyLight 488 in the range of 0.23 to 45 mM. All further experiments were carried out with particles conjugated in the concentration of 10 mM of the probe.
Figure 2.

S1MP modification and physicochemical characteristics. A, Schematic presentation of the fluorescent dye conjugation reaction to S1MPs. B, Zeta potential of the particles following various stages of chemical modification. C, ATR FTIR spectra of oxidized (black), APTES (red), and DyLight 488–conjugated S1MPs (green); inset shows a zoomed-in portion of the spectra from 1,500 to 1,700. D, Flow cytometry data of unmodified (black) and Dylight 488–modified S1MPs (green). E and F, Fluorscent images of Dylight 488–modified S1MPs. A low magnification of S1MP particles in bright field and fluorescence channels (E and F, respectively). Scale bars are at 20 μm.
Analysis of S1MP Size Distribution and Particle Counts
Volume, size distribution, and particle count were obtained using a Z2 Coulter Particle Counter and Size Analyzer (Beckman Coulter, Fullerton, CA). Prior to the analysis, the samples were dispersed in the balanced electrolyte solution (ISOTON II Diluent, Beckman Coulter) and sonicated for 5 seconds to ensure a homogeneous dispersion.
Zeta Potential Analysis
The zeta potential of the silicon particles was analyzed using a Zetasizer nano ZS (Malvern Instruments Ltd., Southborough, MA). For the analysis, 2 μL particle suspension containing at least 2 × 105 particles to give a stable zeta potential evaluation was injected into a sample cell countering filled with phosphate buffer (1.4 mL, pH 7.3). The cell was sonicated for 2 minutes prior to the measurements, and the analysis was conducted at room temperature (23°C) in triplicate.
Fourier Transform Infrared Spectroscopy Analysis
Fourier transform infrared spectroscopy (FTIR) was preformed to assess the multistep covalent attachment of the fluorescent dye onto the surface of the S1MPs. Samples were diluted at a concentration of 1 × 105/μL(≈ 1 mg/mL) and washed in deionized water three times. FTIR was preformed on a Nicolet 6700 (Thermo Scientific, Waltham, MA) using a smart diamond crystal attenuated total reflection (ATR) accessory. A 2 μL drop from each sample was placed on the diamond crystal and subjected to vacuum. Each sample was run for 150 scans at a resolution of 4 wavenumbers.
Fluorescent Dye Conjugation Stability and S1MP Degradation Studies
Analysis of S1MP-Facilitated Biodegradation in Physiologic Media
To evaluate degradation kinetics, 107 particles were added to phosphate-buffered saline (PBS) (1.5 mL, pH 7.2), cell culture medium (Dulbecco's Modified Eagle's Medium B [DMEM], Gibco, BRL (Gaithersburg, MD) containing 10% fetal bovine serum C (FBS; Hyclone, UT), 100% FBS, or fresh heparinized EX mouse blood. The samples (n = 3) were incubated at 37°C and constantly mixed using a rotary shaker until the appropriate time points had elapsed. Aliquots were taken from the tubes: 150 μL were filter-spun (0.45 μm filter) to separate the nondegraded particles from the degradation medium, and the resulting liquid was analyzed for the fluorophore release quantitation by fluorimetry and for silicon (Si) contents by ICPAES.
Evaluation of Fluorescent Intensity by Fluorimetry
For the analysis of fluorescence intensity, the samples were placed on a 96-well plate (Nunclon, Denmark) and EO quantities of DyLight 488 released from the particles surface were determined in triplicate using a FluoSTAR Galaxy (BMG LABTECH GmbH, Offenburg, Germany) microplate variable wavelength fluorescence spectrophotometer (excitation 488 nm, emission 523 nm).
Measurement of S1MP Biodegradation by ICP-AES
Si contents released from the particles during the degradation process were measured using a Vista-Pro ICP–AES (Varian, Walnut Creek, CA). Si was detected at 250.69, 251.43, 251.61, and 288.158 nm. Yttrium (1 ppm) was added to both standards and samples to correct for instrumental drift during the run. A calibration run including the internal control was made before each group of 15 samples. In addition, samples were analyzed in random order to avoid any bias in data acquisition. The detection limit of Si was 15 ppb. To analyze 100% Si contents, the S1MPs were dissolved in 1 N NaOH overnight in 37°C. The results are expressed as a percentage of the silicic acid released to the medium.
Testing the Efficiency of Probe Conjugation by Flow Cytometry
Fluorescence of the DyLight 488–conjugated particles was assessed using a FACScalibur (Becton Dickinson). Bivariate dot-plots defining logarithmic side scatter (SSC) versus logarithmic forward scatter (FSC) were used to evaluate the size and shape of the unlabeled Si particles (3 μm in diameter, 1.5 μm in height) and to exclude nonspecific events from the analysis. A polygonal region (R1) was defined as an electronic gate around the center of the major population of interest for nondegraded particles, which excluded events that were too close to the signal to noise ratio limits of the cytometer. The peaks identified in each sample were analyzed in the corresponding fluorescent histogram, and the geometric mean values were recorded. For particle detection, the detectors used were FSC E-1 and SSC with a voltage setting of 474 V. The fluorescent detector FL1 was set at 800 V and green fluorescence was detected with FL1 using a 530/30 nm bandpass filter. For each analysis, 50,000 to 200,000 gated events were collected. Instrument calibration was carried out before, between, and after each series of experiments for data acquisition using BD Calibrite beads (BD Biosciences, Rockville, MD) (3.5 μm in size).
Evaluation of Biocompatibility of Particles with Human Macrophages and Endothelial Cells
Cell Lines
J774A.1 murine macrophages were obtained from the American Type Culture Collection (ATCC, Manassas, VA) and human umbilical vein endothelial cells (HUVECs) from Lonza Rockland Inc (Rockland, ME). All reagents and medium were purchased from ATCC, Lonza, and Gibco BRL.
Cell Proliferation Assay
A viability assay in the presence of S1MPs at a concentration of 20 particles/cell was performed on HUVECs and J774A.1 cells. Proliferation of the cells was evaluated by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay at 24 to 96 hours using the standard protocol.
Tumor Necrosis Factor a Release from Immune Cells
For tumor necrosis factor α (TNF-α) analysis, J774A.1 immune cells were cultured at 0.4 to 2 × 106 cells/mL in RPMI 1640 containing fetal calf serum (10% w/v), glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) and maintained at 37°C under 5% CO2. The cells were incubated with S1MPs at a ratio of 5 particles/cell for 24 hours. At the end of the experiment, the supernatants were collected and stored at −70°C until the TNF-α analysis using a commercial multiplex kit (Millipore, Billerica, MA).
Intracellular Delivery of Fluorescently Labeled S1MPs
HUVECs were plated at 65,000 cells/cm2 in LabTek Chamber Slides (Thermo Scientific). Cells were allowed to adhere for 5 to 8 hours and then introduced to pSi particles at a ratio of 5 particles/cell. After an overnight incubation, HUVECs were fixed in 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS (Invitrogen, Carlsbad, CA), followed by a brief permeablization in 0.25% Trition X-100. Cells were then blocked in 1% of bovine serum albumin (Sigma-Aldrich, St. Louis, MO) for 30 minutes, followed by a 1-hour incubation with Alexa Fluor 555 phalloidin (Invitrogen). Cells were then washed in PBS, mounted with VECTASHIELD with 4′6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA), and covered with #1.5 coverslips (VWR International Inc, Sugar Land, TX). Fluorescent images were acquired using an inverted TE 2000 Nikon scope fitted with emission and excitation filters capable of exciting fluorochromes with characteristics similar to those of DAPI, fluorescein isothiocyanate (FITC), and Texas red. Images were then analyzed using Nikon Elements and exported as tagged image file format (TIFF) files.
In Vivo Studies
Animals
The animal studies were performed in accordance with the guidelines of the Animal Welfare Act and the Guide for the Care and Use of Laboratory Animals based on the protocols approved by the University of Texas and M.D. Anderson Cancer Center for the Care and Use of Laboratory Animals committees. Healthy 8- to 10-week-old female nu/nu or ICR mice were maintained in a virus antibody–free barrier facility at the M.D. Anderson Cancer Center or Institute of Molecular Medicine, University of Texas Health Science Center, respectively. Mice were injected via the tail vein with 5 ×107 DyLight 750–labeled S1MPs dispersed in 100 μL saline.
In Vivo NIR Imaging
Animals were sedated with isofluorane and imaged on an IVIS Lumina (Xenogen; Caliper Life Sciences, Hopkinton, MA) after intravenous administration of S1MPs. Animals were imaged using the indocyanine green excitation and emission filters at 2, 4, and 24 hours. At each time point, animals were sacrificed and their organs were excised and imaged under the same conditions as live animals to determine the organ distribution of S1MPs. Images and data were analyzed using Living Image 4.0 software (Caliper Life Sciences).
Quantification of S1MP Organ Distribution
At 2, 4, and 24 hours after the injection, the groups of animals (n = 4) were sacrificed and the major organs (liver, spleen, heart, lungs, and kidneys) and tails were excised, imaged separately, weighed, and processed for elemental analysis of Si.29 Briefly, the organs were homogenized in 20% EtOH in 1 N NaOH and left for 48 hours at room temperature for extraction of Si. Then the extracts were centrifuged at 4,200 rpm for 25 minutes and 0.5 mL of the supernatant was withdrawn, diluted with 2.5 mL of deionized water, and analyzed for Si contents. Si content was measured by ICP-AES as described earlier. For measurement of the total Si in each sample (100%), the original particle suspensions were diluted and dissolved in 1 N NaOH for 24 hours at 37°C. Further, all results were recalculated considering the dilutions performed and the amount of the carrier remaining at the site of injection (tail) and were normalized to the percentage of total Si resulting from the number of particles injected or to the individual organ weight.
Results
S1MP Fabrication and Surface Modification
Our manufacturing protocols, described in detail by Chiappini and colleagues,27 allowed for the microfabrication of well-controlled and reproducible S1MPs (see Figure 1, A–D). Statistical analysis by scanning electron microscopy (SEM) of S1MPs from hundreds of independent lots confirmed the accuracy and reproducibility of the process. The analysis showed 3.2 μm diameter (see Figure 1E) and 0.9 μm thickness quasihemispherical S1MPs originating from 2 μm diameter and 0.3 μm deep trenches in the Si (not shown). The tuning of the porous structures at the nanoscale was obtained varying the electrochemical etching parameters during synthesis. The S1MPs used for this study originated from independent production lots relying on the consistent reproducibility demonstrated by a combination of postsynthesis characterization techniques (SEM, dynamic light scattering, N2 isotherms, zeta potential)27 (see Figure 1, E–F).
We developed chemical reactions to conjugate fluorescent probes to the S1MP surface (schematically presented in Figure 2A). After fabrication, S1MPs have a hydrophobic surface and do not disperse in aqueous media. To increase the hydrophilicity of the Si structure, the surface was oxidized in piranha solution. During the oxidation process, the introduction of free hydroxyl groups resulted in a negative zeta potential of S1MPs (−27.6.5 mV; Figure 2B). Further, the hydroxyl surface groups were covalently coupled through silane chemistry to positively charged APTES molecules, reversing the net surface charge of the particles to +8.3 mV. The amine groups of APTES served as a linker for fluorescent probes containing NHS ester bonds to the S1MP surface. In addition to validating the zeta potential, the FTIR spectra (Figure 2C) showed that on modification with APTES, new bands appeared in the range of 1,500 to 1,700 cm−1. On magnification of this range, we noticed that both APTES- and DyLight-modified particles displayed larger absorption bands than oxidized particles (oxidized ⪡ 0.001; APTES and DyLight ≈ 0.001). These bands can be attributed to the bending modes of NH2 and of protonated amines (−NH3+).30 Further, DyLight 488 was used in these initial studies as a model fluorescent probe to enable characterization of the labeled S1MPs with standard techniques such as fluorometry, confocal microscopy, and flow cytometry. On conjugation of the DyLight 488, S1MPs gained fluorescence, as shown in Figure 2. The conjugation of the fluorescent probe on the particle neutralized the APTES-positive charges, resulting in a slightly negative zeta potential (see Figure 2B). Figure 2D shows that, following the modification with DyLight 488, the mean fluorescent intensity of the particles as measured by flow cytometry increased from 4.5 to 234.2 AU. Fluorescent S1MPs, observed under the microscope, displayed a uniform, homogeneous fluorescent signal (as presented in Figure 2, E to F, for DyLight 488-conjugated particles).
Fluorescent Dye Conjugation Stability and S1MP Degradation Studies
To optimize the labeling of S1MPs, we evaluated the effect of dye concentration on the final fluorescence intensity. The net fluorescence intensity of S1MPs consistently saturated at a concentration of 7.5 mM DyLight 488 in the loading solution (concentration range 0–45 mM) This behavior could be attributed to the saturation of the binding sites on the particle surface (Figure 3A). Based on these results, the optimal dye concentration was established at 10 mM. The dye conjugation and fluorescence of S1MPs in an aqueous medium were confirmed stable for at least 20 days in double distilled water.
Figure 3.

A, Stability of the DyLight 488 conjugation to the S1MPs. The particles were kept in deionized water for 20 days, and the fluorescence was followed up by flow cytometry; biode-gradation of S1MPs in various facilitated physiologic conditions was followed up by (B) S1MP fluorescence intensity. C, Release of orthosilicic acid to the medium.
To evaluate the degradation rate of the particles in physiologic media, we measured the release of the fluorescent probe from the S1MP surface and the accumulation of orthosilicic acid (the degradation product of the pSi) in the media (Figure 3, B and C, respectively). Four physiologically relevant environments were tested: PBS (pH 7.2), DMEM with 10% FBS, pure FBS, and fresh heparinized murine blood. In agreement with the independent findings on different pSi structures,17 S1MPs degraded in all environments considered, and although the degradation kinetics were affected by the specific medium, in all conditions, both the fluorescent probe and the S1MPs were completely dissolved in solution within 24 hours.
Biocompatibility of the S1MPs
On systemic administration, delivery vectors encounter two main cell types: endothelial cells lining blood vessels and circulating immune cells. To evaluate the biocompatibility of the S1MPs, we tested the viability of these cells after direct incubation with the Si particles. Proliferation of HUVECs and J774A.1 macrophages in the presence of S1MPs was measured using an MTT proliferation assay at 24 to 72 hours. No significant difference in cell growth was measured for either cell line (Figure 4, A and B). Fluorescent imaging of HUVECs indicated that the S1MPs were found exclusively in the cytoplasm and were internalized through endocytosis and retained in lysosomes.28 The overall morphology of the cell and the actin filaments of the cytoskeleton were not affected following the internalization of S1MPs. Finally, the nuclear compartment showed similar characteristics (size, fluorescent intensity, heterochromatin density) in the presence or absence of the S1MPs. Altogether, these data suggested that there was no underlying cytotoxic effect owing to the internalization of S1MPs (Figure 4C). To further investigate the cellular response to particle internalization, we evaluated the release of the proinflammatory cytokine TNF-α in vitro and in vivo. In vitro experiments were carried out on J744A.1 cells with zymosan as a positive control. S1MPs did not induce release of TNF-α, whereas zymosan caused a very prominent increase in the TNF-α release (Figure 4D). We also tested the plasma levels of TNF-α in mice injected intravenously with S1MPs and found no significant difference between the animals injected with S1MPs and the animals injected with saline (Figure 4E).
Figure 4.

Biocompatibility of S1MPs with endothelial and immune cells in vitro and in vivo. Proliferation assay of the cells exposed to S1MPs at a ratio of 20 particles/cell (n = 4): (A) human umbilical vein endothelial cells (HUVECs); (B) J744 murine macrophages; (C) fluorescent images of the HUVECs incubated with S1MPs for 24 hours (scale bars are at 10 μm); (D) in vitro release of tumor necrosis factor α (TNF-α) 24 hours after incubation of J744 cells with S1MPs (5 particles/cell, n = 4); (E) in vivo plasma levels of TNF-α 24 hours after intravenous injection of the 5 × 108 S1MPs to ICR mice (n = 5).
In Vivo Imaging
To evaluate the fate of the S1MPs following injection in the systemic circulation, we employed an in vivo optical imaging system based on NIR fluorescence detection. The particles were resuspended in a small volume of saline, sonicated to promote dispersion to avoid clusters from obstructing the smaller capillaries, injected in the tail vein, and imaged in vivo (Figure 5). The in vivo biodistribution of NIR S1MPs was followed for up to 24 hours owing to the stable NIR fluorescence of the particles over time. At the earliest time point (15 minutes), a uniform signal was present across the body of the animal, indicating the free circulation of the S1MPs in the bloodstream. A significantly higher signal (fourfold increase) was captured in the bladder and was attributed to the free dye released by the particles on intravenous injection possibly caused by the enzymatic degradation or cleavage of superficial dye molecules on the surface. This phenomenon could also be attributed to the higher shear forces experienced by the particles as they initially interact with blood vessels. At 2 hours, a prominent accumulation of the fluorescent signal was observed in the abdominal area of the mice where the fluorescence signal was restricted to areas corresponding to the liver and spleen as well as in the bladder (although significantly reduced in intensity when compared to the 15-minute time point). At the 4- and 24-hour time points, the most pronounced NIR signal was recorded in the liver and spleen, whereas almost no trace of the dye was present in the bladder (see Figure 5, A and D). For every time point, after the whole-animal imaging, we sacrificed and dissected the animals to isolate the major internal organs (spleen, liver, kidneys, heart, lungs) and muscle tissue for separate NIR imaging (see Figure 5B). The fluorescent signal normalized to the injected dose of S1MPs (see Figure 5C) and to the mass of the organ (data not shown) was quantified and plotted as a function of time elapsed from the S1MP injection (see Figure 5E). Although no trace of the particles was found in the heart, lungs, and muscle, a strong signal was consistently measured in the spleen and liver of the mice. The fluorescence signal in kidneys was significantly higher than the one from other organs at the 2-hour time point but dramatically decreased after 4 hours. This imaging pattern is in agreement with what we observed in the whole animal (early bladder accumulation), indicating that there was an initial burst release of the free dye from the pores of the S1MPs on the intravenous injection rather than urinary clearance of the whole particle. This assumption was further confirmed by the SEM analysis of the urine, which evidenced no presence of S1MPs (data not shown), and by the quantitative analysis of Si in various organs.
Figure 5.

In vivo NIR imaging following intravenous administration of S1MPs tagged with Dylight 750. A, Representative pictures of mice injected with S1MPs and followed for 24 hours in a whole-animal imaging study (time points on the bottom of each image, n = 5). B, Organs collected after sacrificing the animal for ICP analysis (time point 2 hours). C, An NIR image of the vial containing the suspension of particles injected per animal. The scale bars on the right of the images apply to all images of the series. The settings were not changed during acquisition and analysis. D, Quantification of the fluorescence in the abdomen (spleen, liver, and kidneys) and in the bladder. E, Postmortem evaluation of the accumulation of the fluorescence signal in the different organs and tissues.
Quantification of the S1MPs in Organs
The amount of Si was quantified in different organs using ICP-AES. Among the tested tissues, significant amounts of S1MPs were found in the spleen, liver, and lungs (only at 2 hours). The presence of S1MPs in the heart, kidney, and muscle tissues was relatively low or undetectable. Figure 6 presents the analysis of Si in the spleen and liver during the course of the experiment. The data are normalized to the weight of the organ, to the calculated dose injected to each animal subtracting the amount remaining in the site of injection (tail), and to the basal level of Si in tissues analyzed in control animals not injected with S1MPs. These data correlate well with the in vivo NIR imaging of S1MPs, supporting the use of NIR-tagged S1MPs to monitor the in vivo biodistribution of the multistage vector.
Figure 6.
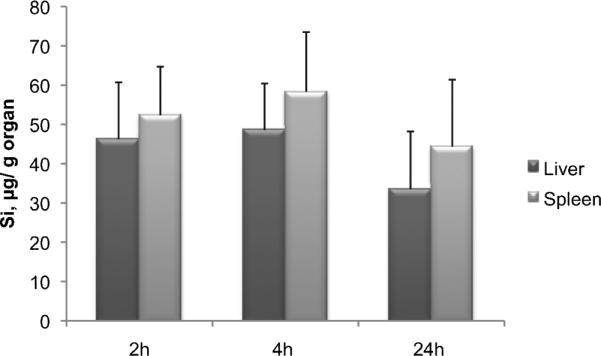
Quantification of orthosilicic acid in the organs of nu/nu mice injected with Dylight 750–conjugated S1MPs through elemental analysis of silicon using ICP-AES.
Discussion
Noninvasive fluorescent imaging holds great promise in providing critical details about the biodistribution of systemically injected therapeutic agents, as well as noninvasive means to assess the disease status, its progression, and the efficacy of the treatment. Owing to the low absorption coefficient of biomolecules in the NIR region, fluorescent reporters that are excited and emit in this region are in great demand. The broad applications of NIR imaging span from their use in diagnosing lymph node recruitment31 to tumor angiogenesis32 and from brain and muscle applications33 to orthopedic research.34 Currently, clinical NIR imaging systems, such as SPY system (Novadaq, Bonita Springs, FL), Photodynamic Eye (Hamamatsu Photonics, Bridgewater, NJ), and Fluorescence-Assisted Resection and Exploration (FLARE) system (Beth Israel Deaconess Medical Center, Boston, MA), are under investigation.31,35–37 However, the true impact of NIR imaging in the drug development arena and clinical applications has yet to be fully realized. Organically based dyes, such as indocyanine green, tend to quench quickly and were reported as not robust enough for long-term monitoring.31 Quantum dots have emerged as powerful alternatives to organic dyes owing to their brightness and robustness. Quantum dots have been used to image lymph node drainage to determine tumor burden, localization, and progression.31,38 However, inorganically based imaging agents, such as quantum dots, are not biodegradable and suffer from poor biocompatibility and clearance issues, thus limiting their use in humans. pSi-based nanostructured materials hold great promise for applications in the biomedical field owing to their biodegradability in physiologic conditions and modifiable physicochemical characteristics (eg, surface chemistry, geometry, and porosity).7,8,39,40 In this article, we are reporting the conjugation of an NIR probe to the surface of nanoporous Si-based multistage drug delivery systems (S1MP) as a means for in vivo monitoring of organ biodistribution. Given that the S1MPs are intended for intravenous administration, we have also tested their degradation profiles and compatibility with the two major cell populations that will be exposed to the S1MPs in circulation. S1MP degradation occurred in all four physiologic media tested, in accordance with previously published studies on pSi materials.15,17,18 The degradationproduct of pSi structures is harmless orthosilicic acid, composed of Si(41)4 units, which is released in the physiologic pH range through hydrolysis of the Si-O bonds8,41 and subsequently excreted in the urine through the kidneys.42 It is noteworthy that in deionized water, the degradation of the probe from the particle surface was negligible for a long period of time, sustaining the nucleophilic nature of the Si-O hydrolysis reaction, requiring the presence of electrolytes in the reaction environment.
Information on the mode of uptake of engineered nanomaterials by macrophages and endothelial cells is still sparse.43–45 The major response of macrophages to stimuli is the release of cytokines, which can promote the cascade of inflammatory response to foreign substances.40,46 We observed that the levels of the proinflammatory cytokine TNF-α following introduction of the particles to immune cells in vitro and in vivo are comparable to the negative control levels. Macrophages and endothelial cells with internalized pSi particles showed normal cellular proliferation, cell morphology, cell viability, and mitotic potential as well as proinflammatory responses, in line with our previous reports.18,47
To allow optimization of the design of emerging nanocarrier systems, reliable and fast techniques for evaluation of their fate in various body organs and tissues should be employed. Most reports in the literature about the in vivo imaging of the biodistribution of biodegradable nanovectors are based on the attachment of radioactive labels for the carries. For example, the biodistribution analysis of radioactively labeled nanoparticles prepared from mixtures of poly(lactide-co-glycolide) (PLGA) with poly(lactide-co-glycolide)-poly(ethylene glycol) (PLGA-PEG) copolymers revealed that the composition of the carrier is important for particle circulation time and that particles predominantly accumulated in the reticuloen-dothelial system organs (liver and spleen).48 A recent study on the distribution of thermally hydrocarbonized pSi nanoparticles functionalized with radioactive 18F label showed that following oral administration, the particles passed intact through the gastrointestinal tract, and after intravenous bolus injection, the particles were rapidly cleared from the circulation and mainly detected in the liver and spleen.49
In our current work, by taking advantage of a fluorescent reporter in the NIR window, we eliminated the need to use radioactive labels and yet achieved submillimeter resolution while maintaining minimal autofluorescence. Our imaging data correlated well with the quantification analysis of Si in the organs, verifying the accuracy of the imaging technique for tracing the biodistribution of S1MPs in the body. NIR imaging has not been able to provide anatomic location of signal owing to the large amount of scattering typically associated with the skin. Previous attempts employed indirect methods using an atlas of animal anatomy to give a general idea of the location. Owing to their inability to provide structurally and anatomically rich information, NIR systems are currently being designed so that they can be coupled with other imaging modalities, such as magnetic resonance imaging (MRI), computed tomography (CT), and positron emission tomography (PET). Using our MSV systems, we can, if required, easily couple the NIR-tagged S1MPs with other clinically relevant imaging contrast agents for MRI, CT, and PET. As an example, we have previously shown the ability to load S1MPs with iron oxide and gold nanoparticles28,47 and with gadolinium constructs for MRI and CT contrast enhancement.50 This coregistration of modalities can be extremely beneficial because the sensitivity of NIR light will allow for the submillimeter resolution to be coupled with the anatomic location of reporters.
We have shown that conjugation of the NIR probe to the surface of MSV carriers based on biocompatible and biodegradable pSi nanovectors enabled precise evaluation of their in vivo biodistribution. This strategy can be further translated for optimization of the targeting of MSV to disease loci as well as for diagnostic imaging of pathologic conditions.
Supplementary Material
Acknowledgments
Financial disclosure of authors: Financial support was received from the following sources: National Institutes of Health (NIH) grants U54CA143837, RO1CA128797, and R33 CA122864; Department of Defense grants DODW81XWH-09-1-0212 and DODW81XWH-07-2-0101; and the State of Texas Emerging Technology Fund. In addition, J.O.M. was supported by Award Number TL1RR024147 from the National Center for Research Resources (NCRR). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCRR or the NIH. The authors also disclose a personal financial interest in Leonardo Biosystems (M.F., E.T., X.L., B.G.), Nanomedical Systems (M.F., X.L.), and Arrowhead Research Corporation (M.F.).
Footnotes
Financial disclosure of reviewers: None reported.
References
- 1.Weissleder R. A clearer vision for in vivo imaging. Nat Biotechnol. 2001;19:316–7. doi: 10.1038/86684. doi:10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 2.Gobin AM, Lee MH, Halas NJ, et al. Near-infrared resonant nanoshells for combined optical imaging and photothermal cancer therapy. Nano Lett. 2007;7:1929–34. doi: 10.1021/nl070610y. doi:10.1021/nl070610y. [DOI] [PubMed] [Google Scholar]
- 3.Chen X, Conti PS, Moats RA. In vivo near-infrared fluorescence imaging of integrin alphavbeta3 in brain tumor xenografts. Cancer Res. 2004;64:8009–14. doi: 10.1158/0008-5472.CAN-04-1956. doi:10.1158/0008-5472.CAN-04-1956. [DOI] [PubMed] [Google Scholar]
- 4.Michalet X, Pinaud FF, Bentolila LA, et al. Quantum dots for live cells, in vivo imaging, and diagnostics. Science. 2005;307:538–44. doi: 10.1126/science.1104274. doi:10.1126/science.1104274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derfus AM, Chan WCW, Bhatia SN. Probing the cytotoxicity of semiconductor quantum dots. Nano Lett. 2003;4:11–8. doi: 10.1021/nl0347334. doi:10.1021/nl0347334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Coffer JL, Chen Y, et al. Transition metal complex-doped hydroxyapatite layers on porous silicon. J Am Chem Soc. 1998;120:11706–9. doi:10.1021/ja9823666. [Google Scholar]
- 7.Salonen J, Laitinen L, Kaukonen AM, et al. Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs. J Control Release. 2005;108:362–74. doi: 10.1016/j.jconrel.2005.08.017. doi:10.1016/j.jconrel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 8.Anglin EJ, Cheng L, Freeman WR, Sailor MJ. Porous silicon in drug delivery devices and materials. Adv Drug Deliv Rev. 2008;60:1266–77. doi: 10.1016/j.addr.2008.03.017. doi:10.1016/j.addr.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu EC, Park JH, Park J, et al. Oxidation-triggered release of fluorescent molecules or drugs from mesoporous Si microparticles. ACS Nano. 2008;2:2401–9. doi: 10.1021/nn800592q. doi:10.1021/nn800592q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prestidge CA, Barnes TJ, Mierczynska-Vasilev A, et al. Loading and release of a model protein from porous silicon powders. Phys Status Solidi A. 2007;204:3361–6. doi:10.1002/pssa.200723112. [Google Scholar]
- 11.Serda RE, Mack A, Pulikkathara M, et al. Cellular association and assembly of a multistage delivery system. Small. 2010;6:1329–40. doi: 10.1002/smll.201000126. doi:10.1002/smll.201000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tasciotti E, Liu X, Bhavane R, et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat Nanotechnol. 2008;3:151–7. doi: 10.1038/nnano.2008.34. doi:10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 13.Bowditch AP, Waters K, Gale H, et al. In-vivo assessment of tissue compatibility and calcification of bulk and porous silicon. Mat Res Soc Symp Proc. 1999;536:149–54. [Google Scholar]
- 14.Park J-H, Gu L, von Maltzahn G, et al. Biodegradable luminescent porous silicon nanoparticles for in vivo applications. Nat Mater. 2009;8:331–6. doi: 10.1038/nmat2398. doi:10.1038/nmat2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Low SP, Voelcker NH, Canham LT, Williams KA. The biocompatibility of porous silicon in tissues of the eye. Biomaterials. 2009;30:2873–80. doi: 10.1016/j.biomaterials.2009.02.008. doi:10.1016/j.biomaterials.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 16.Cheng L, Anglin E, Cunin F, et al. Intravitreal properties of porous silicon photonic crystals: a potential self-reporting intraocular drug-delivery vehicle. Br J Ophthalmol. 2008;92:705–11. doi: 10.1136/bjo.2007.133587. doi:10.1136/bjo.2007.133587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Canham LT. Bioactive silicon structure fabrication through nanoetching techniques. Adv Mater. 1995;7:1033–7. doi:10.1002/adma.19950071215. [Google Scholar]
- 18.Godin B, Gu J, Serda RE, et al. Tailoring the degradation kinetics of mesoporous silicon structures through PEGylation. J Biomed Mater Res A. 2010;94A:1236–43. doi: 10.1002/jbm.a.32807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson SHC, Elliott H, Wallis DJ, et al. Dissolution of different forms of partially porous silicon wafers under simulated physiological conditions. Phys Status Solidi A. 2003;197:331–5. doi:10.1002/pssa.200306519. [Google Scholar]
- 20.Bisi O, Ossicini S, Pavesi L. Porous silicon: a quantum sponge structure for silicon based optoelectronics. Surf Sci Rep. 2000;38:1–126. doi:10.1016/S0167-5729(99)00012-6. [Google Scholar]
- 21.Cunin F, Schmedake TA, Link JR, et al. Biomolecular screening with encoded porous-silicon photonic crystals. Nat Mater. 2002;1:39–41. doi: 10.1038/nmat702. doi:10.1038/nmat702. [DOI] [PubMed] [Google Scholar]
- 22.Canham LT. Silicon quantum wire array fabrication by electro-chemical and chemical dissolution of wafers. Appl Phys Lett. 1990;57:1046–8. doi:10.1063/1.103561. [Google Scholar]
- 23.Herino R. Pore size distribution in porous silicon. In: Canham LT, editor. The properties of porous silicon. Inspec/IEE; London: 1997. pp. 89–96. [Google Scholar]
- 24.Sakamoto J, Annapragada A, Decuzzi P, Ferrari M. Antibiological barrier nanovector technology for cancer applications. Expert Opin Drug Deliv. 2007;4:359–69. doi: 10.1517/17425247.4.4.359. doi:10.1517/17425247.4.4.359. [DOI] [PubMed] [Google Scholar]
- 25.Riehemann K, Schneider SW, Luger TA, et al. Nanomedicine—challenge and perspectives. Angew Chem Int Ed. 2009;48:872–97. doi:10.1002/anie.200802585. [Google Scholar]
- 26.Tanaka T, Mangala LS, Vivas-Mejia PE, et al. Sustained small interfering RNA delivery by mesoporous silicon particles. Cancer Res. 2010;70:3687–96. doi: 10.1158/0008-5472.CAN-09-3931. doi:10.1158/0008-5472.CAN-09-3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chiappini C, Tasciotti E, Fakhoury JR, et al. Tailored porous silicon microparticles: fabrication and properties. Chemphyschem. 2010;11:1029–35. doi: 10.1002/cphc.200900914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferrati S, Mack A, Chiappini C, et al. Intracellular trafficking of silicon particles and logic-embedded vectors. Nanoscale. 2010;2:1512–20. doi: 10.1039/c0nr00227e. doi:10.1039/c0nr00227e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decuzzi P, Godin B, Tanaka T, et al. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release. 2010;141:320–7. doi: 10.1016/j.jconrel.2009.10.014. doi:10.1016/j.jconrel.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Xia B, Xiao S-J, Guo D-J, et al. Biofunctionalisation of porous silicon (PS) surfaces by using homobifunctional cross-linkers. J Mater Chem. 2006;16:570–8. doi:10.1039/b511175g. [Google Scholar]
- 31.Khullar O, Frangioni JV, Grinstaff M, Colson YL. Image-guided sentinel lymph node mapping and nanotechnology-based nodal treatment in lung cancer using invisible near-infrared fluorescent light. Semin Thorac Cardiovasc Surg. 2009;21:309–15. doi: 10.1053/j.semtcvs.2009.11.009. doi:10.1053/j.semtcvs.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perini R, Choe R, Yodh AG, et al. Non-invasive assessment of tumor neovasculature: techniques and clinical applications. Cancer Metastasis Rev. 2008;27:615–30. doi: 10.1007/s10555-008-9147-6. doi:10.1007/s10555-008-9147-6. [DOI] [PubMed] [Google Scholar]
- 33.Wolf M, Ferrari M, Quaresima V. Progress of near-infrared spectroscopy and topography for brain and muscle clinical applications. J Biomed Opt. 2007;12:062104. doi: 10.1117/1.2804899. doi:10.1117/1.2804899. [DOI] [PubMed] [Google Scholar]
- 34.Mayer-Kuckuk P, Boskey AL. Molecular imaging promotes progress in orthopedic research. Bone. 2006;39:965–77. doi: 10.1016/j.bone.2006.05.009. doi:10.1016/j.bone.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Sanchez EQ, Chinnakotla S, Khan T, et al. Intraoperative imaging of pancreas transplant allografts using indocyanine green with laser fluorescence. Proc Bayl Univ Med Cent. 2008;21:258–60. doi: 10.1080/08998280.2008.11928406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reuthebuch O, Haussler A, Genoni M, et al. Novadaq SPY: intraoperative quality assessment in off-pump coronary artery bypass grafting. Chest. 2004;125:418–24. doi: 10.1378/chest.125.2.418. doi:10.1378/chest.125.2.418. [DOI] [PubMed] [Google Scholar]
- 37.Tagaya N, Yamazaki R, Nakagawa A, et al. Intraoperative identification of sentinel lymph nodes by near-infrared fluorescence imaging in patients with breast cancer. Am J Surg. 2008;195:850–3. doi: 10.1016/j.amjsurg.2007.02.032. doi:10.1016/j.amjsurg.2007.02.032. [DOI] [PubMed] [Google Scholar]
- 38.Sampath L, Wang W, Sevick-Muraca EM. Near infrared fluorescent optical imaging for nodal staging. J Biomed Opt. 2008;13:041312. doi: 10.1117/1.2953498. doi:10.1117/1.2953498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salonen J, Kaukonen AM, Hirvonen J, Lehto VP. Mesoporous silicon in drug delivery applications. J Pharm Sci. 2008;97:632–53. doi: 10.1002/jps.20999. doi:10.1002/jps.20999. [DOI] [PubMed] [Google Scholar]
- 40.Cerami A. Inflammatory cytokines. Clin Immunol Immunopathol. 1992;62(Suppl 1):S3–10. doi: 10.1016/0090-1229(92)90035-m. doi:10.1016/0090-1229(92)90035-M. [DOI] [PubMed] [Google Scholar]
- 41.Jugdaohsingh R, Anderson SH, Tucker KL, et al. Dietary silicon intake and absorption. Am J Clin Nutr. 2002;75:887–93. doi: 10.1093/ajcn/75.5.887. [DOI] [PubMed] [Google Scholar]
- 42.Carlisle EM. Silicon: a possible factor in bone calcification. Science. 1970;167:279–80. doi: 10.1126/science.167.3916.279. doi:10.1126/science.167.3916.279. [DOI] [PubMed] [Google Scholar]
- 43.Serda RE, Gu J, Burks JK, et al. Quantitative mechanics of endothelial phagocytosis of silicon microparticles. Cytometry A. 2009;75:752–60. doi: 10.1002/cyto.a.20769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A. 2006;103:4930–4. doi: 10.1073/pnas.0600997103. doi:10.1073/pnas.0600997103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evora C, Soriano I, Rogers RA, et al. Relating the phagocytosis of microparticles by alveolar macrophages to surface chemistry: the effect of 1,2-dipalmitoylphosphatidylcholine. J Control Release. 1998;51:143–52. doi: 10.1016/s0168-3659(97)00149-1. doi:10.1016/S0168-3659(97)00149-1. [DOI] [PubMed] [Google Scholar]
- 46.Kenneth Ward W. A review of the foreign-body response to subcutaneously-implanted devices: the role of macrophages and cytokines in biofouling and fibrosis. J Diabetes Sci Technol. 2008;2:768–77. doi: 10.1177/193229680800200504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serda RE, Ferrati S, Godin B, et al. Mitotic partitioning of silicon microparticles. Nanoscale. 2009;1:250–9. doi: 10.1039/b9nr00138g. doi:10.1039/b9nr 00138g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beletsi A, Panagi Z, Avgoustakis K. Biodistribution properties of nanoparticles based on mixtures of PLGA with PLGA-PEG diblock copolymers. Int J Pharm. 2005;298:233–41. doi: 10.1016/j.ijpharm.2005.03.024. doi:10.1016/j.ijpharm.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 49.Bimbo LM, Sarparanta M, Santos HlA, et al. Biocompatibility of thermally hydrocarbonized porous silicon nanoparticles and their biodistribution in rats. ACS Nano. 2010;4:3023–32. doi: 10.1021/nn901657w. doi:10.1021/nn901657w. [DOI] [PubMed] [Google Scholar]
- 50.Ananta JS, Godin B, Sethi R, et al. Geometrical confinement of gadolinium-based contrast agents in nanoporous particles enhances T1 contrast. Nat Nanotechnol. 2010;5:815–21. doi: 10.1038/nnano.2010.203. doi:10.1038/nnano.2010.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.