

Abstract
Alkyltransferase-like proteins (ATLs) play a role in the protection of cells from the biological effects of DNA alkylation damage. Although ATLs share functional motifs with the DNA repair protein and cancer chemotherapy target O 6-alkylguanine-DNA alkyltransferase, they lack the reactive cysteine residue required for alkyltransferase activity, so its mechanism for cell protection was previously unknown. Here we review recent advances in unraveling the enigmatic cellular protection provided by ATLs against the deleterious effects of DNA alkylation damage. We discuss exciting new evidence that ATLs aid in the repair of DNA O 6-alkylguanine lesions through a novel repair cross-talk between DNA-alkylation base damage responses and the DNA nucleotide excision repair pathway.
Keywords: Alkyltransferase-like protein, ATL, O6-alkylguanine-DNA alkyltransferase, O6-methylguanine-DNA methyltransferase, Nucleotide excision repair, DNA base repair, DNA alkylation, DNA repair
Introduction
Some of the most prevalent cytotoxic and mutagenic DNA lesions arise from alkylation of DNA bases. Consequently, the continual repair of DNA alkylation damage is vital for maintaining genome stability. However, this repair can also provide a major resistance factor for alkylating chemotherapies. Therefore an understanding of the processes that are involved in this repair is crucial.
Alkylating agents that give rise to DNA alkyl lesions can be derived from endogenous sources (such as S-adenosylmethionine) [1], environmental toxins [2], or anticancer chemotherapies [3]. DNA bases are susceptible to alkylation to varying degrees at all of the exocyclic oxygens and most of the ring nitrogens [4]. Alkylating agents generate similar types of damage, but in different proportions depending on their mode of reaction. Most alkylating agents produce the innocuous 7-methylguanine and the highly cytotoxic 3-methyladenine lesions. In addition, bimolecular nucleophilic substitution (SN2) alkylating agents, such as methyl methanesulfonate (MMS) and methyl halides, produce large quantities of such lesions as 3-methyladenine and 3-methylcytosine, which block DNA replication and result in cytotoxicity [4]. The highly mutagenic and cytotoxic O 6-methylguanine (O 6-mG) (Fig. 1) and O 4-methylthymine (O 4-mT) lesions mainly result from unimolecular nucleophilic substitution (SN1) alkylating agents, such as N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) and N-methyl-N-nitrosourea (MNU) [4]. Although O 6-mG and O 4-mT lesions are not usually generated in abundance, they are highly cytotoxic and mutagenic and their repair is critical [4]. O 6-mG lesions are mutagenic because they mispair during replication with thymine, resulting in G:C to A:T transition mutations [5–8]. The high cytotoxicity of these lesions results from the recognition of O 6-mG:T mispairs by the DNA mismatch repair (MMR) pathway, which attempts to repair them by removing the thymine, leading to a futile cycle of nucleotide removal and synthesis that generates DNA single- and double-strand breaks and eventually results in apoptosis (Fig. 2). A thorough understanding of how cells deal with the mutagenic and cytotoxic effects of DNA alkylation damage is thus critical to the development and improvement of cancer prevention strategies.
Fig. 1.

Chemical structures of the lesions recognized by ATL that are discussed in this review. The lesions are abbreviated as follows: O 6-mG, O 6-methylguanine; O 6-bG, O 6-benzylguanine; O 6-pobG, O 6-4-(3-pyridyl)-4-oxobutylguanine; O 6-btG, O 6-(4-bromothenyl)guanine; O 6-HOEtG, O 6-hydroxyethylguanine, O 6-1hpG, O 6-1-hydroxypropylguanine; O 6-2hpG, O 6-2-hydroxypropylguanine
Fig. 2.

Proposed mechanisms for repair of O 6-alkylG (indicated by a red star)
In cells, DNA alkylation damage is mostly repaired through one of two strategies. Most alkyl lesions are removed by lesion-specific DNA glycosylases that excise modified bases to create abasic sites and initiate the base excision repair (BER) pathway [4, 9, 10]. Some lesions are repaired through a direct damage reversal by alkyltransferase proteins (AGTs) [6, 11] or Fe(II)/2-oxoglutarate-dependent dioxygenases of the AlkB family [10, 12]. Recently, catalytically inactive alkyltransferase-like (ATL) proteins were discovered to promote a novel repair mechanism of DNA alkylation damage through cross-talk between DNA-alkylation base damage responses and the DNA nucleotide excision repair (NER) pathway, thereby linking two classically distinct DNA repair pathways. Here we review recent work on deciphering the role of alkyltransferase-like proteins in cellular protection.
Cellular protection against O6-alkylguanine lesions
DNA O 6-alkylguanine (O 6-alkylG) lesions are mostly repaired through the action of alkyltransferase proteins, such as Ada and Ogt in Escherichia coli, and O 6-alkylguanine-DNA alkyltransferase (AGT) (also known as O 6-methylguanine-DNA methyltransferase, MGMT, in humans), which are widespread amongst prokaryotes and eukaryotes [5]. The recent availability of the genome sequences for the moss Physcomitrella patens [13], the green algae Chlamydomonas reinhardtii [14], and the red algae Cyanidioschyzon merolae [15] has allowed us to identify here the first putative plant AGTs (GenBank accession XM_001778913, XM_001778913, and AP006502, respectively) (Fig. 3) (J.L.T. and J.A.T., unpublished observation). AGTs are two-domain α/β fold proteins. The catalytic C-terminal domain possesses an active site -PCHR- motif, which contains the absolutely conserved nucleophilic cysteine residue (Cys145 in humans) [16, 17], along with the nucleotide rotating residues (Arg128 and Tyr114 in humans) [5, 6, 11]. AGT selectively transfers the damaged guanine O 6-alkyl adduct to the nucleophilic cysteine in a stoichiometric, irreversible, damage reversal reaction [5]. AGT specifically repairs O 6-mG lesions, and to a lesser extent O 4-mT. It also repairs, though some less efficiently, larger O 6-alkylG lesions, including O 6-benzylguanine (O 6-bG) [18], O 6-(4-bromothenyl)guanine (O 6-btG) [19], O 6-4-(3-pyridyl)-4-oxobutylguanine (O 6-pobG) [20], and O 6-guanine-alkyl-O 6-guanine interstrand crosslinks [21]. Alteration of residues in and near the AGT active site can have profound effects on the rate and/or ability of AGT to repair various types of damage [22–25]. AGT-mediated repair of O 6-alkylG DNA lesions prevents mutations, but also promotes tumor resistance to therapeutic alkylating agents that are commonly used in cancer treatments [6, 26], making it a prime drug target for alkylation chemotherapies. Structures of human AGT (hAGT) [27, 28] and hAGT in complex with small-molecule [27] or DNA [29, 30] substrates have aided in understanding the mechanism of hAGT-mediated resistance to anticancer therapies [6].
Fig. 3.
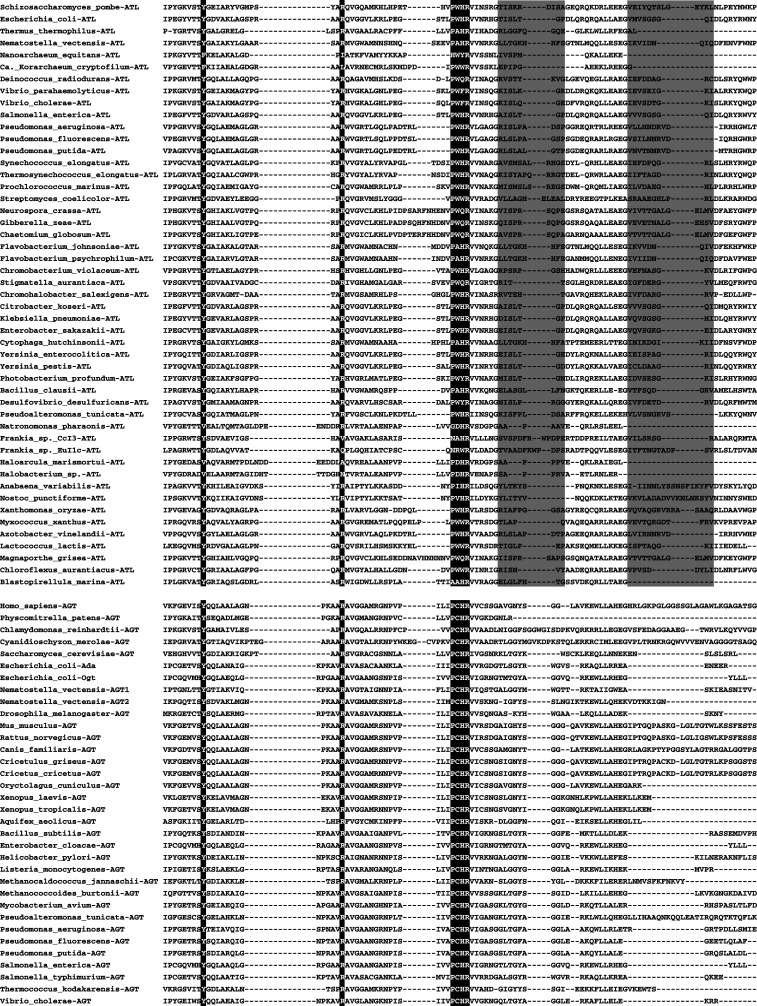
Sequence alignment of select ATLs (top) and AGTs (bottom), showing conservation of key tyrosine, arginine, and binding/active site residues, highlighted in black. The S. pombe Atl1-binding site and C-terminal loop is shaded gray
Alkyltransferase-like proteins are a newly identified family of AGT homologs with sequence similarity to the AGT catalytic domain, with the notable exception that the AGT Cys alkyl acceptor is replaced by tryptophan, alanine, or another residue (Figs. 3, 4) [31, 32]. ATLs co-exist with AGTs in some organisms, and are absent in others [32]. No recognizable ATL has been identified in higher eukaryotes or plants [32]. Most known ATLs are from bacteria, and the three best-studied bacterial ATLs are from E. coli [33–35], Thermus thermophilus [36], and Vibrio parahaemolyticus [37] (Table 1). ATL proteins are also found in some fungi, with the one most characterized from the fission yeast Schizosaccharomyces pombe [38, 39] (Table 1). Interestingly, S. pombe lacks an AGT and only has an ATL, whereas the budding yeast Saccharomyces cerevisiae has an AGT but no ATL [32, 38]. Recently, new ATLs were identified in the Archaea Candidatus Korarchaeum cryptofilum and Nanoarchaeum equitans, ancestral to the two established phyla of Archaea [39]. Also, the first ATL in any multicellular organism, the starlet sea anemone Nematostella vectensis (Table 1), was discovered [39]. Therefore, ATLs exist in all three domains of life.
Fig. 4.

Crystallographic structure of S. pombe Atl1, showing the overall fold. Key residues and loops are mapped to the web logo sequence alignment shown below
Table 1.
Summary for partially and well-characterized ATLs
| Organism | Δatl alkylating agent sensitivity | AGT | Binding site motif | Known recognized lesions | DNA-binding affinity (nM) | Known epistasis with NER proteins | GenBank accession | PDB accession |
|---|---|---|---|---|---|---|---|---|
| E. coli | PNU | Ada, Ogt | PWHR | O6-mG, O6-1hpG, O6-2hpG, O6-HOEtG, AP site | AP site: 3.5 × 10−3 | UvrA, UvrC | AAC73557 | None |
| N. vectensis | Not known | AGT | PAHR | O6-mG | Not known | Not known | XM_001618690 | None |
| S. pombe | SN1 | None | PWHR | O6-mG, O6-bG, O6-btG, O6-HOEtG, O6-pobG |
O6-mG: 0.35 ± 0.04 O6-pobG: 1.6 × 10−2 ± 0.004 AP site: low and/or transient |
Rad13, Swi10 | CAB54827 |
Apo: 3GVA O6-mG-DNA: 3GX4 O6-pobG-DNA: 3GYH |
| T. thermophilus | Methylating | None | PAHR | O6-mG |
Normal: 3.7 × 103 O6-mG: 4.1 × 102 |
Not known | AP008226.1 | None |
| V. parahaemolyticus | Not known | AGT | PWFR | O6-mG | Not known | Not known | AAWQ01000051 | Apo: 2KIF |
ATLs tightly bind single- and double-stranded DNA, but are not alkyltransferases, as no alkyltransferase activity could be detected in vitro for ATLs from S. pombe (Atl1) [38], E. coli (eAtl; also referred elsewhere as the YbaZ protein) [33, 34], or T. thermophilus (TTHA1564) [36]. In addition, eAtl displayed no demethylase, glycosylase, or endonuclease activity [33]. Thus, ATLs do not cleave the alkyl group, base, or oligonucleotide near the lesion. Mutation of Trp to Cys in eAtl was insufficient to restore alkyltransferase activity, indicating other mutations are necessary to restore this activity [33]. The repair of O 6-methylguanine (O 6-mG) by human AGT (hAGT) is inhibited by pre-incubation of alkylated oligonucleotides with Atl1, eAtl, N. vectensis ATL (NvAtl), or Vibrio parahaemolyticus ATL (vpAtl) [33, 37–39], but this inhibition is reversible over time [32, 33].
Overexpression of eAtl in E. coli increased sensitivity of wild-type strains to MNNG, whereas it had no effect on AGT-deficient strains [33]. However, the induced mutation frequency for cells treated with the propylating agent N-propyl-N-nitrosourea (PNU) was approximately two-fold higher for an eAtl deletant E. coli strain as compared to wild-type E. coli [35], suggesting that eAtl provides protection against larger alkylation adducts. Overexpression of S. pombe Atl1 in E. coli cells protected these cells against MNNG-induced alkylation damage, demonstrating that ATLs can act across species [39]. Significantly, inactivation of ATL genes in the AGT-deficient organisms S. pombe and T. thermophilus reduces alkylation damage resistance in these organisms [36, 38]. In S. pombe, atl1 deletants exhibit a marked increase in sensitivity to SN1 versus SN2 alkylating agents [38]. Similarly, in T. thermophilus, Δttha1564 strains showed a five to eight-fold increase in spontaneous mutation frequency measured by His+ G:C to A:T reversions compared to wild-type strains in the presence of methylating agents, suggesting that TTHA1564 aids in the prevention of G:C-to-A:T transition mutations [36]. Also, the spontaneous mutation rate in T. thermophilus Δtttha1564 strains was significantly higher than in WT strains and comparable to that of ΔmutS strains, implicating TTHA1564 in DNA repair in vivo. Together, these results suggested that ATLs protect cells against the deleterious biological effects of DNA alkylation damage.
DNA binding by ATLs
Measurements of ATL-DNA binding have established that ATLs tightly and specifically bind O 6-alkylG. Atl1 binds ssDNA containing O 6-mG, O 6-bG, O 6-btG, or O 6-hydroxyethylG (O 6-HOEtG) [38], and dsDNA containing O 6-mG or O 6-pobG [39], but not dsDNA containing an abasic site (see Fig. 1 for chemical structures) [39]. Surface plasmon resonance experiments measuring Atl1 binding to dsDNA containing O 6-mG or O 6-pobG indicated a dissociation constant (KD) of 0.35 ± 0.04 nM for O 6-mG and 0.016 ± 0.004 nM for O 6-pobG, revealing Atl1 stably binds O 6-mG and larger O 6-alkyl groups [39]. In contrast to Atl1, experiments probing an E. coli proteome chip with dsDNA containing mismatch or abasic site lesions revealed eAtl binds dsDNA containing an abasic site opposite a C, G, A, or T, but not G:T or A:C mismatches or normal, unmethylated DNA [34]. A K D of 3.5 × 10−12 M was determined for eAtl bound to abasic site-containing dsDNA opposite adenine [34]. eAtl also binds short ss- or dsDNA containing O 6-mG with C, T, G, or A opposite an O 6-mG lesion, but not O 4-mT [33]. Neither eAtl nor Atl1 bind DNA containing 8-oxoguanine, 5-hydroxymethylcytosine or ethenoadenine [33, 38], indicating that ATL binding is specific for guanine O 6-alkyl lesions. Gel-shift assays estimated a K D of 3.7 and 0.41 μM for TTHA1564 binding to normal unmethylated or O 6-mG-containing DNA, respectively, revealing that TTHA1564 binds O 6-mG containing DNA with nine-fold higher affinity than normal, unmethylated DNA [36].
Sedimentation equilibrium analysis of Atl1-dsDNA complexes revealed that in solution dominant complexes for 13-mer oligonucleotides containing O 6-mG have a 1:1 stoichiometry, whereas 16-mers and 26-mers form 2:1 and 3:1 limiting complexes, respectively [39]. Saturated Atl1-nonmethylated DNA complexes were formed in one step without the accumulation of intermediates, suggesting Atl1 cooperatively binds nonmethylated DNA. The build-up of cooperative assembly is preceded by specific binding to O 6-mG sites, as indicated by the formation of 1:1 complexes between Atl1 and O 6-mG-containing DNAs before proceeding to saturation in an additional concerted step. The binding site size for Atl1 was estimated to be roughly 8 bp, which is twice the 4 bp/protein-binding site size for AGT [40]. Since the two proteins have similar structures but ATL lacks the N-terminal domain present in AGT, these results suggest that the differences may be due to structures located in AGT’s N-terminal domain and the open-to-closed switch (not seen in AGT) that exposes the C-terminal loop (the ATL structure is discussed in the next section). Therefore, ATLs tightly and specifically bind O 6-alkylGs.
ATL structure and DNA bending
VpAtl solution structures [37] and Atl1 crystal structures without and with damaged DNA containing O 6-mG or O 6-pobG [39] have aided the understanding of ATL–DNA damage interactions. Both VpAtl and Atl1 conserve the fold of the hAGT catalytic domain where DNA binding and alkyl transfer occur (Figs. 3, 4 and 5a) [39]. In addition, identities and roles of key AGT DNA-binding and nucleotide rotating residues are conserved in ATL [37]. Notably lacking in ATLs are the AGT N-terminal domain, active site Cys (Trp in Atl1), and Asn hinge that couples helix-turn-helix (HTH) DNA binding and active site motifs (Figs. 3, 4). The hAGT-binding site loop, proposed to confer substrate specificity, is also present in ATL, although in a different conformation (Fig. 5a). ATL has a C-terminal loop, not seen in AGT. This loop in Atl1 makes additional contacts with DNA (Fig. 5a) [39]. Both the C-terminal and binding site loops in VpAtl are shorter than those for Atl1 [37]. Sequence conservation of other ATL sequences mapped onto Atl1 and VpAtl structures indicate the most conserved residues line the lesion-binding pocket or act in DNA binding, and suggest Atl1 and VpAtl structures are typical of other ATLs [37, 39].
Fig. 5.

Lesion binding by ATL and AGT. a Structural overlay of S. pombe Atl1- (gray, pdb 3gx4) and human AGT- (black, pdb 1t38) O 6-mG-DNA complexes. Key residues, loops, and O 6-mG from the ATL-DNA complex structure are indicated by arrows. b Stereo view of Atl1-O 6-mG DNA complex, with the binding pocket overlayed as a molecular surface. c Stereo view of hAGT-O 6-mG DNA complex, with the binding pocket overlayed as a molecular surface
Atl1 binds the DNA minor groove, like hAGT, via an HTH motif (Fig. 5a) [29, 39], with damaged-strand contacts to the phosphate groups of the O 6-alkylG and two 3′-adjacent nucleotides [39]. Residues from the DNA-binding site loop (Ser67 and Lys70) and C-terminal loop make DNA contacts not found in AGT. The buried surface area for Atl1 is ~1,050 Å2, in comparison to 788 Å2 for hAGT, consistent with Atl1 DNA-binding experiments and supporting the concept of tighter DNA binding.
The Atl1-binding site loop switches conformations between ‘open’ DNA-free and ‘closed’ DNA-bound Atl1 (Fig. 5a) [39]. This loop is flanked by glycines, which suggest flexibility, as was observed for AGT mutants with improved activity toward O 4-mT [25]. The Atl1-binding site loop acts in conjunction with the amino terminus to induce an approximately 45° bend in the DNA (Fig. 5a) [39]. This bend is significantly greater than the ~30° DNA bend caused by hAGT (Fig. 5a) [29]. Therefore, Atl1 induces greater DNA bending than hAGT, although the two proteins have similar folds.
ATL DNA nucleotide flipping and lesion interactions
Fluorescence-measured flipping for a base opposite an abasic site in eAtl [34] or an O 6-mG in TTHA1564 [36], and crystal structures of Atl-DNA complexes [39] established that ATLs use nucleotide flipping from the DNA double helix to access damaged nucleotides, like AGT [29]. However, unlike AGT and most other known DNA nucleotide-flipping proteins, this flipping is not connected to an alkyltransferase activity or any other type of enzymatic activity or catalysis [33, 34, 36, 38, 39], suggesting that it is a switch for pathway activation, rather than catalysis [39].
Atl1 crystal structures showed the flipping of the damaged guanine and established O 6-alkylG interactions within the lesion-binding pocket [39]. Atl1 rotates O 6-mG and O 6-pobG into a specificity pocket containing -PWHR- motif Trp56 (Fig. 5a, b). The extra-helical O 6-alkylG is stabilized by Arg39, whose side chain intercalates the DNA base stack and hydrogen bonds with the orphaned cytosine (Fig. 5a). Trp56 does not π-stack with the O 6-alkylG as originally expected, but rather hydrophobically packs with the alkyl group. Instead, the Arg69 guanidinium group stacks against the O 6-alkylG base in a cation-π interaction. Hydrogen bonds to the O 6-alkylG base side and main chain are conserved from ATL to AGT.
Atl1 has an approximately three times larger binding pocket than hAGT (Fig. 5b, c) [39]. This larger pocket is the result of moving out from the protein core, with respect to AGT, one wall of the binding pocket and the Lys45–Pro55 cap that interacts with larger alkyl groups. Also, the bulky hAGT Tyr158 is replaced by the smaller Atl1 Ile71 to prevent a clash with the Atl1 Trp56 side chain in its DNA-bound, closed position. The VpAtl-binding pocket, partially buried by binding site loop and binding pocket cap residues, is smaller than that for Atl1 [37]. However, VpAtl NMR relaxation data suggest conformational flexibility in this pocket, suggesting VpAtl may adopt conformations in which the substrate binding pocket is more exposed, like for Atl1.
The larger Atl1 pocket readily accommodates the O 6-pobG lesion [39], a bulky and toxicologically relevant adduct that results from metabolically activated tobacco-specific nitrosamines [20]. The pob group only makes hydrophobic protein interactions to Trp56 and Pro50 of the binding cap, which is ~5.3 Å further out than the comparable AGT Pro140 (Cα to Cα distance). No major structural changes between O 6-mG- and O 6-pobG-bound Atl1 were observed in the DNA conformation or lesion-binding site. The pob conformation observed in the Atl1 structure would clash with the hAGT-binding site loop and is incompatible with smaller E. coli AGT (Ada-C and Ogt) active site pockets [39]. The backbone conformational dynamics observed for the VpAtl guanine-lesion recognition cavity may account for the broader recognition of various alkyl guanine lesions for ATLs versus AGT [37]. Therefore, ATLs flip damage into a large binding pocket that accommodates a broader range of DNA lesions than AGTs.
ATL connection to nucleotide excision repair
One of the most puzzling mysteries surrounding ATLs was that they paradoxically protect cells from the biological effects of DNA alkylation damage, despite lacking the AGT reactive cysteine and alkyltransferase activity. Yet, it was clear that ATL must be playing a role in protection in vivo, since organisms without an AGT, such as S. pombe and T. thermophilus, were rendered more sensitive to alkylating reagents upon inactivation of their ATL genes [36, 38]. Tight binding affinities [34, 36] for, yet inability to repair [33, 38] O 6-alkylG lesions led to the conclusion that ATLs are damage sensors or play a role in the DNA nucleotide excision repair (NER) pathway [32, 35, 36, 38, 39], which excises bulky, DNA-distorting lesions. Through a series of genetic and biochemical experiments described below, Atl1 function was indeed discovered to be linked to NER.
The existence of in vivo ATL-DNA complex binding partners was suggested by the tight binding of ATLs to O 6-alkylG-containing oligonucleotides and the resulting conformational change that exposes the C-terminal loop for possible intermolecular interactions [39]. In vitro, eAtl interacts with E. coli NER proteins UvrA [35, 39] and UvrC [39], but not UvrB [35, 39]. Similarly, TTHA1564 interacts with T. thermophilus UvrA with a KD of 30 nM, indicating this interaction is significant [36]. Pulldown experiments with TTHA1564 identified other possible binding partners, including the α, β, and β′ subunits of RNA polymerase, the PIWI motif protein, putative Rad52 homologue, and UvrD helicase, although the number of matched peptides for UvrD was not significant. An interaction was also identified between eAtl and E. coli DNA repair helicase IV (HelD) [34], which is involved in the RecF pathway of DNA recombination repair, DNA methylation-based damage, and possibly limited UV damage repair in combination with the Rep protein [41]. Intriguingly, S. pombe Atl1 interacts with E. coli UvrA in vitro, suggesting conservation of ATL features for NER recognition [39].
ATL’s connection to NER is also supported by both microbial and eukaryotic genetic evidence. In S. pombe, ∆rad13 cells exhibit an increased spontaneous mutation rate that is strikingly suppressed to wild-type levels in Δatl1 Δrad13 mutants [39]. Reversion rate of the ade6-485 mutation and spot and clonogenic assays revealed Atl1 is epistatic with Rad13 and Swi10, but not Rhp14 or Rad2 (homologs of human XPG, ERCC1, XPA and Fen-1, respectively) for MNNG toxicity [39]. In humans, XPG and ERCC1 are part of the NER pathway, where XPG is the endonuclease that is responsible for the 3′ incision of DNA lesions and the XPF-ERCC1 endonuclease complex cuts at the 5′ side. Atl1’s epistasis with Rad13 and Swi10, therefore indicate Atl1 has a role in the NER pathway. In E. coli, chromosomal mutagenesis data revealed that eAtl and UvrA are epistatic, and a complementary relationship was discovered between the AGT and NER pathways for repairing O 6-alkyl adducts roughly related to size [35]. Quantitative determination of the fraction of mutations induced by adducts of varying size in mutant E. coli strains with inactive AGT and/or NER revealed that O 6-mG was repaired by AGT, whereas O 6-1-hydroxypropylguanine (O 6-1hpG) and O 6-2-hydroxypropylguanine (O 6-2hpG) (see Fig. 1) were repaired by NER, and O 6-HOEtG was repaired by both repair pathways [35]. Together, these data strongly suggest that ATL function is connected to NER.
Proposed mechanisms for ATL-mediated DNA repair pathway cross-talk
NER is a versatile repair mechanism that removes bulky, unrelated, helix-distorting lesions through the excision of a long DNA patch that contains the lesion [42]. The large variety of structurally unrelated lesions processed by NER has led to the proposal that the repair machinery recognizes the distortion of the helical structure of the DNA induced by the presence of the lesion, such as DNA bending or base pair disruption, rather than the lesion itself [43]. In higher eukaryotes, the global genomic repair (GGR) subpathway of NER is initiated by XPC recognition of bulky lesions and results in damage removal on non-transcribed DNA [43]. The efficient repair of lesions from actively transcribed DNA is carried out by the NER transcription-coupled repair (TCR) subpathway [42, 44]. TCR is recruited to sites of transcription arrest of a stalled elongating RNA polymerase, and subsequently engages downstream damage recognition components of GGR for lesion removal in higher eukaryotes [42, 44]. Both these mechanisms of NER-mediated repair of damaged DNA are distinct from the classical alkylated DNA base damage repair processes by direct damage reversal proteins or BER.
Previous studies suggested that NER may play a role in the processing of O 6-alkylG lesions [45–47], but the extent of this role and how it was accomplished was unknown. Particularly puzzling was how weakly distorting O 6-mG lesions [45–47], which are also insufficient to stall transcription [48], could recruit NER, especially since NER recognition of damage appears to be dependent on large distortions in the DNA [43]. Atl1 structures provided a plausible explanation, by showing a conformational switch accompanied by large DNA bending upon binding O 6-alkylG damage [39], which could be suitable to recruit NER. These structures, together with ATL genetic experiments and the other recent ATL studies described in the previous sections of this review, provide a basis to aid in understanding how ATLs promote a cross-talk DNA repair pathway connection between base repair and NER to recruit NER to sites of O 6-alkyG damage.
Specific NER complex protein–protein and protein–DNA interactions govern the coordinated assembly of NER protein factors required for effective DNA lesion recognition and repair. Initial structures existing for part of the NER pathway are providing structural and mechanistic insights into this process. In eukaryotic NER systems, initial damage recognition by S. cerevisiae XPC orthologue, Rad4 reveals DNA binding by the normal strand with the damaged strand evidently exposed for recognition [49]. This is consistent with the DNA-damaged strand binding observed for the DDB2 subunit of the UV-DDB complex of mammalian GGR [50], which binds UV-induced and other weakly distorting DNA lesions to aid recruitment of the XPC complex. Structures for the XPB [51] and XPD [52] helicases suggest specific recognition motifs and domains to distinguish damaged sites and transcription sites. The highly specific and stable complex formed upon association of XPA and ERCC1 for recruitment of the ERCC1–XPF nuclease is mediated by only a small region of XPA that perfectly fits into a central domain groove of ERCC1 [53]. In bacterial NER systems, structures of bacterial UvrA reveal conformational flexibility, asymmetry, and a likely open-to-closed DNA-binding mechanism [54, 55]. Conformational flexibility was also observed for UvrC and is likely significant for its function [56]. Geobacillus stearothermophilus UvrA and UvrB interact through a polar interface that contains several highly conserved charged residues [57]. The E. coli transcription repair coupling factor Mfd likely recruits UvrA recruitment at stalled transcription forks by structural but not catalytic mimicry of UvrB [58].
Protein–DNA interactions for the recognition and cleavage of ds-/ssDNA junctions by crenarchaeal XPF from Aeropyrum pernix are mediated by coupling movement of its nuclease and helix-hairpin-helix DNA-binding domains through a flexible linker to accommodate the large distortion of its DNA substrates to promote strand cleavage [59]. Similarly, structural and biochemical studies on the XPF-related dimeric Pyrococcus furiosus Hef nuclease that cleaves the 5′ side of nicked Holliday junctions showed a bipartite binding mode that is likely conserved in the XPF/Rad1/Mus81 nuclease family [60]. DNA binding-induced dramatic conformational changes of extended loops within the ssDNA-binding protein RPA DNA-binding domain imply DNA binding regulation for and/or by protein–protein interactions [61]. Overall, these NER structures suggest a general theme of conformational flexibility and specific binding partner interactions to drive repair, which appears to be preserved in ATLs.
ATLs may invoke the GGR subpathway of NER (Fig. 6). ATLs present damage to NER for processing in a way that resembles the UV-DDB complex of mammalian GGR. Atl1 binds the DNA damaged strand [39], as also seen for DDB2 [50]. DNA damaged strand binding by ATL is accompanied by open-to-closed conformational flexibility of a loop, somewhat reminiscent of UvrA. This damaged strand binding could increase the single-stranded nature of the undamaged strand to favor recruitment and binding of proteins that initiate repair by NER [35], such as mammalian XPC. Alternatively, ATLs may directly recruit the nucleases XPG and ERCC1/XPF to damage sites to initiate NER, independently of XPC, and facilitate the remainder of the response.
Fig. 6.

Possible participation of ATL in NER responses
ATLs may also recruit NER by binding O 6-alkylG lesions to block elongating RNA polymerase and initiate TCR (Fig. 6). TTHA1564 interactions with both UvrA and RNA polymerase led to the proposal that TTHA1564 may play a role analogous to transcription-repair coupling factor (TRCF) in TCR, where TTHA1564-O 6-mG may stall RNA polymerase at the damaged site [36]. Such a role could be reflected in the similar binding affinities for T. thermophilus UvrA with TRCF and TTHA1564 [36]. Similarly, the eAtl-UvrA interaction was suggested to recruit the UvrA2·UvrB complex in a way analogous to the E. coli transcription-repair coupling factor Mfd [35]. Further experiments are needed, however, to establish ATL’s role in GGR and/or TCR.
Although presentation of damage to NER by ATLs is similar to NER lesion recognition factors, ATLs recognize and bind lesions similarly to AGT and BER glycosylases. Notably, ATLs use a positive channel for lesion binding and 180° nucleotide flipping [39], which allows protein handoffs without release of toxic and mutagenic DNA intermediates that are characteristic of BER and recombination repair pathways [62–66]. Yet ATLs are not alkyltransferases, glycosylases, or long-patch BER or alternative UV excision repair proteins, and in S. pombe, increased spontaneous mutation rates for ∆rad13 strains are drastically reduced in ∆atl1 ∆rad13 strains, indicating that ATL redirects endogenous damage from other repair pathways to NER. Thus, ATLs recognize damage as base repair but present it as NER, thereby adopting elements of damage recognition and processing from both base repair and NER pathways [39] and supporting their role in cross-talk between these two pathways [35, 39].
ATL could promote cross-talk in some organisms by blocking AGT-mediated recognition and repair of O 6-alkylG and redirecting this repair to NER. Such an action was discovered in E. coli, where O 6-alkyl adducts are repaired by the AGT or NER pathways according to size [35]. This observation prompted the proposal of an “enhanced-NER” pathway in E. coli, where eAtl binds large O 6-alkylG adducts to improve their repair by NER through a direct interaction between eAtl and UvrA, whereas O 6-mG is repaired by AGT [35]. To explain the observation that ATL does not promote O 6-mG repair by NER when AGT is inactivated in E. coli, eAtl was proposed to bind O 6-mG with a weaker binding affinity than larger O 6-alkylGs [35]. Although such a hypothesis would have to be confirmed by further binding affinity experiments on various O 6-alkylG lesions, it is supported by the fact that Atl1 has a higher affinity for bulky O 6-pobG than for O 6-mG [39]. ATL-mediated cross-talk between AGTs and NER is further supported by Atl1-O 6-pobG DNA crystal structures, where the observed conformation of pob would conflict with the smaller active site pockets of AGTs [39]. This is consistent with the poor repair of O 6-pobG by E. coli AGTs, and could explain why organisms with multiple AGTs, like E. coli, would need an ATL. It is worth noting that this proposed ATL-enhanced repair by NER in E. coli would differ in organisms without an AGT, like T. thermophilus and S. pombe.
Interestingly, ATLs from ancestral Archaea exist as fusion proteins with BER nuclease Endo V, suggesting that ATL may be an ancient link between BER and NER [39, 67]. Such a connection is implied from recent structures of Endo V [68] and the UV-DDB complex [50], where a mutual, wedge-based binding mechanism that would allow recognition of minimally distorting DNA lesions was discovered [69]. This binding mechanism uses a 3- or 4-amino-acid wedge to extract the lesion from the DNA helix into a shallow binding pocket that can accommodate a variety of damaged bases, reminiscent of DNA binding by ATLs. ATL-Endo V fusions in Archaea suggest that in these organisms ATL and Endo V may act together in a coordinated pathway [67], where Endo V may have an XPG-like function. Similar AGT-Endo V fusion proteins have also been discovered, and these proteins retain both activities [70]. It will be interesting to see if these ATL- and AGT-EndoV fusion proteins are biologically significant.
Overall, ATL studies suggest a general repair mechanism (Figs. 2 and 6) in which ATLs bind weakly distorting O 6-alkylG adducts to sculpt them into bulky lesions that are redirected to and repaired by the NER pathway [35, 39]. In this way, ATLs make visible DNA lesions that are more likely to be overlooked by DNA repair machinery [71]. In some organisms, like E. coli, that contain AGT and ATL, small lesions are repaired by AGT, whereas ATL targets larger, bulky lesions for processing by NER nucleases and promotes competition for O 6-alkylG repair between the AGT and NER pathways, which otherwise function independently in the absence of ATL [35]. Therefore, ATLs use non-enzymatic nucleotide flipping as a switch to control DNA pathway activation, by channeling DNA base damage into the nucleotide excision repair pathway [39].
Conclusions and outlook
The discovery of ATLs presented the puzzling challenge of how these proteins, which bind DNA alkylation damage but do not repair it, are able to protect cells against the detrimental effects of DNA alkylation damage. Recent studies on ATLs have provided valuable insight into how they function by revealing that they bridge alkylated DNA base damage responses with NER: two DNA repair pathways that were previously thought to function independently from each other. ATL’s connection to NER was an unexpected discovery, since alkylated DNA base damage was classically thought to be recognized and repaired by direct damage reversal proteins like AGT or by lesion-specific DNA glycosylases, which excise modified bases to create abasic sites and initiate the BER pathway of DNA repair.
However, despite the advances in understanding the role of ATLs in DNA repair, key questions remain to be answered. For example, do ATLs engage NER through TCR or GGR or both? What protein or proteins act immediately downstream of ATL? Do NER proteins participate in protein–protein interactions with ATL for a direct lesion handoff or in protein–DNA interactions with ATL-DNA complexes for an indirect lesion handoff? If protein–protein interactions are relevant, what is the interaction interface and how easily can it be disrupted? Does ATL have higher affinity for certain types of O 6-alkylG damage, e.g., larger or bulkier alkyl groups, over AGT? The answers to these questions are eagerly anticipated.
Also of interest is whether ATLs or their analogs are present in higher eukaryotes, including mammals and humans. It is unclear why no recognizable ATL has been identified in humans, plants, or other higher eukaryotes. Nevertheless, the possibility of discovering ATL functional analogs in these organisms is not excluded. Since the function of ATL is to bind DNA damage to bridge DNA repair pathways that otherwise function independently from one another, it is possible that another protein performs this role in higher eukaryotes. Interestingly, a similar question was raised for the existence of archaeal NER proteins XPA and ERCC1, because although homodimeric XPF-like endonucleases exist in Archaea, homologs for XPA and ERCC1 have only been identified in eukaryotes, and the ERCC1–XPA interaction is less conserved in lower eukaryotes, such as yeast [53]. The proposal that this observation for XPA and ERCC1 may be attributed to added complexity and distinct functional organization for eukaryotic NER in comparison to bacterial NER [53], may also extend to ATL. However, the recent discovery of a multi-cellular eukaryotic ATL in N. vectensis, whose genome is more similar to vertebrates than invertebrates and has aided in genome characterization of the long-extinct last common ancestor of all eumetazoans [72], is promising. Although it is possible that mammalian cells may use lesion recognition factors, such as the DDB complex, to enhance NER-mediated repair of O 6-alkylG lesions [35], the existence of ATL in a multi-cellular eukaryote, yeast, bacteria, and ancestral Archaea shows ATL is present in all three domains of life and argues ATL interactions are ancestral to present-day repair pathways. Together, these observations suggest that although no ATL has yet been discovered in higher eukaryotes and mammals, it is likely that an ATL or similar protein functional analog exists in these organisms.
Equally intriguing is why ATLs lack alkyltransferase activity. As mentioned previously, mutating in an active site cysteine was insufficient to restore repair activity, raising the question, “What other protein elements are required for alkyltransferase function?” Unfortunately, available ATL structures do not identify obvious candidates for mutation to restore this activity. However, these structures do suggest that ATL is missing two AGT elements that may be essential for activity. The first is an AGT-like N-terminal domain. Although the precise function of the AGT N-terminal domain remains unknown, experiments where the two hAGT domains were separately characterized indicated the C-terminal domain required the N-terminal domain and Zn2+ for activity, whereas the N-terminal domain surprisingly had weak AGT activity that required Zn2+in the absence of the C-terminal domain [73]. From these data, the N-terminal domain was proposed to play an important structural role in orienting the C-terminal domain for efficient alkyltransfer [73]. The second is a catalytically essential well-ordered water molecule that is part of a hydrogen bonding network with the catalytic cysteine, a histidine, and a glutamate. In AGT, this water participates in four, nearly tetrahedral hydrogen bonds to nearby side and main chain atoms [27]. This water is absent in Atl1 structures, likely because of steric hindrance by the active site tryptophan side chain and the removal of two of the water’s hydrogen bonding partners by the moving out of a loop. In AGT, this loop is anchored through contacts with the N-terminal domain, again suggesting that the absence of this domain in ATL may contribute to ATL’s inactivity. It will be interesting to see if a functional ATL can be designed.
In summary, ATL uses non-enzymatic nucleotide flipping to redirect the processing and repair of specific base damage, i.e., O 6-alkylG, into the general damage NER pathway. As the mystery of ATLs is beginning to be unraveled, a better understanding of ATL function is emerging. There are many areas where this new knowledge could be applied. One exciting possibility is to use it to assist in understanding tumor cell resistance to alkylating chemotherapies. If ATLs are found in humans, these results could significantly aid the development of more effective cancer treatments. For example, inhibiting ATL could improve the effectiveness of alkylating chemotherapies, or increasing ATL could help to protect bone marrow and other tissue sensitive to cancer treatments. Therefore, a thorough understanding of ATL function could impact our understanding of genomic integrity and responses to base damage relevant to pathogens, cancer development, and potential improved cancer interventions by alkylating agents and repair pathway control.
Acknowledgments
This work was supported by National Institutes of Health Grant R01CA097209 (J.A.T.) and The Skaggs Institute for Chemical Biology (J.L.T.).
Abbreviations
- AGT
O6-alkylguanine-DNA alkyltransferase
- ATL
Alkyltransferase-like protein
- Atl1
S. pombe ATL
- BER
Base excision repair
- eAtl
E. coli ATL
- GGR
Global genomic repair
- hAGT
Human AGT
- HelD
E. coli DNA repair helicase IV
- HTH
Helix-turn-helix
- MGMT
O6-methylguanine-DNA methyltransferase
- MMR
Mismatch repair
- MMS
Methyl methanesulfonate
- MNNG
N-methyl-N′-nitro-N-nitrosoguanidine
- MNU
N-methyl-N-nitrosourea
- NER
Nucleotide excision repair
- nvAtl
N. vectensis ATL
- O4-mT
O4-methylthymine
- O6-alkylG
O6-alkylguanine
- O6-bG
O6-benzylguanine
- O6-btG
O6-(4-bromothenyl)guanine
- O6-HOEtG
O6-hydroxyethylguanine
- O6-1hpG
O6-1-hydroxypropylguanine
- O6-2hpG
O6-2-hydroxypropylguanine
- O6-mG
O6-methylguanine
- O6-pobG
O6-4-(3-pyridyl)-4-oxobutylguanine
- PNU
N-propyl-N-nitrosourea
- TCR
Transcription-coupled repair
- TRCF
Transcription-repair coupling factor
- TTHA1564
T. thermophilus ATL
References
- 1.Rydberg B, Lindahl T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-l-methionine is a potentially mutagenic reaction. EMBO J. 1982;1:211–216. doi: 10.1002/j.1460-2075.1982.tb01149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beranek DT. Distribution of methyl and ethyl adducts following alkylation with monofunctional alkylating agents. Mutat Res. 1990;231:11–30. doi: 10.1016/0027-5107(90)90173-2. [DOI] [PubMed] [Google Scholar]
- 3.Colvin DM. Alkylating agents and platinum antitumor compounds. In: Holland JF, Frei E, Bast RC Jr, Kufe DW, Morton DL, Weichselbaum RR, editors. Cancer medicine. Media: Willams & Wilkins; 1997. pp. 949–975. [Google Scholar]
- 4.Sedgwick B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol. 2004;5:148–157. doi: 10.1038/nrm1312. [DOI] [PubMed] [Google Scholar]
- 5.Pegg AE. Repair of O 6-alkylguanine by alkyltransferases. Mutat Res. 2000;462:83–100. doi: 10.1016/S1383-5742(00)00017-X. [DOI] [PubMed] [Google Scholar]
- 6.Tubbs JL, Pegg AE, Tainer JA. DNA binding, nucleotide flipping, and the helix-turn-helix motif in base repair by O 6-alkylguanine-DNA alkyltransferase and its implications for cancer chemotherapy. DNA Repair. 2007;6:1100–1115. doi: 10.1016/j.dnarep.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loechler EL, Green CL, Essigmann JM. In vivo mutagenesis by O 6-methylguanine built into a unique site in a viral genome. Proc Natl Acad Sci USA. 1984;81:6271–6275. doi: 10.1073/pnas.81.20.6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pauly GT, Hughes SH, Moschel RC. Comparison of mutagenesis by O 6-methyl- and O 6-ethylguanine and O 4-methylthymine in Escherichia coli using double-stranded and gapped plasmids. Carcinogenesis. 1998;19:457–461. doi: 10.1093/carcin/19.3.457. [DOI] [PubMed] [Google Scholar]
- 9.Huffman JL, Sundheim O, Tainer JA. DNA base damage recognition and removal: new twists and grooves. Mutat Res. 2005;577:55–76. doi: 10.1016/j.mrfmmm.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 10.Hitomi K, Iwai S, Tainer JA. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair. 2007;6:410–428. doi: 10.1016/j.dnarep.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Daniels DS, Tainer JA. Conserved structural motifs governing the stoichiometric repair of alkylated DNA by O 6-alkylguanine-DNA alkyltransferase. Mutat Res. 2000;460:151–163. doi: 10.1016/s0921-8777(00)00024-0. [DOI] [PubMed] [Google Scholar]
- 12.Sundheim O, Vagbo CB, Bjoras M, Sousa MM, Talstad V, Aas PA, Drablos F, Krokan HE, Tainer JA, Slupphaug G. Human ABH3 structure and key residues for oxidative demethylation to reverse DNA/RNA damage. EMBO J. 2006;25:3389–3397. doi: 10.1038/sj.emboj.7601219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud P-F, Lindquist EA, Kamisugi Y, Tanahashi T, Sakakibara K, Fujita T, Oishi K, Shin-I T, Kuroki Y, Toyoda A, Suzuki Y, Hashimoto S-i, Yamaguchi K, Sugano S, Kohara Y, Fujiyama A, Anterola A, Aoki S, Ashton N, Barbazuk WB, Barker E, Bennetzen JL, Blankenship R, Cho SH, Dutcher SK, Estelle M, Fawcett JA, Gundlach H, Hanada K, Heyl A, Hicks KA, Hughes J, Lohr M, Mayer K, Melkozernov A, Murata T, Nelson DR, Pils B, Prigge M, Reiss B, Renner T, Rombauts S, Rushton PJ, Sanderfoot A, Schween G, Shiu S-H, Stueber K, Theodoulou FL, Tu H, Van de Peer Y, Verrier PJ, Waters E, Wood A, Yang L, Cove D, Cuming AC, Hasebe M, Lucas S, Mishler BD, Reski R, Grigoriev IV, Quatrano RS, Boore JL. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. 2008;319:64–69. doi: 10.1126/science.1150646. [DOI] [PubMed] [Google Scholar]
- 14.Merchant SS, Prochnik SE, Vallon O, Harris EH, Karpowicz SJ, Witman GB, Terry A, Salamov A, Fritz-Laylin LK, Marechal-Drouard L, Marshall WF, Qu LH, Nelson DR, Sanderfoot AA, Spalding MH, Kapitonov VV, Ren Q, Ferris P, Lindquist E, Shapiro H, Lucas SM, Grimwood J, Schmutz J, Cardol P, Cerutti H, Chanfreau G, Chen CL, Cognat V, Croft MT, Dent R, Dutcher S, Fernandez E, Fukuzawa H, Gonzalez-Ballester D, Gonzalez-Halphen D, Hallmann A, Hanikenne M, Hippler M, Inwood W, Jabbari K, Kalanon M, Kuras R, Lefebvre PA, Lemaire SD, Lobanov AV, Lohr M, Manuell A, Meier I, Mets L, Mittag M, Mittelmeier T, Moroney JV, Moseley J, Napoli C, Nedelcu AM, Niyogi K, Novoselov SV, Paulsen IT, Pazour G, Purton S, Ral JP, Riano-Pachon DM, Riekhof W, Rymarquis L, Schroda M, Stern D, Umen J, Willows R, Wilson N, Zimmer SL, Allmer J, Balk J, Bisova K, Chen CJ, Elias M, Gendler K, Hauser C, Lamb MR, Ledford H, Long JC, Minagawa J, Page MD, Pan J, Pootakham W, Roje S, Rose A, Stahlberg E, Terauchi AM, Yang P, Ball S, Bowler C, Dieckmann CL, Gladyshev VN, Green P, Jorgensen R, Mayfield S, Mueller-Roeber B, Rajamani S, Sayre RT, Brokstein P, Dubchak I, Goodstein D, Hornick L, Huang YW, Jhaveri J, Luo Y, Martinez D, Ngau WC, Otillar B, Poliakov A, Porter A, Szajkowski L, Werner G, Zhou K, Grigoriev IV, Rokhsar DS, Grossman AR. The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science. 2007;318:245–250. doi: 10.1126/science.1143609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuzaki M, Misumi O, Shin IT, Maruyama S, Takahara M, Miyagishima SY, Mori T, Nishida K, Yagisawa F, Yoshida Y, Nishimura Y, Nakao S, Kobayashi T, Momoyama Y, Higashiyama T, Minoda A, Sano M, Nomoto H, Oishi K, Hayashi H, Ohta F, Nishizaka S, Haga S, Miura S, Morishita T, Kabeya Y, Terasawa K, Suzuki Y, Ishii Y, Asakawa S, Takano H, Ohta N, Kuroiwa H, Tanaka K, Shimizu N, Sugano S, Sato N, Nozaki H, Ogasawara N, Kohara Y, Kuroiwa T. Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature. 2004;428:653–657. doi: 10.1038/nature02398. [DOI] [PubMed] [Google Scholar]
- 16.Mitra S, Kaina B. Regulation of repair of alkylation damage in mammalian genomes. Prog Nucleic Acid Res Mol Biol. 1993;44:109–142. doi: 10.1016/S0079-6603(08)60218-4. [DOI] [PubMed] [Google Scholar]
- 17.Pegg AE, Dolan ME, Moschel RC. Structure, function and inhibition of O 6-alkylguanine-DNA alkyltransferase. Prog Nucleic Acid Res Mol Biol. 1995;51:167–223. doi: 10.1016/S0079-6603(08)60879-X. [DOI] [PubMed] [Google Scholar]
- 18.Goodtzova K, Kanugula S, Edara S, Pauly GT, Moschel RC, Pegg AE. Repair of O 6-benzylguanine by the Escherichia coli Ada and Ogt and the human O 6- alkylguanine-DNA alkyltransferase. J Biol Chem. 1997;272:8332–8339. doi: 10.1074/jbc.272.13.8332. [DOI] [PubMed] [Google Scholar]
- 19.Shibata T, Glynn N, McMurry TB, McElhinney RS, Margison GP, Williams DM. Novel synthesis of O 6-alkylguanine containing oligodeoxyribonucleotides as substrates for the human DNA repair protein, O 6-methylguanine DNA methyltransferase (MGMT) Nucleic Acids Res. 2006;34:1884–1891. doi: 10.1093/nar/gkl117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L, Spratt TE, Liu XK, Hecht SS, Pegg AE, Peterson LA. Pyridyloxobutyl adduct O 6-[4-oxo-4-(3-pyridyl)butyl]guanine is present in 4-(acetoxymethylnitrosamino)-1-(3-pyridyl)-1-butanone-treated DNA and is a substrate for O 6-alkylguanine-DNA alkyltransferase. Chem Res Toxicol. 1997;10:562–567. doi: 10.1021/tx9602067. [DOI] [PubMed] [Google Scholar]
- 21.Fang QM, Noronha AM, Murphy SP, Wilds CJ, Tubbs JL, Tainer JA, Chowdhury G, Guengerich FP, Pegg AE. Repair of O-6-G-alkyl-O-6-G interstrand cross-links by human O 6-alkylguanine-DNA alkyltransferase. Biochemistry. 2008;47:10892–10903. doi: 10.1021/bi8008664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu-Welliver M, Pegg AE. Point mutations at multiple sites including highly conserved amino acids maintain activity but render O 6-alkylguanine-DNA alkyltransferase insensitive to O 6-benzylguanine. Biochem J. 2000;347:519–526. doi: 10.1042/0264-6021:3470519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu-Welliver M, Kanugula S, Pegg AE. Isolation of human O 6-alkylguanine-DNA alkyltransferase mutants highly resistant to inactivation by O 6-benzylguanine. Cancer Res. 1998;58:1936–1945. [PubMed] [Google Scholar]
- 24.Liu L, Watanabe K, Fang Q, Williams KM, Guengerich FP, Pegg AE. Effect of alterations of key active site residues in O 6-alkylguanine-DNA alkyltransferase on its ability to modulate the genotoxicity of 1,2-dibromoethane. Chem Res Toxicol. 2007;20:155–163. doi: 10.1021/tx600257g. [DOI] [PubMed] [Google Scholar]
- 25.Fang Q, Kanugula S, Tubbs JL, Tainer JA, Pegg AE. Repair of O 4-alkylthymine by O 6-alkylguanine-DNA alkyltransferases. J Biol Chem. 2010;285:8185–8195. doi: 10.1074/jbc.M109.045518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margison GP, Santibáñez-Koref MF. O6-Alkylguanine-DNA alkyltransferase: role in carcinogenesis and chemotherapy. Bioessays. 2002;24:255–266. doi: 10.1002/bies.10063. [DOI] [PubMed] [Google Scholar]
- 27.Daniels DS, Mol CD, Arvai AS, Kanugula S, Pegg AE, Tainer JA. Active and alkylated human AGT structures: a novel zinc site, inhibitor and extrahelical base binding. EMBO J. 2000;19:1719–1730. doi: 10.1093/emboj/19.7.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wibley JEA, Pegg AE, Moody PCE. Crystal structure of the human O 6-alkylguanine-DNA alkyltransferase. Nucleic Acids Res. 2000;28:393–401. doi: 10.1093/nar/28.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daniels DS, Woo TT, Luu KX, Noll DM, Clarke ND, Pegg AE, Tainer JA. DNA binding and nucleotide flipping by the human DNA repair protein AGT. Nat Struct Mol Biol. 2004;11:714–720. doi: 10.1038/nsmb791. [DOI] [PubMed] [Google Scholar]
- 30.Duguid EM, Rice PA, He C. The structure of the human AGT protein bound to DNA and its implications for damage detection. J Mol Biol. 2005;350:657–666. doi: 10.1016/j.jmb.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 31.Margison GP, Povey AC, Kaina B, Koref MFS. Variability and regulation of O 6-alkylguanine-DNA alkyltransferase. Carcinogenesis. 2003;24:625–635. doi: 10.1093/carcin/bgg005. [DOI] [PubMed] [Google Scholar]
- 32.Margison GP, Butt A, Pearson SJ, Wharton S, Watson AJ, Marriott A, Caetano CM, Hollins JJ, Rukazenkova N, Begum G, Santibanez-Koref MF. Alkyltransferase-like proteins. DNA Repair. 2007;6:1222–1228. doi: 10.1016/j.dnarep.2007.03.014. [DOI] [PubMed] [Google Scholar]
- 33.Pearson SJ, Ferguson J, Santibanez-Koref M, Margison GP. Inhibition of O 6-methylguanine-DNA methyltransferase by an alkyltransferase-like protein from Escherichia coli . Nucleic Acids Res. 2005;33:3837–3844. doi: 10.1093/nar/gki696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen CS, Korobkova E, Chen H, Zhu J, Jian X, Tao SC, He C, Zhu H. A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli . Nat Methods. 2008;5:69–74. doi: 10.1038/nmeth1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mazon G, Philippin G, Cadet J, Gasparutto D, Fuchs RP. The alkyltransferase-like ybaZ gene product enhances nucleotide excision repair of O 6-alkylguanine adducts in E. coli . DNA Repair. 2009;8:697–703. doi: 10.1016/j.dnarep.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 36.Morita R, Nakagawa N, Kuramitsu S, Masui R. An O 6-methylguanine-DNA methyltransferase-like protein from Thermus thermophilus interacts with a nucleotide excision repair protein. J Biochem (Tokyo) 2008;144:267–277. doi: 10.1093/jb/mvn065. [DOI] [PubMed] [Google Scholar]
- 37.Aramini JM, Tubbs JL, Kanugula S, Rossi P, Ertekin A, Maglaqui M, Hamilton K, Ciccosanti CT, Jiang M, Xiao R, Soong TT, Rost B, Acton TB, Everett JK, Pegg AE, Tainer JA, Montelione GT. Structural basis of O 6-alkylguanine recognition by a bacterial alkyltransferase-like DNA repair protein. J Biol Chem. 2010;285:13736–13741. doi: 10.1074/jbc.M109.093591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pearson SJ, Wharton S, Watson AJ, Begum G, Butt A, Glynn N, Williams DM, Shibata T, Santibanez-Koref MF, Margison GP. A novel DNA damage recognition protein in Schizosaccharomyces pombe . Nucleic Acids Res. 2006;34:2347–2354. doi: 10.1093/nar/gkl270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tubbs JL, Latypov V, Kanugula S, Butt A, Melikishvili M, Kraehenbuehl R, Fleck O, Marriott A, Watson AJ, Verbeek B, McGown G, Thorncroft M, Santibanez-Koref MF, Millington C, Arvai AS, Kroeger MD, Peterson LA, Williams DM, Fried MG, Margison GP, Pegg AE, Tainer JA. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasimas JJ, Pegg AE, Fried MG. DNA-binding mechanism of O 6-alkylguanine-DNA alkyltransferase. Effects of protein and DNA alkylation on complex stability. J Biol Chem. 2003;278:7973–7980. doi: 10.1074/jbc.M211854200. [DOI] [PubMed] [Google Scholar]
- 41.Mendonca VM, Kaiser-Rogers K, Matson SW. Double helicase II (uvrD)-helicase IV (helD) deletion mutants are defective in the recombination pathways of Escherichia coli . J Bacteriol. 1993;175:4641–4651. doi: 10.1128/jb.175.15.4641-4651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 43.Sugasawa K, Okamoto T, Shimizu Y, Masutani C, Iwai S, Hanaoka F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–521. doi: 10.1101/gad.866301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellon I. Transcription-coupled repair: a complex affair. Mutat Res. 2005;577:155–161. doi: 10.1016/j.mrfmmm.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 45.Samson L, Thomale J, Rajewsky MF. Alternative pathways for the in vivo repair of O 6-alkylguanine and O 4-alkylthymine in Escherichia coli: the adaptive response and nucleotide excision repair. EMBO J. 1988;7:2261–2267. doi: 10.1002/j.1460-2075.1988.tb03066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voigt JM, Van Houten B, Sancar A, Topal MD. Repair of O 6-methylguanine by ABC excinuclease of Escherichia coli in vitro. J Biol Chem. 1989;264:5172–5176. [PubMed] [Google Scholar]
- 47.Edara S, Kanugula S, Pegg AE. Expression of the inactive C145A mutant human O 6-alkylguanine-DNA alkyltransferase in E. coli increases cell killing and mutations by N-methyl-N’-nitro-N-nitrosoguanidine. Carcinogenesis. 1999;20:103–108. doi: 10.1093/carcin/20.1.103. [DOI] [PubMed] [Google Scholar]
- 48.Viswanathan A, Doetsch PW. Effects of nonbulky DNA base damages on Escherichia coli RNA polymerase-mediated elongation and promoter clearance. J Biol Chem. 1998;273:21276–21281. doi: 10.1074/jbc.273.33.21276. [DOI] [PubMed] [Google Scholar]
- 49.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 50.Scrima A, Konickova R, Czyzewski BK, Kawasaki Y, Jeffrey PD, Groisman R, Nakatani Y, Iwai S, Pavletich NP, Thoma NH. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fan L, Arvai AS, Cooper PK, Iwai S, Hanaoka F, Tainer JA. Conserved XPB core structure and motifs for DNA unwinding: implications for pathway selection of transcription or excision repair. Mol Cell. 2006;22:27–37. doi: 10.1016/j.molcel.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 52.Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsodikov OV, Ivanov D, Orelli B, Staresincic L, Shoshani I, Oberman R, Scharer OD, Wagner G, Ellenberger T. Structural basis for the recruitment of ERCC1-XPF to nucleotide excision repair complexes by XPA. EMBO J. 2007;26:4768–4776. doi: 10.1038/sj.emboj.7601894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Timmins J, Gordon E, Caria S, Leonard G, Acajjaoui S, Kuo MS, Monchois V, McSweeney S. Structural and mutational analyses of Deinococcus radiodurans UvrA2 provide insight into DNA binding and damage recognition by UvrAs. Structure. 2009;17:547–558. doi: 10.1016/j.str.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 55.Pakotiprapha D, Inuzuka Y, Bowman BR, Moolenaar GF, Goosen N, Jeruzalmi D, Verdine GL. Crystal structure of Bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol Cell. 2008;29:122–133. doi: 10.1016/j.molcel.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karakas E, Truglio JJ, Croteau D, Rhau B, Wang L, Van Houten B, Kisker C. Structure of the C-terminal half of UvrC reveals an RNase H endonuclease domain with an Argonaute-like catalytic triad. EMBO J. 2007;26:613–622. doi: 10.1038/sj.emboj.7601497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pakotiprapha D, Liu Y, Verdine GL, Jeruzalmi D. A structural model for the damage-sensing complex in bacterial nucleotide excision repair. J Biol Chem. 2009;284:12837–12844. doi: 10.1074/jbc.M900571200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Assenmacher N, Wenig K, Lammens A, Hopfner KP. Structural basis for transcription-coupled repair: the N terminus of Mfd resembles UvrB with degenerate ATPase motifs. J Mol Biol. 2006;355:675–683. doi: 10.1016/j.jmb.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 59.Newman M, Murray-Rust J, Lally J, Rudolf J, Fadden A, Knowles PP, White MF, McDonald NQ. Structure of an XPF endonuclease with and without DNA suggests a model for substrate recognition. EMBO J. 2005;24:895–905. doi: 10.1038/sj.emboj.7600581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishino T, Komori K, Ishino Y, Morikawa K. Structural and functional analyses of an archaeal XPF/Rad1/Mus81 nuclease: asymmetric DNA binding and cleavage mechanisms. Structure. 2005;13:1183–1192. doi: 10.1016/j.str.2005.04.024. [DOI] [PubMed] [Google Scholar]
- 61.Bochkareva E, Belegu V, Korolev S, Bochkarev A. Structure of the major single-stranded DNA-binding domain of replication protein A suggests a dynamic mechanism for DNA binding. EMBO J. 2001;20:612–618. doi: 10.1093/emboj/20.3.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 63.Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/S0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 64.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc Natl Acad Sci USA. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcin ED, Hosfield DJ, Desai SA, Haas BJ, Bjoras M, Cunningham RP, Tainer JA. DNA apurinic-apyrimidinic site binding and excision by endonuclease IV. Nat Struct Mol Biol. 2008;15:515–522. doi: 10.1038/nsmb.1414. [DOI] [PubMed] [Google Scholar]
- 66.Williams RS, Moncalian G, Williams JS, Yamada Y, Limbo O, Shin DS, Groocock LM, Cahill D, Hitomi C, Guenther G, Moiani D, Carney JP, Russell P, Tainer JA. Mre11 dimers coordinate DNA end bridging and nuclease processing in double-strand-break repair. Cell. 2008;135:97–109. doi: 10.1016/j.cell.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein–protein interactions from genome sequences. Science. 1999;285:751–753. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 68.Dalhus B, Arvai AS, Rosnes I, Olsen OE, Backe PH, Alseth I, Gao HH, Cao WG, Tainer JA, Bjoras M. Structures of endonuclease V with DNA reveal initiation of deaminated adenine repair. Nat Struct Mol Biol. 2009;16:138–143. doi: 10.1038/nsmb.1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Scharer OD, Campbell AJ. Wedging out DNA damage. Nat Struct Mol Biol. 2009;16:102–104. doi: 10.1038/nsmb0209-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kanugula S, Pauly GT, Moschel RC, Pegg AE. A bifunctional DNA repair protein from Ferroplasma acidarmanus exhibits O 6-alkylguanine-DNA alkyltransferase and endonuclease V activities. Proc Natl Acad Sci USA. 2005;102:3617–3622. doi: 10.1073/pnas.0408719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reissner T, Schorr S, Carell T. Once overlooked, now made visible: ATL proteins and DNA repair. Angew Chem Int Ed Engl. 2009;48:7293–7295. doi: 10.1002/anie.200904042. [DOI] [PubMed] [Google Scholar]
- 72.Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, Jurka J, Genikhovich G, Grigoriev IV, Lucas SM, Steele RE, Finnerty JR, Technau U, Martindale MQ, Rokhsar DS. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317:86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- 73.Fang Q, Kanugula S, Pegg AE. Function of domains of human O 6-alkylguanine-DNA alkyltransferase. Biochemistry. 2005;44:15396–15405. doi: 10.1021/bi051460d. [DOI] [PubMed] [Google Scholar]