


Abstract
We established a database of alternative splice forms (ASforms) for nine eukaryotic organisms. ASforms are defined by comparing high-scoring ESTs with mRNA sequences using BLAST, taking known exon–intron information (from the Ensembl database). Filtering programs compare the ends of each aligned sequence pair for deletions or insertions in the EST sequence, which indicate the existence of alternative splice forms with respect to the exon–intron boundaries. Moreover, we defined the alternative splice profile of each human sequence. It indicates the number of alternatively spliced ESTs (NAE), the number of constitutively spliced ESTs (NCE) as well as the number of alternative splice sites (NSS) per mRNA. NAE and NCE correspond to the EST coverage and can be used as a quality indicator for the predicted alternative splice variants. The NSS value specifies the splice propensity of a gene. Additionally, the tissue type information of all ESTs was included. This allows (i) restriction of the search to certain tissues and (ii) calculation of the tissue-NAEs, tissue-NCEs and tissue-NSS. These scores are suitable for the estimation of tissue specificity of certain ASforms. Furthermore, the developmental stage and disease information of the ESTs is available. EASED is accessible at http://eased.bioinf.mdc-berlin.de/.
INTRODUCTION
The concept of alternative splicing as a mechanism to create a high diversity of functional proteins in mammals has received increasing evidence and support with the progress of the Human Genome Project (1). Investigations based on human sequence material (experimental data) and computational methods suggest about half of the identified genes to be alternatively spliced in conjunction with cellular processes (1–3).
Various approaches to the detection of alternative spliceforms (ASforms) computationally are based on expressed sequence tags (ESTs) [(4–12) see also the review by Modrek and Lee (13)]. Determining the conditions under which an mRNA was isolated involves the collection and classification of information belonging to the corresponding EST, e.g. the tissue origin, the developmental stage or the association with diseases such as cancer.
DESCRIPTION OF EASED
The Extended Alternatively Spliced EST Database is an online compendium of ASforms for several organisms (Arabidopsis thaliana, Bos taurus, Caenorhabditis elegans, Drosophila melanogaster, Danio rerio, Homo sapiens, Mus musculus, Rattus norvegicus, Xenopus laevis). At the moment, the additional information described here (alternative splice profile, tissue types, developmental stage, disease, classification of splice events) are only available for human.
PREDICTION ALGORITHM
ASforms are defined by comparing high-scoring ESTs with mRNA sequences [both from GenBank (14)] using BLAST (15). The exon–intron information for human sequences was obtained from the Ensembl database (16). (For the other organisms the exon–intron information is not yet included.) Repetitive sequences of all mRNAs were previously masked by MaskerAid (17). The algorithm used to identify ASforms takes the currently available mRNA sequences and aligns these sequences to all available ESTs using the BLASTN program. A matching pair of mRNA and EST has to fulfil certain criteria to be considered as an ASform. The alignment has to show at least two high-scoring pairs (HSPs). Filtering programs with defined parameters (gap length ≥ 30 nucleotides; HSP length ≥ 100 nucleotides and percentage of identity of each HSP ≥ 98%) compare the ends of each aligned sequence pair for deletions or insertions in the EST sequence, which suggest the existence of ASforms. All predicted ASforms are stored in a database.
Alternative Splice Profile (ASP)
We defined the so-called alternative splice profile (ASP) of each human sequence. It indicates the number of alternatively spliced ESTs (NAEs), the number of constitutively spliced ESTs (NCEs) as well as the number of alternative splice sites (NSSs) per mRNA. NAE and NCE correspond to the EST coverage and can be used as a quality indicator for the predicted alternative splice variants. The NSS value specifies the splice propensity of a gene.
Tissue types
Another useful feature for human sequences is the tissue type information. This information was derived from the MeSH (Medical Subject Headings) tree (http://www.nlm.nih.gov/mesh/meshhome.html). We use the tissue type classification for human in the second layer, which contains 43 different human tissues. This allows the search to be restricted to certain tissues and the calculation of the tissue-NAEs, tissue-NCEs and tissue-NSSs.
Additional information
All available information concerning the developmental stage (‘embryo’, ‘newborn’, ‘juvenile’ and ‘adult’) as well as the disease status (‘health’, ‘cancer’ and ‘disease’ meaning all other diseases except those marked as ‘cancer’) was additionally included. As previously mentioned for the tissue-specific parameter, this information also enables the calculation of the ASPs for selected developmental stages or diseases.
Classification of the types of alternative splice events
We classify the types of alternative splice events in terms of the location of the HSP boundaries compared with the given exon–intron boundaries. We define an exact match of a HSP boundary to an exon–intron boundary with an assumed 10 bp ‘uncertainty’. In doing this we assumed three possible donor as well as acceptor splice site events (Fig. 1): the HSP start or end lies on the exon–intron boundary (xas or exact alternative splice; Fig. 1a), the HSP boundary lies within an exon (eas, alternative splice within an exon; Fig. 1b) or the HSP boundary lies within an intron (ias, alternative splice within an intron; Fig. 1c). For the donor site, the alternative splice events are termed 5xas, 5eas or 5ias. For the acceptor site the classification gives rise to 3xas 3eas or 3ias splice sites. Using this classification, we can mark all splice sites as (i) alternative 3′ splice sites (3eas or 3ias), (ii) alternative 5′ splice site (5eas or 5ias), (iii) cassette exons (3xas and 5xas) and (iv) retained introns [exact 3′ and 5′ splice sites (3xas and 5xas); the inserted nucleotides originate from the intron sequence].
Figure 1.
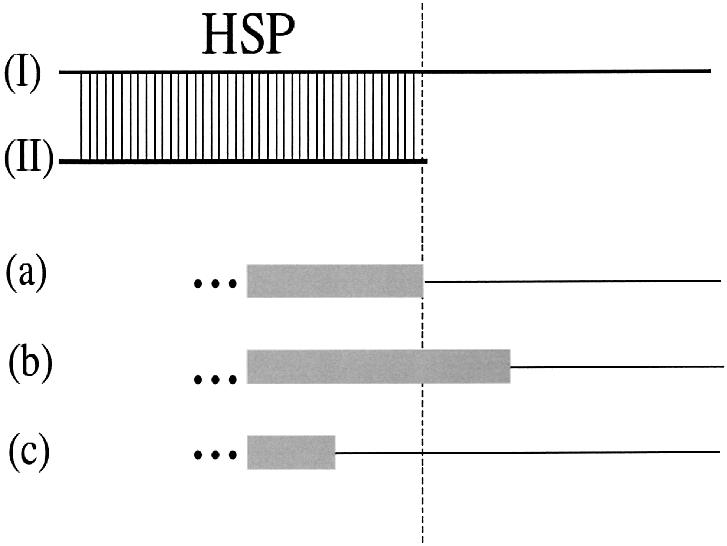
Classification of the types of alternative splice events. Exons are shown as boxes, introns as a horizontal line. The HSP was found by comparing two sequences (I and II) using BLAST. If (a) the end of the HSP (marked by the dotted line) lies on the exon–intron boundary, we name it xas. In the case of the end of HSP lying within an exon it is an eas event (b) or otherwise within an intron, it is termed ias (c). This illustration shows only the 5′ splice site.
Additionally, the type of alternative splicing is given as a ‘skip’ (the EST sequence is shorter than the mRNA sequence and a gap between two HSPs was found on the mRNA) or ‘insert’ (vice versa).
DATABASE PRESENTATION
Query interface
The database for human alternative splice forms is accessible via http://eased.bioinf.mdc-berlin.de/. EASED provides a number of possibilities to search for ASforms:
(i) In the simplest mode, users can query for ASforms by mRNA, CDS or EST accession numbers, the Ensembl gene ID or Ensembl transcript ID.
(ii) A keyword search mode for all organisms, which will identify a gene via a full text search of the gene’s name and description.
(iii) The search for other identifiers includes GO numbers (insert e.g. ‘GO:0007048’), Swiss-Prot entry name, RefSeq ID, EC number (if indicated in the description line), chromosome number (insert e.g. ‘7’ or ‘Y’) and protein ID.
(iv) The restriction to splice sites with a defined (e.g. high) number or percentage of ESTs can be used to filter out those splice sites with a high coverage of ESTs.
(v) The tissue type search allows all ASforms to be extracted with a predetermined number or percentage of ESTs from one of the 43 human tissues.
(vi) Moreover, one can search for splice sites with a defined fraction of a selected developmental stage (adult, embryo, newborn, juvenile) or disease (cancer, healthy, other diseases).
(vii) A further restriction to splice sites with or without exact exon–intron boundaries and to skipped or inserted splicing events is possible.
All these search options can be combined in one query.
The result of a query is summarized in a table with selectable links to the full information entries. The main features are listed in this table: the gene name, entry number, organism name and the number of splice sites (NSS). Detailed information is available by clicking on the entry name (Fig. 2).
Figure 2.

The result of a query is summarized in tabular form with selectable links to the full information entries. In this table, the main features are listed: the entry number, organisms name, gene name and the number of alternative splice sites (NSS).
Detailed information
For each ASform, we stored the following information from other databases: GenBank ID, CDS ID, Swiss-Prot ID (if available), length, taxonomy, mRNA entry name. The calculated alternative splice site profile denotes the number of alternatively or constitutively spliced ESTs as mentioned, as well as the number of alternative splice sites. In the splice site view, a graphical overview of the location of the matching (alternatively spliced) ESTs is given. The EST information (tissue type, disease status and developmental stage) can be obtained for the whole transcript (section ‘Alternative Splice Frequency’) or for each splice site separately. The alternative splice event is classified by its length, the type (skip or insert) and the localization of the HSP compared with the exon–intron boundaries. To ease searching for the most interesting information, a color code for the tissue types and developmental stages and a bar code for the number of matching ESTs were established (also see http://eased.bioinf.mdc-berlin.de/eased_legend.html) (Fig. 3a and b).
Figure 3.


(a) The detailed view of an entry in the EASED database for the gene aldehyde dehydrogenase. In the section ‘General Information’, selected features (gene ID, description, organism, transcript information and similarity matches) from the Ensembl database are shown. The second section describes ‘Alternative Splice Frequency’ for the whole transcript, i.e. for all alternative splice sites. It displays the alternative, as well as constitutive splice profiles with respect to cancer and to distinct tissues and developmental stages. Each color represents a certain tissue and developmental stage. The number of bars in the lower area of the chart correlates with the number of ESTs. The ‘Splice Site View’ gives an illustration of the localization of the splice site events within the genomic sequence. The inserted or skipped events are shown in red. In the example presented, the following types of alternative splice events were found: (i) an alternative 3′ splice site that leads to an inserted intron sequence, (ii) alternative 5′ and 3′ that leads to two truncated exons, (iii) + (iv) completely skipped exons and (v) an alternative 5′ splice site with a truncated exon and a skipped exon. (b) The detailed view of an entry in the EASED database for the gene aldehyde dehydrogenase (continued). In contrast to the information in ‘Alternative Splice Frequency’ (a) the section ‘Splice Site Profile’ describes the ASP for each splice site separately. The color and bar codes are similar to those in (a).
FUTURE DIRECTIONS
EASED is an ongoing project. The features mentioned (ASP, tissue type, developmental stage, disease status and exon–intron information), which describe human sequences, will be added for the eight other organisms in the near future. A number of new features are currently in development to expand the scope and usability of the resource. To this end, the algorithm that predicts potential ASforms will be improved and upgraded. As an important feature, it is planned to add evolutionary information, which will enable crosslinking of results from orthologous genes.
CONCLUSIONS
The EASED project is establishing a comprehensive database of alternatively spliced mRNAs from the (freely accessible) sequence pool of humans and eight model organisms. At the time of writing, EASED consists of nearly 30 000 alternatively spliced transcripts.
Moreover, EASED includes useful biological information, e.g. tissue type and developmental stage notation. This can be useful to biologists in several ways. Its main advantage is in providing the possibility to search for biologically relevant data. This feature is not yet included in any other alternative splice database and facilitates, e.g. extended statistical studies.
Another focus of EASED relates to finding candidate genes for the origin of diseases. Using combined query parameters (e.g. the number of ESTs expressed in cancer tissue and in a certain tissue) enables the user to filter out sequences of interest. As a result of the parametrization of the alternative splice profile, a ranking of the queried sequences is possible. EASED will be updated regularly and will be extended in the coming months.
AVAILABILITY
EASED is freely available on the web at http://eased.bioinf.mdc-berlin.de/.
REFERENCES
- 1.McPherson J.D., Marra,M., Hillier,L., Waterston,R.H., Chinwalla,A., Wallis,J., Sekhon, M.,Wylie,K., Mardis,E.R., Wilson,R.K. et al. International Human Genome Sequencing Consortium (2001) A physical map of the human genome. Nature, 409, 934–941. [DOI] [PubMed] [Google Scholar]
- 2.Brett D., Pospisil,H., Valcarcel,J., Reich,J. and Bork,P. (2002) Alternative splicing and genome complexity. Nature Genet., 30, 29–30. [DOI] [PubMed] [Google Scholar]
- 3.Mironov A.A., Fickett,J.W. and Gelfand,M.S. (1999) Frequent alternative splicing of human genes. Genome Res., 9, 1288–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thanaraj T.A. (1999) A clean data set of EST-confirmed splice sites from Homo sapiens and standards for clean-up procedures. Nucleic Acids Res., 27, 2627–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coward E., Haas,S.A. and Vingron,M. (2002) SpliceNest: visualization of gene structure and alternative splicing based on EST clusters. Trends Genet., 18, 53–55. [Google Scholar]
- 6.Huang Y.H., Chen,Y.T., Lai,J.J., Yang,S.T. and Yang,U.C. (2002) PALS db: Putative Alternative Splicing database. Nucleic Acids Res., 30, 186–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thanaraj T.A., Clark,F. and Muilu,J. (2003) Conservation of human alternative splice events in mouse. Nucleic Acids Res., 31, 2544–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kan Z., Rouchka,E.C., Gish,W.R. and States D.J. (2001) Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res., 11, 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Modrek B., Resch,A., Grasso,C. and Lee,C. (2001) Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res., 29, 2850–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee C., Atanelov,L., Modrek,B. and Xing,Y. (2003) ASAP: the Alternative Splicing Annotation Project. Nucleic Acids Res., 31, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croft L., Schandorff,S., Clark,F., Burrage,K., Archtander,P. and Mattick,J.S. (2000) ISIS, the intron information system, reveals the prevalence of alternative splicing in the human genome. Nature Genet., 24, 340–341. [DOI] [PubMed] [Google Scholar]
- 12.Kent W.J. and Zahler,A.M. (2000) The intronerator: exploring introns and alternative splicing in Caenorhabditis elegans. Nucleic Acids Res., 28, 91–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Modrek B. and Lee,C. (2002) A genomic view of alternative splicing. Nature Genet., 30, 13–19. [DOI] [PubMed] [Google Scholar]
- 14.Benson D.A., Karsch-Mizrachi,I., Lipman,D.J., Ostell,J. and Wheeler,D.L. (2003) GenBank. Nucleic Acids Res., 31, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Altschul S.F., Madden,T.L., Schäffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clamp M., Andrews,D., Barker,D., Bevan,P., Cameron,G., Chen,Y., Clark,L., Cox,T., Cuff,J., Curwen,V. et al. (2003) Ensembl 2002: accommodating comparative genomics. Nucleic Acids Res., 31, 38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bedell J.A., Korf,I. and Gish,W. (2000) MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics, 16, 1040–1041. [DOI] [PubMed] [Google Scholar]