

Abstract
Inflammation and airway remodeling occur in a variety of airway diseases. Modeling aspects of the inflammatory and fibrotic processes following repeated exposure to particulate matter may provide insights into a spectrum of airway diseases, as well as prevention/treatment strategies. An agent-based model (ABM) was created to examine the response of an abstracted population of inflammatory cells (nominally macrophages, but possibly including other inflammatory cells such as lymphocytes) and cells involved in remodeling (nominally fibroblasts) to particulate exposure. The model focused on a limited number of relevant interactions, specifically those among macrophages, fibroblasts, a pro-inflammatory cytokine (TNF-α), an anti-inflammatory cytokine (TGF-β1), collagen deposition, and tissue damage. The model yielded three distinct states that were equated with (1) self-resolving inflammation and a return to baseline, (2) a pro-inflammatory process of localized tissue damage and fibrosis, and (3) elevated pro- and anti-inflammatory cytokines, persistent tissue damage, and fibrosis outcomes. Experimental results consistent with these predicted states were observed in histology sections of lung tissue from mice exposed to particulate matter. Systematic in silico studies suggested that the development of each state depended primarily upon the degree and duration of exposure. Thus, a relatively simple ABM resulted in several, biologically feasible, emergent states, suggesting that the model captures certain salient features of inflammation following exposure of the lung to particulate matter. This ABM may hold future utility in the setting of airway disease resulting from inflammation and fibrosis following particulate exposure.
INTRODUCTION
Particulate inhalation from tobacco smoke [1,2,3] and, to a lesser extent, occupational exposure [4,5,6], as well as exposure to air pollutants [3,7,8,9,10], have been identified as being among the primary causes of chronic airway diseases such as chronic obstructive pulmonary disease (COPD) in the general population. However, the degree and duration of the exposure and composition of the particulate to which the lung is exposed may determine the exact inflammatory response elicited [11,12]. While fibrosis and increased cellularity are often observed in the lungs of smokers and those regularly exposed to occupational or environmental pollution [13,14,15], chronic airway disease does not necessarily result from repeated exposure. Instead, chronic airway disease develops over time through a complex set of inflammatory and tissue repair mechanisms that are not yet fully understood [1,16]. Other factors such as age [17,18] and genetic disposition [19,20,21,22,23,24] have also been shown to contribute to inflammatory lung diseases such as COPD.
The inflammatory response is known to play a central role in the development of diseases such as COPD [25]. One central inflammatory cell type involved in this response is the macrophage. Macrophages contribute to the homeostasis of self-resolving return to baseline lung tissue via the clearance of pathogens and inhaled particulate from the lung. These same macrophages, however, are also thought to orchestrate some of the pathophysiology of COPD [26]. In addition, other inflammatory cells such as lymphocytes and neutrophils may be involved in various inflammatory diseases of the lung, including COPD [27,28,29]. In a prototypical inflammatory lung disease such as COPD, the response of macrophages following the inhalation of pathogens and particulate matter is intensified, leading to elevated levels of inflammatory cells and the production of high levels of pro-inflammatory (TH1) mediators, which in turn lead to a “feed-forward” loop of inflammation, fibrosis, and tissue damage [1,16,26]. This “feed-forward” loop is thought to be key in the development of the limited airflow as well as the altered structure and function of the airways, pulmonary vasculature, and lung parenchyma that are characteristic of COPD. Similar mechanisms have been suggested in some studies of pulmonary fibrosis [30]; however, pulmonary fibrosis can also be driven primarily by anti-inflammatory (TH2), pro-fibrotic cytokine secretion from epithelial cells with little involvement of inflammatory cells [31,32,33]. These mechanisms are distinct from a number of other airway diseases, such as asthma, in which macrophages are not necessarily the primary drivers of the disease process [25,34].
A number of in vitro, ex vivo and in vivo models have been used to study chronic airway disease. While these experimental models have yielded interesting insights in an efficient and controlled manner, no model appears capable of fully recapitulating the development of COPD in humans [35,36,37]. For example, in vitro and ex vivo models do not contain the full complement of circulating cells that are repeatedly recruited and activated in chronic inflammatory processes such as COPD. These models, however, can provide a quick and relatively inexpensive way to answer focused questions about the mechanisms that underlie the behavior of specific cell types in this disease. Animal models of COPD are both more expensive and time consuming than their in vitro and ex vivo counterparts, but allow for a more in depth examination of the mechanisms underlying the development of COPD in a multi-organ system. However, animal models may lack multiple components of the anatomy, physiology, genetics, and immune system of human patients depending on the model [35,36,37].
Recent developments in mathematical modeling (so-called “in silico” studies) of inflammation and related processes in the settings of sepsis, trauma, and wound healing have suggested a novel experimental pathway in the study of complex diseases [38,39,40,41,42]. Therefore, an in silico approach to modeling specific aspects of airway diseases, that both mimics behavior which is physiologically relevant to the human case and allows for the rapid modulation of a number of variables simultaneously, may provide an ideal approach to studying specific questions regarding immune activity in the development of a spectrum of airway diseases.
In this study, a relatively simple agent-based model (ABM), equivalent to a “lumped-parameter model” in which some of the parameters theoretically represent an algebraic combination of several quantities, was created in order to examine the role of macrophages and fibroblasts in the inflammatory and fibrotic response to particulate exposure. The model focuses on a limited number of biologically relevant, well-vetted interactions, specifically those among macrophages, fibroblasts, a rapidly produced pro-inflammatory cytokine (e.g. TNF-α), a slowly produced anti-inflammatory cytokine (e.g. TGF-β1), collagen deposition (a marker for both tissue healing and fibrosis), and lung tissue damage. The model was partially validated against an in vivo experimental paradigm of cigarette smoke exposure in mice. The results of this study show that this ABM can simulate several biologically feasible emergent states. These studies suggest that the dynamics of this simple computational model describe biologically relevant aspects of immune activity following exposure to particulates. Moreover, the mechanisms in this model may be expanded to serve as a platform for the investigation of the processes of inflammation and fibrosis that result from particulate exposure in the lung.
METHODS
Software
In this study, Netlogo v4.0 freeware (Center for Connected Learning and Computer Based Modeling, Northwestern University, Evanston, IL) was utilized to design a simple ABM that simulates inflammation in the lung following particulate exposure. Netlogo allows for the utilization of variables that can be described as “patches” or “agents”. Patch variables have a fixed location within the simulated environment and contain specific information about the local environment. Agents are allowed to move freely within the environment (i.e. from patch to patch) and respond to the information contained locally within each patch as well as other agents within the system.
Agent-Based Model
In this model, agent variables were used to represent particulate matter, inflammatory cells, as well as fibroblasts. Patch variables were used to represent the local levels of pro- (TNF-α) and anti-inflammatory (TGF-β1) cytokines produced by macrophages as well as tissue damage resulting from macrophage activation. Additionally, a patch variable was used to represent collagen deposition by fibroblasts during tissue repair.
The present simulation was a highly simplified (“lumped parameter”) model, created to determine if a relatively small set of consensus interactions could account for a variety of behaviors observed in lung inflammation. Therefore, all inflammatory cells (neutrophils, macrophages, and lymphocytes) were subsumed under a single variable (nominally macrophages), all pro-inflammatory cytokines under a single variable (nominally TNF-α) and all anti-inflammatory/pro-fibrotic cytokines and growth factors under another single variable (nominally TGF-β1). Thus, some of the parameters in our model theoretically represent an algebraic combination of several quantities and do not have a value that can be determined experimentally. However, the interactions within the model were governed by a set of rules which were based upon known interactions between cells and cytokines in inflammation and fibrosis leading to chronic airway disease as well as insights derived from prior models of inflammation and wound healing [1,16,25,26,43,44]. These interactions are outlined in Figure 1, and the specific details of each rule are listed in the Supplementary Materials (Supplemental Table 1). The rules of the ABM were based on known biology, including conserved inflammation/healing interactions modeled previously [43,44].
Figure 1. Model Interactions.
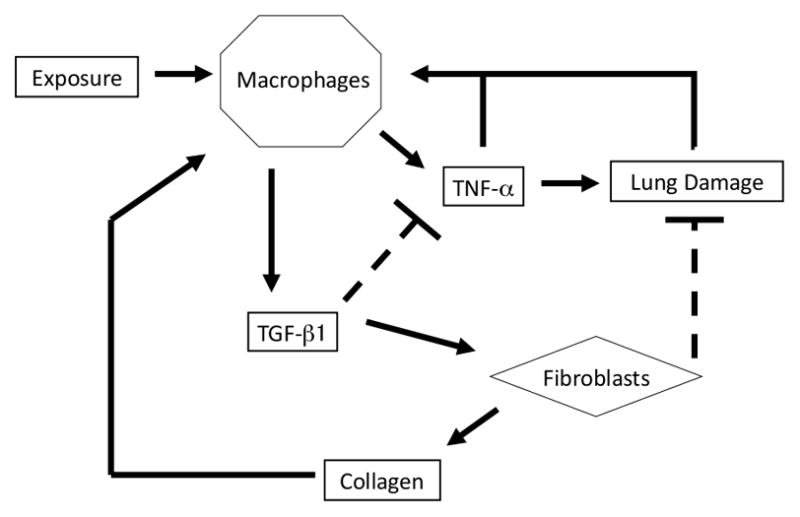
This figure illustrates the interactions of components of the inflammation/healing ABM. Briefly, the system is perturbed from its base state via exposure to particulate matter. Upon encountering particulate matter, macrophages become activated to produce TNF-α (pro-inflammatory cytokines), the production of which is antagonized by the amount of anti-inflammatory cytokine present within the environment. TNF-α acts in part as a chemoattractant, allowing additional macrophages to enter the environment and to move in order to aid in the clearance of particulate matter. In this model, TNF -α also acts a surrogate for proteases and reactive oxygen species which cause tissue damage. Following clearance of particulate matter, macrophages cease to produce TNF-α and instead produced TGF-β1; this is analogous to the transition from M1 to M2 macrophage phenotype. TGF-β1 acts as both an antagonist of TNF-α production and as a chemoattractant, allowing additional fibroblasts to enter the system. Fibroblasts move toward and repair areas of tissue damage while also depositing collagen. High collagen levels also activate macrophages within the environment, a process that is a surrogate for the pro-inflammatory effects of fibrosis. For a detailed description of the ABM rules, please see Supplementary Materials.
The model environment consisted of 2500 patches on a torus arranged in a grid (50 patches × 50 patches). The environment was populated with 30 randomly distributed macrophages and 10 randomly distributed fibroblasts prior to execution of the model. In the absence of a stimulus, cells were allowed to move into an adjacent patch along a randomly generated trajectory, thus “patrolling” the environment. This is equivalent to the monitoring of blood vessels and tissues by CX3CR1 mononuclear cells, a process which has recently been described to play an important role in the response to particulate exposure of the lungs [45,46]. The system was perturbed from this inactive state via the addition of randomly distributed particulate matter to the environment, simulating particulate exposure via inhalation. Upon initiation of the exposure regimen, 5, 10, 15, or 20 simulated particles were added to the model environment, again at random intervals. Addition of particulates occurred at intervals of 50, 100, and 200 time steps, or iterations, of the model. The number of times the particulate was added was also varied (50, 100 and 200 times). The values were chosen to represent different levels and frequencies exposure, both of which are thought to be linked to the likelihood of development of chronic lung disease [1,6,8]. While cessation of smoking or particulate exposure has been shown to slow or prevent the development of chronic inflammatory processes of the lung, there is little evidence that cessation is capable of reversing the progression of chronic inflammatory processes of the lung once they have begun [16,47,48,49]. Therefore, the model was allowed to run following the end of the exposure regimen for 10,000 time steps in order to examine the progression of the model following cessation of exposure in order to determine if recovery from observed pro-inflammatory or disease states was possible.
Upon encountering particulate matter within the environment, macrophages ceased their movement until they cleared the particles and became activated (Supplemental Table 1). The amount of time to clearance depended upon the number of macrophages interacting with the particle (Supplemental Table 1), Activated macrophages produced pro-inflammatory cytokines (TNF-α) [1,50,51,52]. The degree of pro-inflammatory cytokine production was antagonized by the presence of anti-inflammatory cytokines within the patch where the macrophage was located [53,54] (Supplemental Table 1). TNF-α acted both as a chemoattractant, allowing additional macrophages to enter the environment and to move towards sites of inflammation in order to aid in the clearance of particulate matter, and as a surrogate for a number of TNF-α-induced proteases and reactive oxygen species secreted by macrophages leading to tissue damage and the release of “alarm/danger signals”, also known as “Damage-associated Molecular Patterns” [DAMPs]) [53,54,55,56] (Supplemental Table 1). This behavior was programmed to resemble the cellular activation, chemotactic and inflammatory mediator production, and subsequent tissue damage that occurs following particulate exposure in the lung [15,16,19,26,28,57,58,59,60,61,62]. Upon clearance of the particulate matter, and after the initial period of macrophage activation, macrophages ceased to produce TNF-α and instead produced TGF-b1 for a short period of time, indicating the “deactivation” of the macrophage [52,63] (Supplemental Table 1). TGF-b1 acted as both an antagonist of TNF-a production and a chemoattractant, allowing additional fibroblasts to enter the system [64,65] (Supplemental Table 1). Both TNF-α and TGF-β1 were allowed to diffuse from patch to patch and were subject to degradation [66,67,68] (Supplemental Table 1). In this manner, the macrophages in this model could exhibit features of both M1 (pro-inflammatory, TNF-a-producing) and M2 (regulatory, tissue remodeling, TGF-β1-producing) macrophages [52,63]. Fibroblasts moved toward and repaired areas of tissue damage caused by the activation of macrophages and secretion of TNF-a while depositing collagen [64,65] (Supplemental Table 1), but did not secrete either pro- or anti-inflammatory cytokines in the present, simplified model. Collagen deposition continued until tissue life was restored to 100% (Supplemental Table 1). This represents the default mammalian response of scar tissue formation following tissue injury [65,69]. Finally, high collagen levels, simulating areas of fibrosis, also acted as activators of macrophages within the environment [30] (Supplemental Table 1). Thus, this ABM is theoretically capable of simulating both healing and fibrotic long-term outcomes of an inflammatory response. For a more detailed description of the ABM rules and assumptions, please see Supplementary Materials (Supplemental Table 1).
The model was run 40 times each at all combinations of the exposure degrees (0, 5, 10, 15, 20), intervals (50, 100, 200), and number of exposures in a full factorial design with a total of 1800 model runs. The results of each run were evaluated for the appearance of the graphical output of the model and the numerical outputs for each of the variables and agents were exported and evaluated in Microsoft Excel®. These outputs were then used to group the results into three distinct and easily identifiable categories, which included 1) self-resolving return to baseline tissue, 2) localized tissue damage and fibrosis, and 3) systemic damage and fibrosis. The specifics of each state are described in detail below.
In Vivo Exposure Model
All animal experiments were performed according to the 1996 NIH Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of Pittsburgh. Twelve to fourteen week-old female BALB/c mice (Harlan-Sprague, Indianapolis, IN, USA) were exposed to cigarette smoke (3R4F non-filtered cigarettes; University of Kentucky, Lexington, KY, USA) using a smoking apparatus as previously described by Shapiro, et al. [70] During the first week of exposure, the animals received one cigarette in the morning and one in the afternoon. Following the second week, the animals received four cigarettes per day, two in the morning and two in the afternoon. This experimental protocol was carried out for five weeks. Animals that developed respiratory distress during exposure to cigarette smoke were removed and allowed to recover for one day. If the problem continued consistently, the animal was removed from the study. Animals that were not exposed to smoke inhalation were used as controls. Three animals, respectively, were exposed to each of the control and experimental conditions described above.
Following the 5-week exposure period, the animals were euthanized and the lung tissues of each animal were harvested and fixed for histological examination. Tissues were embedded in paraffin and sectioned prior to staining with hematoxylin and eosin. All slides were then investigated for signs of tissue damage, extracellular matrix deposition, and cellular aggregation and histological images were assessed by a trained, blinded pathologist and compared to the graphical output of the model.
RESULTS
The output of the ABM used in this study included both graphical and numerical data. The graphical and the numerical data were considered together, leading to the identification of three distinct, easily identifiable model end states: (1) return to baseline of both pro- and anti-inflammatory mediators concomitant with tissue healing, (2) elevated pro-inflammatory cytokines concomitant with localized tissue damage and fibrosis, and (3) simultaneously elevated pro- and anti-inflammatory cytokines concomitant with extensive tissue damage and fibrosis.
Graphical Output and Model Validation
Graphical Output
The following variables were included in the graphical output of the model: macrophages, fibroblasts, tissue life, and collagen deposition. During analysis of the graphical output, three distinct states were observed (Figure 2a–c). The first state, suggesting self-a resolving inflammatory and tissue damage response and return of the system to baseline, was characterized by the presence of little to no tissue damage and minimal collagen deposition. This state was accompanied by baseline numbers of randomly distributed, non-activated macrophages and fibroblasts (Figure 2a). The second state, which could be characterized as localized tissue damage and fibrosis, was characterized by localized tissue damage/death/high DAMP levels, along with collagen deposition. This state was accompanied by the presence of activated macrophages and fibroblasts, both of which appeared to be localized to the areas of tissue damage and collagen deposition (Figure 2b). Further, it was observed that, in general, the majority of the macrophages and fibroblasts were localized at the periphery of the areas of tissue damage, as was the majority of the collagen deposition. Pro-inflammatory cytokines remained elevated in this state, but anti-inflammatory mediators were absent. The third state, suggestive of a major regulatory derangement of inflammation in which both pro- and anti-inflammatory cytokines were present along with systemic damage and fibrosis, was characterized by widespread tissue damage and random deposition of large amounts of collagen. This state was accompanied by elevated numbers of both activated macrophages and fibroblasts. In this state, neither macrophages nor fibroblasts appeared to be localized in any distinguishable pattern.
Figure 2. Graphical Representation of Model Output and Partial Experimental Validation of Resultant States.

Representative graphical output of the model for the self-resolving (A), localized tissue damage and fibrosis (B), and persistent, extensive tissue damage and fibrosis (C) outcomes. Panel D-F are histological sections from mice subjected to experimental smoke inhalation as described in the Materials and Methods. Representative photomicrographs of the histological appearance of control animals (D) and animals exposed to smoke for 5 weeks (E and F) are shown. Some animals exposed to smoke exhibited small areas of cellular aggregation (E) adjacent to the small airways of the lung. Slightly larger aggregates were observed in other animals (F). Extensive areas of tissue damage and fibrosis were not observed.
Partial Validation Against Histological Data
BALB/c mice were exposed to cigarette smoke for five weeks as described in the Materials and Methods. Photomicrographs of the histological appearance of the lungs of control mice (Figure 2d) and smoke-exposed mice (Figure 2e and f) are shown. No cellular aggregates were observed in control animals that were not exposed to smoke inhalation (Figure 2d). In the lungs of animals exposed to smoke, a small number of inflammatory cell aggregates (likely lymphocytes) were observed adjacent to the small airways of the lung (Figure 2e and f). The size of the aggregate differed between smoke-exposed animals, with some animals exhibiting small aggregates (Figure 2e) and some expressing larger aggregates (Figure 2f); however, no other lung pathology was observed in any animal. The aggregates observed in Figure 2e were similar in appearance to those that developed in the localized tissue damage and fibrosis state in the computational model (Figure 2b). While diffuse damage and fibrosis was not observed histologically, larger aggregates and a small amount of extracellular matrix deposition suggestive of more extensive tissue damage were observed histologically in some animals (Figure 2f).
Numerical Output
Numerical output of the model included both agent and patch variables. Output for the agent variables represented a count of the number of agents (macrophages, fibroblasts) present within the global environment. Output for the patch variables (tissue life, pro- and anti-inflammatory cytokines, and collagen deposition) was calculated as an average value for all of the patches within the global environment. These outputs were examined for each of the states described above.
Tissue Life
The first variable investigated was the tissue life during each simulation. A slight decrease (< 5%) in tissue life was observed during all simulations in the first 25% of the period of exposure of the system to particulate matter. The model was observed to have three distinct states thereafter. In the first state, tissue life returned to baseline levels (~100%; i.e. tissue damage returned to 0) after the initial 25% of the period of exposure, even during the continued exposure of the system to particulate matter. In the second state, a gradual decrease in tissue life was observed following the first 25% of the period of exposure. This gradual decrease was seen to continue even after exposure had ceased, and no signs of recovery were observed during the remainder of the simulation. In the third state, there was a similar period of gradual decrease in tissue life; however, this period was followed by a more rapid decrease in tissue life, eventually approaching 0%. Averaged results generated from 10 simulations representative of each state are presented in Figure 3a. Each outcome was the result of the same simulation with the same initial conditions, indicating a high level of stochastic variation present within the model. The three observed states were found to correspond to the states identified as 1) healing and return to baseline; 2) pro-inflammation, localized tissue damage (seen in the context of cellular aggregates), and fibrosis; and 3) simultaneous pro- and anti-inflammation, extensive damage, and fibrosis in the evaluation of the graphical output of the model.
Figure 3. Tissue Life, Macrophage Number, Fibroblast Number, and Collagen Deposition.

Averaged tissue life (A), macrophage number (B), fibroblast number (C), and collagen deposition (D) results from 10 simulations resulting in each state (self-resolving state = solid green line, localized damage and fibrosis state = dashed blue line, persistent global tissue damage and fibrosis = dotted red line). Tissue life and collagen deposition results reported in arbitrary units. All results are reported as the mean ± standard deviation of the mean.
Macrophage Levels
Macrophage levels during the initial 25% of the period of exposure of the system were similar in each of the three states and were characterized by a spike in of the number of macrophages in the system from baseline (30 cells) to approximately 75 cells, followed by a decrease to approximately 50 thereafter. After this initial response period, the simulations could again be grouped into one of three states as described for tissue life. The self-resolving state (return to baseline) was characterized by a gradual drop in macrophage number back to baseline levels prior to the cessation of exposure of the system. Macrophage numbers in those simulations that resulted in the state characterized by localized tissue damage along with fibrosis were seen to decrease in a similar manner; however, slightly elevated macrophage numbers were observed for the duration of the period of exposure. The final state, characterized by the simultaneous presence of both pro- and anti-inflammatory mediators, extensive damage, and fibrosis, was also characterized by a rapid increase in macrophage numbers to the predetermined model maximum (300 cells). Once this rapid increase was initiated, macrophage numbers did not return to baseline levels in any simulation, even if the addition of particulate matter to the system was terminated. For this reason, we refer to this last state as “self-sustaining.” Averaged results from 10 simulations representative of each state are presented in Figure 3b.
Fibroblast Levels
Fibroblast numbers remained at baseline levels (10 cells) in both the self-resolving and pro-inflammation/localized tissue damage/fibrosis states throughout the duration of the simulations. In the extensive damage and fibrosis state, fibroblast numbers remained at baseline during the initial period of the simulation, increased rapidly from baseline to 60–70 cells, and then decreased thereafter, with slightly elevated levels for the duration of the simulation. The initiation of the rapid increase in fibroblast level in the extensive damage and fibrosis state appeared to correspond with the start of the rapid increase in the number of macrophages. Averaged results from 10 simulations representative of each state are presented in Figure 3c.
Collagen Levels
The amount of collagen deposited increased slightly from baseline in all three states during the initial 25% of the period of exposure of the system. After this initial period, the levels of collagen followed one of three distinct patterns that were associated with the three states described above. Briefly, following the initial 25% of the perturbation period, the level of collagen for those simulations resulting in the self-resolving state slowly decreased back to baseline levels. The pro-inflammation/localized tissue damage/fibrosis state was characterized by a slow but steady increase in collagen levels throughout the simulation, even after exposure of the system to particulate matter had ceased. The extensive damage and fibrosis state was characterized by a rapid increase in the level of collagen deposited following the first 25% of the perturbation period, with a slight decrease in collagen levels after the addition of particulate matter was ceased. The timing of the changes in the levels of collagen deposition in all three states appears to correspond to the timing of the change in macrophage and fibroblast numbers. Averaged results from 10 simulations representative of each state are presented in Figure 3d.
Cytokine Levels
The levels of the pro- and anti-inflammatory cytokines were assessed in simulations resulting in each of the three described states. The self-resolving state was characterized by slightly increased levels of pro- and anti-inflammatory cytokines during the initial period of exposure of the system to particulate matter. The initial increase in the level of the pro-inflammatory cytokine was larger than that observed for the anti-inflammatory cytokine. The levels of both cytokines decreased towards a basal state characterized by low levels of both pro- and anti-inflammatory cytokines, with a slight predominance of the anti-inflammatory cytokines. The localized tissue damage and fibrosis state was characterized by an initial increase levels of both pro- and anti-inflammatory cytokines, similar to that observed in the self-resolving state. However, in contrast to the self-resolving state, the longer-term outcomes of the localized tissue damage and fibrosis state included elevated levels of the pro-inflammatory cytokine, which persisted past the initial period of exposure and lasted for the duration of the simulation. The extensive damage and fibrosis state was characterized by an increase in both pro- and anti-inflammatory cytokines throughout the duration of the simulation. This increase was larger in magnitude than the increases observed in the localized tissue damage and fibrosis state. The predominance of the pro-inflammatory cytokine as compared to the anti-inflammatory cytokine was much larger in extensive damage and fibrosis state than in the localized tissue damage state. In the extensive damage and fibrosis state, the rapid increase in the pro-inflammatory cytokine corresponded to the rapid increase in macrophage number, and spikes in the anti-inflammatory cytokine corresponded with the spikes in fibroblast numbers. Averaged results for the global cytokine levels in 10 simulations representative of each state are presented in Figures 4a and 4b.
Figure 4. Cytokine Levels.
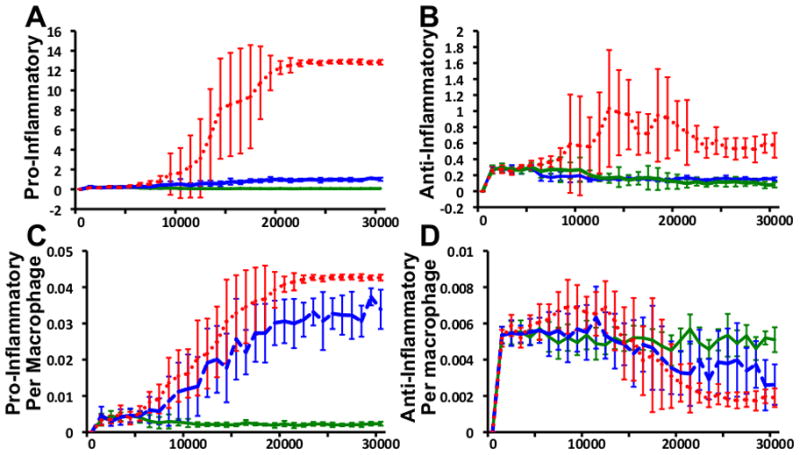
Averaged pro-inflammatory (A) and anti-inflammatory (B) cytokine level results from 10 simulations resulting in each state (self-resolving state = solid green line, localized damage and fibrosis state = dotted blue line, persistent global tissue damage and fibrosis = dotted red line). Pro-inflammatory (C) and anti-inflammatory (D) cytokine levels per macrophage. All results reported in arbitrary units. All results are reported as the mean ± standard deviation of the mean.
The levels of pro- and anti-inflammatory cytokines were also investigated on a per-macrophage basis. The per-macrophage levels of the pro-inflammatory cytokine were elevated in those simulations that resulted in the localized tissue damage and fibrosis and extensive damage and fibrosis states when compared to the per-macrophage levels in the self-resolving state. Paradoxically, the per-macrophage levels of the anti-inflammatory cytokine were decreased in those simulations that resulted in the localized tissue damage and fibrosis and extensive damage and fibrosis states when compared to the per-macrophage levels in the self-resolving state, despite the elevated global levels characteristic of this state. Per-macrophage cytokine levels are presented in Figures 4c and 4d.
System Variability
In order to determine which factors most influenced the development of the states described above the following system inputs were varied: the amount of simulated particulate delivered to the system during each exposure, the frequency of exposure, the duration of exposure, and the length of macrophage and fibroblast life.
Variation of Exposure
The number of particles delivered to random locations within the environment in each simulated exposure was set to 0, 5, 10, 15, and 20. The interval between these exposures was also varied. Intervals included one exposure every 50, 100, or 200 iterations of the model. Finally, the number of times the system was exposed to particulate matter was also varied. The system was perturbed 50, 100, and 200 times. Simulations were run to include all possible combinations of the variables described. Each combination was run 40 times and the resultant development of a healing and return to baseline, localized tissue damage and fibrosis, or extensive damage and fibrosis end state was characterized based upon the criteria described above and recorded. The results for each variable combination are presented in Table 1. The results suggest that complex interactions between the amount of particulate, the frequency of exposure, and the duration of exposure can influence the formation of the described states. Higher amounts of particulate, shorter intervals, and increased frequency were all related to the formation of localized tissue damage and fibrosis and extensive damage and fibrosis states.
Table 1.
Effects of Varying Exposure Upon the Formation of Self-Resolving, Localized Tissue Damage and Fibrosis, and Persistent Tissue Damage and Fibrosis States
| Interval Between Exposures | Number of Times Exposed | Number of Particles Per Exposure | ||||
|---|---|---|---|---|---|---|
| 0 | 5 | 10 | 15 | 20 | ||
| 50 | 40, 0, 0 | 40, 0, 0 | 30, 10, 0 | 10, 30, 0 | 0, 40, 0 | |
| 50 | 100 | 40, 0, 0 | 40, 0, 0 | 24, 16, 0 | 0, 39, 1 | 0, 7, 33 |
| 200 | 40, 0, 0 | 40, 0, 0 | 20, 20, 0 | 0, 3, 37 | 0, 0, 40 | |
| 50 | 40, 0, 0 | 39, 1, 0 | 39, 1, 0 | 34, 6, 0 | 29, 11, 0 | |
| 100 | 100 | 40, 0, 0 | 39, 1, 0 | 38, 2, 0 | 29, 11, 0 | 17, 22, 1 |
| 200 | 40, 0, 0 | 39, 1, 0 | 34, 5, 1 | 22, 12, 6 | 2, 14, 24 | |
| 50 | 40, 0, 0 | 40, 0, 0 | 39, 1, 0 | 40, 0, 0 | 39, 1, 0 | |
| 200 | 100 | 40, 0, 0 | 40, 0, 0 | 40, 0, 0 | 38, 1, 1 | 37, 2, 1 |
| 200 | 40, 0, 0 | 40, 0, 0 | 39, 0, 1 | 38, 1, 1 | 25, 8, 7 | |
Number of simulations out of 40 resulting in self-resolving (S), localized damage and fibrosis (L), and persistent damage and fibrosis (P) states expressed as S, L, P.
Variation of Macrophage and Fibroblast Life
The amount of time that macrophages and fibroblasts remain in the simulation before they “die” and are removed from the system was varied to observe the effects of cell life span upon the formation of the three observed states. Both macrophage and fibroblast life (number of iterations before a cell “dies” and is removed from the system) were set to 300, 400, and 500 time steps and simulations were run 10 times. Either macrophage life or fibroblast life was altered, while the other was set at 300 iterations. The results were then grouped into healing and return to baseline, localized tissue damage and fibrosis, and extensive damage and fibrosis, and the maximum macrophage levels for each state were assessed. The maximum number of macrophages was similar in both the self-resolving and localized tissue damage and fibrosis states, while the maximum for the extensive damage and fibrosis state always reached the user-defined maximum of 300. The proportionality of the developed states remained similar for all variations of macrophage and fibroblast life, suggesting that the ABM is robust to this variation. Maximum macrophage levels for the self-resolving and localized tissue damage and fibrosis state for each macrophage and fibroblast life are presented in Figure 5a and b, respectively.
Figure 5. Variation of Macrophage and Fibroblast Life.
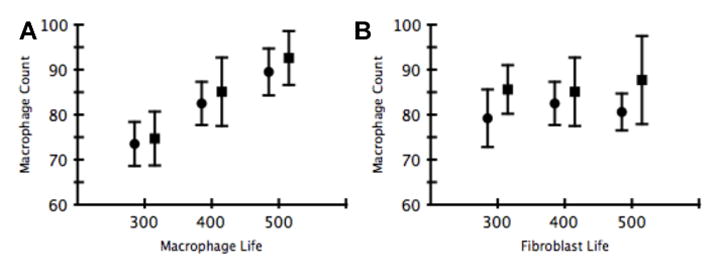
Effects of variation of macrophage and fibroblast life upon maximum macrophage levels in self-resolving (circle) and localized damage and fibrosis (square) states (10 simulations for each state). All simulations which resulted in the global tissue damage and fibrosis state resulted in macrophage levels reaching the user-defined maximum of 300. All results are reported as the mean ± standard deviation of the mean.
DISCUSSION
The present study describes the development of a simple ABM capable of reproducing multiple aspects of innate immune activity following simulated exposure to particulate matter. In many ways, this computational model has many generic features of tissue inflammation and healing. The model includes interactions among prototypical inflammatory cells (referred to generically in the present reduced model as “macrophages”, but including other inflammatory cell types that play a role in lung inflammation [such as lymphocytes and neutrophils]), prototypical reparative cells (referred to as “fibroblasts,” but including also myofibroblasts which can also lead to over-exuberant, harmful healing [i.e. fibrosis]), a canonical early pro-inflammatory cytokine (TNF-α), a canonical anti-inflammatory cytokine (TGF-β1), collagen deposition, and tissue damage/dysfunction/DAMPs. Though the “wiring” of this relatively simple ABM is not inherently specific to the lung, these simulations predicted the existence of three biologically relevant, stable states: (1) healing and return to baseline, (2) localized tissue damage and fibrosis, and (3) extensive damage and fibrosis. However, we note that the state of the model at early time points (<25% of model run time) was relatively similar for each of the subsequent end states. This finding suggests that many of the trajectories lie close to a manifold that divides the attracting states and that there may be an early intermediate state which leads to each of the observed end states thereafter. However, following this initial period we note that the second state is not merely a stage of progression on the way to the third state, but rather that each state is characterized by distinct features not programmed into the simulation. Each of these states was characterized by distinct patterns of cell behavior and cytokine production, as well as tissue damage and repair. The development of each state was linked directly to the amount, frequency, and duration of exposure. Notably, these simulations suggest that increased particulate exposure, at shorter intervals and at higher frequencies, increase the likelihood of the development of either the localized tissue damage and fibrosis or systemic damage and fibrosis state.
It is widely accepted that macrophages, which may account for as much as 95% of the leukocyte population within the lung, play a pivotal role in the development and orchestration of the disease processes that underlie COPD [26,71]. Studies have shown increased numbers of macrophages in the lungs of smokers and individuals with COPD and other airway diseases [14,15]. Further, these macrophages have been shown to produce higher levels of pro-inflammatory cytokines as well as proteases, leading to additional cell recruitment and tissue destruction [26,59,60,61,62,72]. This “feed-forward” cycle of inflammation, characteristic in chronic airway disease, was also observed in the present study. Those simulations that resulted in the localized tissue damage and fibrosis and systemic damage and fibrosis states exhibited not only larger numbers of macrophages and higher overall levels of pro-inflammatory cytokines, but also increased levels of pro-inflammatory cytokine production per macrophage present within the system. It is unclear whether the number of macrophages, the global level of cytokines, the level of cytokine production per cell in the system, or some combination thereof is the determining factor for the development of the pathologic states observed in this study. Likely, it is the complex interactions and interrelation of all of these factors which accounts for the observed behavior rather than any individual factor.
In addition to performing a pro-inflammatory role in the clearance of pathogens, macrophages can also serve to maintain tissue homeostasis or resolve an ongoing inflammatory response through the secretion of anti-inflammatory cytokines [73,74]. Following pro-inflammatory activation and pathogen clearance, and/or subsequent to defined cytokine signals, a failure to transition to an anti-inflammatory phenotype may be a determining factor in the healing process. A failure to do so may push a system towards a pathologic chronic inflammatory state, characterized by tissue fibrosis due to a failure to suppress pro-inflammatory stimuli below a certain threshold. In patients with COPD, the release of certain anti-inflammatory cytokines as well as inhibitors of protease activity by macrophages is decreased, thus failing to provide “control signals” for the pro-inflammatory process. In turn, this aberrant signaling leads to the deterioration of lung tissue and a chronic, vicious cycle of inflammation→damage→inflammation [57,58,75,76].
The role of TGF-β1 in COPD and other airway diseases that lead to fibrosis of the small airways is highly complex [77,78]. Experimental studies have shown that while macrophages of COPD patients produce fewer anti-inflammatory cytokines and anti-proteases, levels of TGF-β1 in the lung as a whole are increased leading to the stimulation of fibrosis through elevated TGF-β1 activation [79,80,81,82]. Pulmonary fibrosis, a distinct disease, is also associated with increased levels of TGF-β1 within the lung as a whole, however the source of TGF-β1 in pulmonary fibrosis is not necessarily of an inflammatory cell origin. A similar trend was observed in this study. The per-cell production of TGF-β1 was decreased in the systemic damage and fibrosis state; however, due to an increase in the number of TGF-β1 secreting macrophages in the system, the global level of TGF-β1 in the system was found to be higher than was observed in either the localized tissue damage and fibrosis or self-resolving states. Again, the overall cytokine level as well as the relative level of expression per cell in the system may be determinants of which state develops with time.
In patients with COPD, neutrophils and macrophages secrete proteases including neutrophil elastase, MMP-9, and MMP-12, among others [58]. It is recognized that this process eventually leads to an imbalance of the protease-anti-protease system present within the lung, leading to a complex cycle of ECM destruction, cell recruitment and activation, cytokine and effector molecule secretion, and further protease production [58]. A recent study utilized a mathematical model to investigate the failure of protease-anti-protease homeostasis in COPD [83]. That model predicted that the system could result in equilibrium behaviors that included a “normal” equilibrium in which the parameters returned to their baseline levels or a “COPD” equilibrium in which protease levels remained high even after smoking cessation. The development of the “COPD” equilibrium was attributed to a “feed forward” loop in which levels never returned to baseline. In that study, the secretion of the pro-inflammatory cytokine acted as a surrogate for the more complex process of tissue damage resulting from protease activity during an inflammatory process. Similar to the previously described study, the present study observed a “feed forward” loop of inflammation stemming from an imbalance of the signals that either promoted or prevented tissue damage. In addition, the levels of tissue damage (DAMPs), collagen, and cytokines remained high once the “feed forward” loop leading to the extensive damage and fibrosis state was initiated, even after the addition of particulate matter to the system had ceased. We have observed similar dynamics in our computational simulations of sepsis as well as the response to anthrax, in which the inflammation→damage→inflammation loop took over as the driver of inflammation even after the original impetus for the inflammatory response (i.e. bacterial infection) was no longer present[84,85].
In the present study, a third, localized tissue damage and fibrosis, state was also observed to exist in addition to the self-resolving and extensive damage and fibrosis states. In this state, tissue damage and collagen deposition increased over time, the level of the pro-inflammatory cytokine TNF-α was elevated, and the number of macrophages and overall level of the anti-inflammatory cytokine TGF-β1 all returned to baseline. This observation is in contrast to the extensive damage and fibrosis state, in which tissue damage and collagen levels increased more rapidly than in the localized tissue damage and fibrosis state, while the number of macrophages and levels of TNF-α remained elevated throughout the simulation. Interestingly, the levels of TGF-β1 remained low, and an increase in the number of fibroblasts was not observed. That the ABM includes spatio-temporal effects in contrast to a well-mixed model such as one based on ordinary differential equations may largely be responsible for the presence of this state. The spatial aspect of ABM causes a pattern-forming instability, which allows the localized regions of damage to be self-sustaining while not affecting the system as a whole[86,87]. In a well-mixed system, the low levels of tissue damage and other markers would be in the basin of attraction for the fixed point corresponding to the basal state. In the in vivo data, we found both large and small, localized aggregations of inflammatory cells. This finding, as well as prior studies showing localized aggregates/granulomas in lung infections [88], supports the need to include in future models a spatial aspect such as that provided by an ABM or partial differential equation model. The findings from the current ABM lead us to hypothesize that the basins of attraction of the three stable states may be sufficiently close so that due to stochasticity certain exposure protocols lead to all three states.
Experimental studies in a mouse model of smoke inhalation support, though do not fully prove, this hypothesis. A cigarette smoke exposure study was performed in an attempt to validate aspects of our ABM against histological data obtained from an in vivo experimental paradigm of particulate exposure. The results of the animal study showed that after 5 weeks of cigarette smoke inhalation, mice exhibited aggregation of inflammatory cells adjacent to the small airways. This finding, while somewhat similar to the outcomes suggested by the localized tissue damage and fibrosis state as well as extensive damage and fibrosis state, did not match exactly with any of the end states observed in our model. Our ABM did not consider lung architecture or physiological function. Moreover, due to the short time course of exposure used in the in vivo study, the appearance of the lung tissue likely represents a state somewhere within the first 25% of the exposure period observed in the in silico simulation. However, the tri-stability of the present model demonstrates the possible outcomes that may result stochastically from further exposure of the animal to particulate inhalation. Further experiments are needed to assess the validity of this hypothesis.
While this relatively simple model was capable of reproducing a number of complex biological phenomena and a non-intuitive emergent state (localized tissue damage and fibrosis tissue with localized damage and fibrosis), there are a number of limitations associated with its use. First, the model represents a simplification of the immune response to particulate matter in the lung, and as such does not include the full complement of cells known to be involved in the pathogenesis of airway disease. Future iterations of the model might also include interactions between neutrophils, T cells, eosinophils, and epithelial cell components, which may be important when attempting to relate the present model to disease specific mechanisms of inflammation and fibrosis. In addition to cellular populations, a number of other components that play an important role in the development of airway disease have been omitted from this simplified model. The present model used the actions one pro-inflammatory and one anti-inflammatory/pro-healing cytokine to simulate actions that, in reality, involve complex relationships among multiple cytokines, chemokines, proteases, and other mediators of the inflammatory process. Future models may also include more specific interactions between proteases, anti-proteases, and components of the ECM during the tissue destruction and remodeling process in the lung. ECM fragments often act as DAMPs during inflammatory processes, and interactions among these components and their cellular receptors may represent an important variable in the modeling of inflammatory process of the lung [89,90].
The present model also does not account for the varied tissue structures present within the lung (e.g. airway, vasculature, parenchyma, etc.), and therefore cannot recreate some of the tissue-specific localization of the disease processes involved in some airway diseases. Further, this model does not contain outputs for predicting restriction of airflow. However, a number of models have been reported for the evaluation of airway dynamics and expiratory flow [91,92,93]. Incorporation of this type of model with the present model may provide a valuable tool for more in-depth studies of the interaction of tissue level inflammatory processes, tissue remodeling, and airway dynamics.
Finally, only a limited amount of histological data was used in an attempt to validate the model. The model was designed using arbitrary units in order to observe the net behavior of the system rather than specific numerical outputs calibrated to experimental data. The calibration and validation of in silico models can be a highly complex process due to the highly abstract nature of some physiologic processes [94], yet is clearly required in order for this ABM to have clinically translational utility. Indeed, ABMs are capable of predicting clinically relevant and accurate outcomes based on patient specific, individualized inputs to a system calibrated using experimental data As such, the calibration of the current model to experimentally obtained data and sensitivity analysis may increase its utility in the prediction of complex behavior during inflammation of the lung. There are a number of methods available for this type of analysis, and a full sensitivity and uncertainty analysis will would be addressed in a part of subsequent iterations of the model which may that include additional components and disease-specific details.[95]
In summary, despite these limitations, the present ABM can reproduce a number of biologically feasible phenomena and intriguing emergent states through the utilization of a simple set of rules based upon known biology. It is, in fact the very simplicity of the model and the resultant complex behaviors (that to some degree match experimental data) that highlight the power of mechanistic computational modeling for deriving major insights into biological processes. For example, in prior work from our group [84] we showed that a simple (three-variable) mathematical model of infection-induced inflammation, which contained the positive feedback loop of inflammation→damage→inflammation mentioned above, could explain diverse qualitative outcomes of sepsis patients and suggest why “one size fits all” therapies for sepsis are unlikely to work. The same basic structure, expanded greatly, underlies other mathematical models of inflammation that are calibrated with, and validated against, prospectively-gathered experimental data [96,97,98]. The findings presented herein suggest that the dynamics of the current model, though as yet not calibrated to experimental or clinical data and lacking certain physiologic aspects important to the study of inflammation of the lung, describe aspects of inflammatory activity and fibrosis following particulate exposure in a biologically plausible manner. We suggest that this model may hold future utility in the investigation of inflammatory conditions of the lung.
Supplementary Material
Acknowledgments
The authors would like to thank Yungchien Chu, Nagarjun Konduru, and Gilles Clermont for their contributions to this work as part of the “Systems Approach to Inflammation” course at the University of Pittsburgh (http://www.pitt.edu/~cler/MSCMP3780/syllabus2006.htm). The authors would also like to acknowledge NIH R33-HL-089082 (Vodovotz) for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Yoshida T, Tuder RM. Pathobiology of cigarette smoke-induced chronic obstructive pulmonary disease. Physiol Rev. 2007;87:1047–1082. doi: 10.1152/physrev.00048.2006. [DOI] [PubMed] [Google Scholar]
- 2.Sethi JM, Rochester CL. Smoking and chronic obstructive pulmonary disease. Clin Chest Med. 2000;21:67–86. viii. doi: 10.1016/s0272-5231(05)70008-3. [DOI] [PubMed] [Google Scholar]
- 3.Lopez AD Disease Control Priorities Project. Global burden of disease and risk factors. New York, NY Washington, DC: Oxford University Press; World Bank; 2006. p. xxix.p. 475. [Google Scholar]
- 4.Boschetto P, Quintavalle S, Miotto D, Lo Cascio N, Zeni E, et al. Chronic obstructive pulmonary disease (COPD) and occupational exposures. J Occup Med Toxicol. 2006;1:11. doi: 10.1186/1745-6673-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trupin L, Earnest G, San Pedro M, Balmes JR, Eisner MD, et al. The occupational burden of chronic obstructive pulmonary disease. Eur Respir J. 2003;22:462–469. doi: 10.1183/09031936.03.00094203. [DOI] [PubMed] [Google Scholar]
- 6.Oxman AD, Muir DC, Shannon HS, Stock SR, Hnizdo E, et al. Occupational dust exposure and chronic obstructive pulmonary disease. A systematic overview of the evidence. Am Rev Respir Dis. 1993;148:38–48. doi: 10.1164/ajrccm/148.1.38. [DOI] [PubMed] [Google Scholar]
- 7.Liu S, Zhou Y, Wang X, Wang D, Lu J, et al. Biomass fuels are the probable risk factor for chronic obstructive pulmonary disease in rural South China. Thorax. 2007;62:889–897. doi: 10.1136/thx.2006.061457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu Y, Lee K, Perez-Padilla R, Hudson NL, Mannino DM. Outdoor and indoor air pollution and COPD-related diseases in high- and low-income countries. Int J Tuberc Lung Dis. 2008;12:115–127. [PubMed] [Google Scholar]
- 9.Medina-Ramon M, Zanobetti A, Schwartz J. The effect of ozone and PM10 on hospital admissions for pneumonia and chronic obstructive pulmonary disease: a national multicity study. Am J Epidemiol. 2006;163:579–588. doi: 10.1093/aje/kwj078. [DOI] [PubMed] [Google Scholar]
- 10.Schikowski T, Sugiri D, Ranft U, Gehring U, Heinrich J, et al. Long-term air pollution exposure and living close to busy roads are associated with COPD in women. Respir Res. 2005;6:152. doi: 10.1186/1465-9921-6-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maier KL, Alessandrini F, Beck-Speier I, Hofer TP, Diabate S, et al. Health effects of ambient particulate matter--biological mechanisms and inflammatory responses to in vitro and in vivo particle exposures. Inhal Toxicol. 2008;20:319–337. doi: 10.1080/08958370701866313. [DOI] [PubMed] [Google Scholar]
- 12.Green FH, Vallyathan V, Hahn FF. Comparative pathology of environmental lung disease: an overview. Toxicol Pathol. 2007;35:136–147. doi: 10.1080/01926230601132055. [DOI] [PubMed] [Google Scholar]
- 13.Fraig M, Shreesha U, Savici D, Katzenstein AL. Respiratory bronchiolitis: a clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am J Surg Pathol. 2002;26:647–653. doi: 10.1097/00000478-200205000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, et al. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med. 1998;158:1277–1285. doi: 10.1164/ajrccm.158.4.9802078. [DOI] [PubMed] [Google Scholar]
- 15.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 16.Larsson K. Aspects on pathophysiological mechanisms in COPD. J Intern Med. 2007;262:311–340. doi: 10.1111/j.1365-2796.2007.01837.x. [DOI] [PubMed] [Google Scholar]
- 17.Mannino DM, Davis KJ. Lung function decline and outcomes in an elderly population. Thorax. 2006;61:472–477. doi: 10.1136/thx.2005.052449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundback B, Lindberg A, Lindstrom M, Ronmark E, Jonsson AC, et al. Not 15 but 50% of smokers develop COPD?--Report from the Obstructive Lung Disease in Northern Sweden Studies. Respir Med. 2003;97:115–122. doi: 10.1053/rmed.2003.1446. [DOI] [PubMed] [Google Scholar]
- 19.Kent L, Smyth L, Clayton C, Scott L, Cook T, et al. Cigarette smoke extract induced cytokine and chemokine gene expression changes in COPD macrophages. Cytokine. 2008 doi: 10.1016/j.cyto.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 20.Siafakas NM, Tzortzaki EG. Few smokers develop COPD. Why? Respir Med. 2002;96:615–624. doi: 10.1053/rmed.2002.1318. [DOI] [PubMed] [Google Scholar]
- 21.Kim KM, Park SH, Kim JS, Lee WK, Cha SI, et al. Polymorphisms in the Type IV Collagen Alpha3 Gene and the Risk of COPD. Eur Respir J. 2008 doi: 10.1183/09031936.00076207. [DOI] [PubMed] [Google Scholar]
- 22.Celedon JC, Lange C, Raby BA, Litonjua AA, Palmer LJ, et al. The transforming growth factor-beta1 (TGFB1) gene is associated with chronic obstructive pulmonary disease (COPD) Hum Mol Genet. 2004;13:1649–1656. doi: 10.1093/hmg/ddh171. [DOI] [PubMed] [Google Scholar]
- 23.Keatings VM, Cave SJ, Henry MJ, Morgan K, O’Connor CM, et al. A polymorphism in the tumor necrosis factor-alpha gene promoter region may predispose to a poor prognosis in COPD. Chest. 2000;118:971–975. doi: 10.1378/chest.118.4.971. [DOI] [PubMed] [Google Scholar]
- 24.Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet. 2005;365:2225–2236. doi: 10.1016/S0140-6736(05)66781-5. [DOI] [PubMed] [Google Scholar]
- 25.Barnes PJ. Immunology of asthma and chronic obstructive pulmonary disease. Nat Rev Immunol. 2008;8:183–192. doi: 10.1038/nri2254. [DOI] [PubMed] [Google Scholar]
- 26.Barnes PJ. Alveolar macrophages as orchestrators of COPD. Copd. 2004;1:59–70. doi: 10.1081/COPD-120028701. [DOI] [PubMed] [Google Scholar]
- 27.Alcorn JF, Crowe CR, Kolls JK. TH17 cells in asthma and COPD. Annu Rev Physiol. 72:495–516. doi: 10.1146/annurev-physiol-021909-135926. [DOI] [PubMed] [Google Scholar]
- 28.Cornwell WD, Kim V, Song C, Rogers TJ. Pathogenesis of inflammation and repair in advanced COPD. Semin Respir Crit Care Med. 31:257–266. doi: 10.1055/s-0030-1254066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lane N, Robins RA, Corne J, Fairclough L. Regulation in chronic obstructive pulmonary disease: the role of regulatory T-cells and Th17 cells. Clin Sci (Lond) 119:75–86. doi: 10.1042/CS20100033. [DOI] [PubMed]
- 30.Prasse A, Pechkovsky DV, Toews GB, Jungraithmayr W, Kollert F, et al. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am J Respir Crit Care Med. 2006;173:781–792. doi: 10.1164/rccm.200509-1518OC. [DOI] [PubMed] [Google Scholar]
- 31.Harari S, Caminati A. IPF: new insight on pathogenesis and treatment. Allergy. 65:537–553. doi: 10.1111/j.1398-9995.2009.02305.x. [DOI] [PubMed] [Google Scholar]
- 32.Selman M, Pardo A. The epithelial/fibroblastic pathway in the pathogenesis of idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2003;29:S93–97. [PubMed] [Google Scholar]
- 33.Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc. 2006;3:364–372. doi: 10.1513/pats.200601-003TK. [DOI] [PubMed] [Google Scholar]
- 34.Ichinose M. Differences of inflammatory mechanisms in asthma and COPD. Allergol Int. 2009;58:307–313. doi: 10.2332/allergolint.09-RAI-0106. [DOI] [PubMed] [Google Scholar]
- 35.Wright JL, Churg A. Animal models of COPD: Barriers, successes, and challenges. Pulm Pharmacol Ther. 2008 doi: 10.1016/j.pupt.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 36.Martorana PA, Cavarra E, Lucatteli M, Lungarella G. Models for COPD involving cigarette smoke. Drug Discovery Today: Disease Models. 2006;3:225–230. [Google Scholar]
- 37.Fujita M, Nakanishi Y. The pathogenesis of COPD: lessons learned from in vivo animal models. Med Sci Monit. 2007;13:RA19–24. [PubMed] [Google Scholar]
- 38.Vodovotz Y, Clermont G, Chow C, An G. Mathematical models of the acute inflammatory response. Curr Opin Crit Care. 2004;10:383–390. doi: 10.1097/01.ccx.0000139360.30327.69. [DOI] [PubMed] [Google Scholar]
- 39.Vodovotz Y, Csete M, Bartels J, Chang S, An G. Translational systems biology of inflammation. PLoS Comput Biol. 2008;4:e1000014. doi: 10.1371/journal.pcbi.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.An G, Faeder J, Vodovotz Y. Translational systems biology: introduction of an engineering approach to the pathophysiology of the burn patient. J Burn Care Res. 2008;29:277–285. doi: 10.1097/BCR.0b013e31816677c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vodovotz Y, Constantine G, Rubin J, Csete M, Voit EO, et al. Mechanistic simulations of inflammation: current state and future prospects. Math Biosci. 2009;217:1–10. doi: 10.1016/j.mbs.2008.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.An G, Mi Q, Dutta-Moscato J, Solovyev A, Mikheev M, et al. Agent-based models in translational systems biology. Wiley Interdisciplinary Reviews - Systems Biology. 2009 doi: 10.1002/wsbm.45. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li NY, Verdolini K, Clermont G, Mi Q, Rubinstein EN, et al. A patient-specific in silico model of inflammation and healing tested in acute vocal fold injury. PLoS One. 2008;3:e2789. doi: 10.1371/journal.pone.0002789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mi Q, Riviere B, Clermont G, Steed DL, Vodovotz Y. Agent-based model of inflammation and wound healing: insights into diabetic foot ulcer pathology and the role of transforming growth factor-beta1. Wound Repair Regen. 2007;15:671–682. doi: 10.1111/j.1524-475X.2007.00271.x. [DOI] [PubMed] [Google Scholar]
- 45.Xiong Z, Leme AS, Ray P, Shapiro SD, Lee JS. CX3CR1+ Lung Mononuclear Phagocytes Spatially Confined to the Interstitium Produce TNF-{alpha} and IL-6 and Promote Cigarette Smoke-Induced Emphysema. J Immunol. 2011;186:3206–3214. doi: 10.4049/jimmunol.1003221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- 47.Fletcher C, Peto R, Tinker R, Speizer FE. The Natural History of Chronic Bronchitis and Emphysema. Oxford: Oxford University Press; 1976. [Google Scholar]
- 48.Wise RA, Kanner RE, Lindgren P, Connett JE, Altose MD, et al. The effect of smoking intervention and an inhaled bronchodilator on airways reactivity in COPD: the Lung Health Study. Chest. 2003;124:449–458. doi: 10.1378/chest.124.2.449. [DOI] [PubMed] [Google Scholar]
- 49.Hodge S, Hodge G, Holmes M, Reynolds PN. Increased airway epithelial and T-cell apoptosis in COPD remains despite smoking cessation. Eur Respir J. 2005;25:447–454. doi: 10.1183/09031936.05.00077604. [DOI] [PubMed] [Google Scholar]
- 50.Alexis NE, Lay JC, Zeman K, Bennett WE, Peden DB, et al. Biological material on inhaled coarse fraction particulate matter activates airway phagocytes in vivo in healthy volunteers. J Allergy Clin Immunol. 2006;117:1396–1403. doi: 10.1016/j.jaci.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 51.Imrich A, Ning Y, Lawrence J, Coull B, Gitin E, et al. Alveolar macrophage cytokine response to air pollution particles: oxidant mechanisms. Toxicol Appl Pharmacol. 2007;218:256–264. doi: 10.1016/j.taap.2006.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 53.Cavaillon JM. Cytokines and macrophages. Biomed Pharmacother. 1994;48:445–453. doi: 10.1016/0753-3322(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 54.Nathan C. Points of control in inflammation. Nature. 2002;420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 55.Lotze MT, Zeh HJ, Rubartelli A, Sparvero LJ, Amoscato AA, et al. The grateful dead: damage-associated molecular pattern molecules and reduction/oxidation regulate immunity. Immunol Rev. 2007;220:60–81. doi: 10.1111/j.1600-065X.2007.00579.x. [DOI] [PubMed] [Google Scholar]
- 56.Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 57.Chung KF, Adcock IM. Multifaceted mechanisms in COPD: inflammation, immunity, and tissue repair and destruction. Eur Respir J. 2008;31:1334–1356. doi: 10.1183/09031936.00018908. [DOI] [PubMed] [Google Scholar]
- 58.Djekic UV, Gaggar A, Weathington NM. Attacking the multi-tiered proteolytic pathology of COPD: New insights from basic and translational studies. Pharmacol Ther. 2008 doi: 10.1016/j.pharmthera.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lim S, Roche N, Oliver BG, Mattos W, Barnes PJ, et al. Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10. Am J Respir Crit Care Med. 2000;162:1355–1360. doi: 10.1164/ajrccm.162.4.9910097. [DOI] [PubMed] [Google Scholar]
- 60.Russell RE, Culpitt SV, DeMatos C, Donnelly L, Smith M, et al. Release and activity of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by alveolar macrophages from patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2002;26:602–609. doi: 10.1165/ajrcmb.26.5.4685. [DOI] [PubMed] [Google Scholar]
- 61.Russell RE, Thorley A, Culpitt SV, Dodd S, Donnelly LE, et al. Alveolar macrophage-mediated elastolysis: roles of matrix metalloproteinases, cysteine, and serine proteases. Am J Physiol Lung Cell Mol Physiol. 2002;283:L867–873. doi: 10.1152/ajplung.00020.2002. [DOI] [PubMed] [Google Scholar]
- 62.Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, et al. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest. 2000;117:684–694. doi: 10.1378/chest.117.3.684. [DOI] [PubMed] [Google Scholar]
- 63.Porcheray F, Viaud S, Rimaniol AC, Leone C, Samah B, et al. Macrophage activation switching: an asset for the resolution of inflammation. Clin Exp Immunol. 2005;142:481–489. doi: 10.1111/j.1365-2249.2005.02934.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cockbill S. Wounds: the healing process. Hosp Pharmacist. 2002;9:225–260. [Google Scholar]
- 65.Witte MB, Barbul A. General principles of wound healing. Surg Clin North Am. 1997;77:509–528. doi: 10.1016/s0039-6109(05)70566-1. [DOI] [PubMed] [Google Scholar]
- 66.Roberts AB, Sporn MB. Transforming growth factor-β. In: Clark RAF, editor. The Molecular and Cellular Biology of Wound Repair. New York: Plenum Press; 1996. pp. 275–308. [Google Scholar]
- 67.Roberts AB, Sporn MB. The transforming growth factor-βs. In: Sporn MB, Roberts AB, editors. Peptide Growth Factors and their Receptors. Berlin: Springer-Verlag; 1990. pp. 419–472. [Google Scholar]
- 68.Matthews N, Watkins JF. Tumour-necrosis factor from the rabbit. I. Mode of action, specificity and physicochemical properties. Br J Cancer. 1978;38:302–309. doi: 10.1038/bjc.1978.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bowler PG. Wound pathophysiology, infection and therapeutic options. Ann Med. 2002;34:419–427. doi: 10.1080/078538902321012360. [DOI] [PubMed] [Google Scholar]
- 70.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–2004. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 71.Martin TR, Frevert CW. Innate immunity in the lungs. Proc Am Thorac Soc. 2005;2:403–411. doi: 10.1513/pats.200508-090JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aldonyte R, Jansson L, Piitulainen E, Janciauskiene S. Circulating monocytes from healthy individuals and COPD patients. Respir Res. 2003;4:11. doi: 10.1186/1465-9921-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 74.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 75.Pons AR, Sauleda J, Noguera A, Pons J, Barcelo B, et al. Decreased macrophage release of TGF-beta and TIMP-1 in chronic obstructive pulmonary disease. Eur Respir J. 2005;26:60–66. doi: 10.1183/09031936.05.00045504. [DOI] [PubMed] [Google Scholar]
- 76.Takanashi S, Hasegawa Y, Kanehira Y, Yamamoto K, Fujimoto K, et al. Interleukin-10 level in sputum is reduced in bronchial asthma, COPD and in smokers. Eur Respir J. 1999;14:309–314. doi: 10.1034/j.1399-3003.1999.14b12.x. [DOI] [PubMed] [Google Scholar]
- 77.Camoretti-Mercado B, Solway J. Transforming growth factor-beta1 and disorders of the lung. Cell Biochem Biophys. 2005;43:131–148. doi: 10.1385/CBB:43:1:131. [DOI] [PubMed] [Google Scholar]
- 78.Koli K, Myllarniemi M, Keski-Oja J, Kinnula VL. Transforming growth factor-beta activation in the lung: focus on fibrosis and reactive oxygen species. Antioxid Redox Signal. 2008;10:333–342. doi: 10.1089/ars.2007.1914. [DOI] [PubMed] [Google Scholar]
- 79.Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, et al. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD) Am J Respir Crit Care Med. 2001;163:1476–1483. doi: 10.1164/ajrccm.163.6.9908135. [DOI] [PubMed] [Google Scholar]
- 80.Hodge SJ, Hodge GL, Reynolds PN, Scicchitano R, Holmes M. Increased production of TGF-beta and apoptosis of T lymphocytes isolated from peripheral blood in COPD. Am J Physiol Lung Cell Mol Physiol. 2003;285:L492–499. doi: 10.1152/ajplung.00428.2002. [DOI] [PubMed] [Google Scholar]
- 81.Beghe B, Bazzan E, Baraldo S, Calabrese F, Rea F, et al. Transforming growth factor-beta type II receptor in pulmonary arteries of patients with very severe COPD. Eur Respir J. 2006;28:556–562. doi: 10.1183/09031936.06.00077105. [DOI] [PubMed] [Google Scholar]
- 82.de Boer WI, van Schadewijk A, Sont JK, Sharma HS, Stolk J, et al. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158:1951–1957. doi: 10.1164/ajrccm.158.6.9803053. [DOI] [PubMed] [Google Scholar]
- 83.Anthony Tony Cox L., Jr A Mathematical Model of Protease-Antiprotease Homeostasis Failure in Chronic Obstructive Pulmonary Disease (COPD) Risk Anal. 2008 doi: 10.1111/j.1539-6924.2008.01152.x. [DOI] [PubMed] [Google Scholar]
- 84.Kumar R, Clermont G, Vodovotz Y, Chow CC. The dynamics of acute inflammation. J Theor Biol. 2004;230:145–155. doi: 10.1016/j.jtbi.2004.04.044. [DOI] [PubMed] [Google Scholar]
- 85.Kumar R, Chow CC, Bartels JD, Clermont G, Vodovotz Y. A mathematical simulation of the inflammatory response to anthrax infection. Shock. 2008;29:104–111. doi: 10.1097/SHK.0b013e318067da56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walgraef D. Spatio-temporal pattern formation : with examples from physics, chemistry, and materials science. New York: Springer; 1997. p. x.p. 306. [Google Scholar]
- 87.Murray JD. Mathematical biology. New York: Springer; 2002. [Google Scholar]
- 88.Segovia-Juarez JL, Ganguli S, Kirschner D. Identifying control mechanisms of granuloma formation during M. tuberculosis infection using an agent-based model. J Theor Biol. 2004;231:357–376. doi: 10.1016/j.jtbi.2004.06.031. [DOI] [PubMed] [Google Scholar]
- 89.Taylor KR, Trowbridge JM, Rudisill JA, Termeer CC, Simon JC, et al. Hyaluronan fragments stimulate endothelial recognition of injury through TLR4. J Biol Chem. 2004;279:17079–17084. doi: 10.1074/jbc.M310859200. [DOI] [PubMed] [Google Scholar]
- 90.Sarir H, Henricks PA, van Houwelingen AH, Nijkamp FP, Folkerts G. Cells, mediators and Toll-like receptors in COPD. Eur J Pharmacol. 2008;585:346–353. doi: 10.1016/j.ejphar.2008.03.009. [DOI] [PubMed] [Google Scholar]
- 91.Barbini P, Brighenti C, Cevenini G, Gnudi G. A dynamic morphometric model of the normal lung for studying expiratory flow limitation in mechanical ventilation. Ann Biomed Eng. 2005;33:518–530. doi: 10.1007/s10439-005-2511-6. [DOI] [PubMed] [Google Scholar]
- 92.Barbini P, Cevenini G, Avanzolni G. Nonlinear mechanisms determining expiratory flow limitation in mechanical ventilation: a model-based interpretation. Ann Biomed Eng. 2003;31:908–916. doi: 10.1114/1.1590665. [DOI] [PubMed] [Google Scholar]
- 93.Yang XL, Liu Y, Luo HY. Respiratory flow in obstructed airways. J Biomech. 2006;39:2743–2751. doi: 10.1016/j.jbiomech.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 94.Vodovotz Y. Deciphering the complexity of acute inflammation using mathematical models. Immunol Res. 2006;36:237–245. doi: 10.1385/IR:36:1:237. [DOI] [PubMed] [Google Scholar]
- 95.Marino S, Hogue IB, Ray CJ, Kirschner DE. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol. 2008;254:178–196. doi: 10.1016/j.jtbi.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chow CC, Clermont G, Kumar R, Lagoa C, Tawadrous Z, et al. The acute inflammatory response in diverse shock states. Shock. 2005;24:74–84. doi: 10.1097/01.shk.0000168526.97716.f3. [DOI] [PubMed] [Google Scholar]
- 97.Prince JM, Levy RM, Bartels J, Baratt A, Kane JM, 3rd, et al. In silico and in vivo approach to elucidate the inflammatory complexity of CD14-deficient mice. Mol Med. 2006;12:88–96. doi: 10.2119/2006-00012.Prince. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lagoa CE, Bartels J, Baratt A, Tseng G, Clermont G, et al. The role of initial trauma in the host’s response to injury and hemorrhage: insights from a correlation of mathematical simulations and hepatic transcriptomic analysis. Shock. 2006;26:592–600. doi: 10.1097/01.shk.0000232272.03602.0a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.