

Abstract
Biological and positional evidence supports the involvement of the GAD1 and distal-less homeobox genes (DLXs) in the etiology of autism. We investigated 42 SNPs in these genes as risk factors for autism spectrum disorders (ASD) in a large family-based association study of 715 nuclear families. No single marker showed significant association after correction for multiple testing. A rare haplotype in the DLX1 promoter was associated with ASD (p-value = 0.001). Given the importance of rare variants to the etiology of autism revealed in recent studies, the observed rare haplotype may be relevant to future investigations. Our observations, when taken together with previous findings, suggest that common genetic variation in the GAD1 and DLX genes is unlikely to play a critical role in ASD susceptibility.
Keywords: Autism spectrum disorder, genetic association, candidate gene study, DLX homeobox, GAD1
Autism spectrum disorders (ASD) are heterogeneous and complex developmental disorders characterized by pervasive impairments in social interactions, reciprocal communication, and repetitive and stereotyped patterns of behaviors and interests. Genetic factors are thought to play a significant role in the manifestation of ASD (Folstein and Rutter 1977; Steffenburg et al. 1989; Bailey et al. 1995) with complex inheritance (Risch et al. 1999; Folstein and Rosen-Sheidley 2001; Santangelo and Tsatsanis 2005), but linkage and candidate gene association studies have had limited success in identifying or replicating common genetic variants.
One intriguing model of autism etiology posits an imbalance between excitation and inhibition in the neural circuits that mediate language development and social behaviors (Rubenstein and Merzenich 2003). A hyperexcitable state of the brain could result from multiple molecular and genetic mechanisms underlying abnormal neurotransmitter and receptor systems, neural development, and regulators of gene expression in the developing cortex (Rubenstein and Merzenich 2003). Among these, the glutamic acid decarboxylase 1 (GAD1) gene and the distal-less homeobox (DLX) genes are attractive candidates based on their biological functions.
The GAD1 gene encodes 67kDa glutamic acid decarboxylase isoform (GAD67), the rate-limiting enzyme responsible for γ-aminobutyric acid (GABA) biosynthesis from glutamatic acid and the major GAD isoform in the human brain for early brain development (Feldblum et al. 1993). A good deal of evidence has accumulated indicating that disturbance in the GABAergic (inhibitory) or glutamatergic (excitatory) signaling system or the imbalance between both, including altered neurotransmitter levels, receptor levels and receptor polymorphisms may be involved in the etiology of autism (Blatt et al. 2001; Hussman 2001; Menold et al. 2001; Purcell et al. 2001; Buxbaum et al. 2002; Dhossche et al. 2002; Jamain et al. 2002; Rubenstein and Merzenich 2003; Shuang et al. 2004; Ma et al. 2005; Collins et al. 2006; Kim et al. 2006; Shinohe et al. 2006; Strutz-Seebohm et al. 2006; DeVito et al. 2007), as the GABAergic system plays an important role in regulating the migration of neuronal percursors, acceleration of neural maturation, and learning, while the glutamatergic system is crucial in brain formation and information processing. The mechanism by which GAD1 may contribute to autism risk is not well understood, but a significant decrease (40-61%) of the key synthesizing enzyme GAD67 mRNA and protein levels in parietal and cerebellar cortices, specifically in Purkinje cells (Fatemi et al. 2002; Yip et al. 2007), has been demonstrated in postmortem studies of autistic brains.
DLX genes encode homeodomain transcription factors which are key regulators in forebrain and basal ganglia development. In the mammalian genome, six DLX genes are arranged in three coherently transcribed bigene pairs (i.e., DLX1/2, DLX3/4, and DLX5/6). They are primarily expressed in the basal ganglia, reticular nucleus of the thalamus, and cortical local circuit neurons, the regions that regulate forebrain memory and the motor system, as well as the amygdala and hypothalamus, the regions that participate in modulating emotional and social behaviors (Rubenstein and Merzenich 2003). Sequence comparisons of mouse and zebrafish reveal that the Dlx3/4 cluster has a more distant phylogenetic relationship and diverse expression pattern relative to the other two Dlx clusters, which are extensively expressed during development of the majority of forebrain GABAergic neurons (Panganiban and Rubenstein 2002). The first evidence for expression of either Dlx3 or Dlx4 in the CNS of a vertebrate was only recently demonstrated (Zhu and Bendall 2006).
The GAD1 and DLX1/2 genes are located on chromosome 2q31, and the DLX3/4 and DLX5/6 clusters reside on chromosomes 17q21 and 7q22, respectively. Intriguingly, all of these regions coincide with linkage peaks in several genome-wide linkage scans for autism (2001a; 2001b; Cantor et al. 2005), providing positional support for their potential involvement. Nevertheless, association studies of these genes are few, with mixed results, and almost all of them examined only one or a few single nucleotide polymorphisms (SNPs) in one gene or were restricted to rare variations in the coding regions. Genetic variation of DLX3/4 has not yet been examined for ASD association.
Knockout mouse models showed no apparent phenotypic abnormalities in mice homozygous for one Dlx mutant allele when other Dlx genes were still normally co-expressed in the same regions, but detectable phenotypic changes were observed when mice lacked at least two functional Dlx genes, suggesting biological epistasis between various combinations of mutations in different Dlx genes may be important (Panganiban and Rubenstein 2002; Depew et al. 2005). Moreover, the expression of GAD67 and GAD65 (another GAD isoform encoded by the GAD2 gene) have been observed to be controlled by the Dlx genes. For example, in Dlx1/2 double mutant mice, GAD67 expression is abolished, and differentiation and migration defects in the development of telencephalic GABAergic neurons have been detected (Anderson et al. 1997a; Anderson et al. 1997b; Pleasure et al. 2000). Further, ectopic Dlx2 and Dlx5 expression in the developing cerebral cortex was shown to induce expressions of both GAD genes (Stuhmer et al. 2002). Both loss- and gain-of-function experiments illustrate the role of Dlx genes in regulating GAD expression and their involvement in controlling the specification, differentiation, and migration of forebrain GABAergic neurons. In aggregate, these data indicate that combinations of functional mutations in GAD1 and DLX genes may alter the inhibition/excitation ratio in some parts of the brain. Therefore, we chose to investigate the individual and joint effects of GAD1, DLX1/2, DLX3/4, and DLX5/6 genes on ASD susceptibility.
The sample was comprised of 2,837 individuals from the Autism Genetic Resource Exchange (AGRE) who were included in a genomewide association scan (GWAS) (Weiss et al. 2009). Most of the sample has a nuclear family structure with ≥ 2 affected offspring. The male to female ratio among 1,372 ASD cases is nearly 4:1. Families with fragile X syndrome and known chromosomal abnormalities were excluded.
The primary phenotype in the study was autism spectrum disorders (ASD) (coded as a binary variable), assessed using the Autism Diagnostic Interview-Revised (ADI-R) (Lord et al. 1994). Six ASD cases in the sample lacked ADI-R questionnaire data so their phenotypic status was defined by their score on the Autism Diagnostic Observation Schedule (ADOS) (Lord et al. 2000). Because some previous studies have found stronger linkage signals to language delay phenotypes than for diagnostic status (Bradford et al. 2001; Buxbaum et al. 2001; Alarcon et al. 2002; Alarcon et al. 2005), we also investigated two qualitative language phenotypes, “delayed acquisition of first words” and “delayed acquisition of phrase speech”, derived from items in the ADI-R. Children were coded as being delayed in single word use if they began using single words later than 24 months; the age cutoff for delayed phrase speech was 33 months.
Because this candidate gene study builds on a GWAS investigation (Weiss et al. 2009), two sets of SNPs were analyzed. Quality control was conducted separately on each set of SNPs before combining the data. The first panel included all SNPs (N=17) from the genomic regions of GAD1 and DLX1/2, DLX3/4, DLX5/6 genes in the GWAS. But because the GWAS was designed to capture genetic variation across the entire genome, there was not sufficient coverage of any of our candidate genes. Hence a second panel of pairwise tag SNPs was selected to provide adequate coverage (r2 > 0.8) using the haplotype structure of the 30 HapMap CEPH trio samples, as the majority of the ASD families are of European descent. Thirty SNPs that were in Hardy-Weinberg equilibrium (HWE) among the founders and had minor allele frequency (MAF) ≥ 5% were selected for genotyping on the Sequenom iPLEX platform (Sequenom Inc., San Diego, CA).
Quality filtering and dataset merging processes were conducted using the PLINK toolset (Purcell et al. 2007). Twenty-seven individuals and 5 SNPs were ultimately discarded from the analyses because they failed at least one of the final quality control criteria, including MAF > 3%, SNP genotyping call rate > 90%, individual genotyping success rate > 80%, family and SNP Mendelian error rate < 5% and < 1%, respectively, and HWE p-value > 0.001. The overall genotyping accuracy was 99.0%. The final analyzed dataset includes 42 SNP markers (first panel: 15, second panel: 27) and 2,810 subjects from 715 nuclear families.
Tests for single-marker allelic associations were carried out using the Family-Based Association Test (FBAT) program (Laird et al. 2000) under the null hypothesis (H0) of “no linkage and no association.” We also used empirical variance estimation (fbat -e option) to account for potential linkage in a secondary analysis. Haplotypic associations with 2-, 3-, and 4-marker sliding windows were examined using the “hbat -e” function in FBAT. Global and individual haplotype tests of association were performed under the “multiallelic” and “biallelic” modes, respectively. All association tests were performed under an additive model.
Given the evidence of biological epistasis between DLX and GAD1 genes in studies of knockout mice, we also screened for potential multi-locus (2- and 3-locus) interactions using the Multifactor Dimensionality Reduction Pedigree Disequilibrium Test (MDR-PDT) program (Martin et al. 2006), a within-family measure of association between genotype and disease. Statistical significance of the maximum n-locus MDR-PDT statistic is determined by permutation testing.
To correct for multiple testing of single-markers, the effective number of independent tests was determined using the method proposed by Nyholt (2004) and modified by Li and Ji (2005), as implemented in the SNP Spectral Decomposition (SNPSpD) software. Following this procedure, the 42 SNPs examined here are equivalent to 32 independent tests, yielding a corrected significance level of 0.00156 to reject H0.
Results of single-marker association tests were shown in Table 1. Eight markers had nominal evidence of association with ASD in either GAD1 or DLX5/6, with the smallest p-value observed at marker rs2241164 (p = 0.0089) in intron 4 of GAD1. This marker is one of four SNPs examined in GAD1 that span 20 kb from introns 4 to 12 and have a moderate to high pairwise correlation with each other (r2=0.68-0.95), suggesting that these SNPs may tag the same disease susceptibility allele. Results were similar for the delayed language development subphenotypes (Table 1). Although the observed number of SNPs with nominal p-values < 0.05 (8 SNPs) is greater than expected (3 SNPs), none of the single-marker tests remain statistically significant after correction. Results are similar when taking potential linkage into account and when restricting the analyses to individuals of European descent (data not shown).
Table 1. Results of Single Marker Association Tests.
| CHR | SNP rs# | MAF | SNP position (UCSC hg17) |
Closest gene | Gene Region | PASD | PDWORD | PDPHRASE |
|---|---|---|---|---|---|---|---|---|
| 2 | rs6755814 | 0.390 | 171492504 | GAD1 | Promoter | 0.4650 | 0.9571 | 0.4124 |
| 2 | rs3791878 | 0.313 | 171497698 | GAD1 | Promoter | 0.0475 | 0.1891 | 0.0700 |
| 2 | rs3762555 | 0.242 | 171497902 | GAD1 | Promoter | 0.2930 | 0.9290 | 0.8287 |
| 2 | rs3828275 | 0.419 | 171508247 | GAD1 | Intron 3 | 0.6535 | 0.5649 | 0.4928 |
| 2 | rs2241164 | 0.335 | 171512066 | GAD1 | Intron 4 | 0.0089 | 0.0357 | 0.0111 |
| 2 | rs10191129 | 0.247 | 171514100 | GAD1 | Intron 5 | 0.2887 | 0.6306 | 0.3836 |
| 2 | rs769407 | 0.253 | 171519215 | GAD1 | Intron 6 | 0.0128 | 0.0419 | 0.0054 |
| 2 | rs7561581 | 0.305 | 171521979 | GAD1 | Intron 6 | 0.0173 | 0.0489 | 0.0150 |
| 2 | rs4668331 | 0.252 | 171531754 | GAD1 | Intron 12 | 0.0139 | 0.0455 | 0.0064 |
| 2 | rs16858996 | 0.122 | 171544609 | GAD1 | Downstream | 0.1025 | 0.8545 | 0.5627 |
| 2 | rs17701824 | 0.433 | 171544753 | GAD1 | Downstream | 0.1227 | 0.1192 | 0.0739 |
| 2 | rs4389303 | 0.094 | 171544949 | GAD1 | Downstream | 0.8041 | 0.4937 | 0.4831 |
| 2 | rs788160 | 0.191 | 172771822 | DLX1 | Promoter | 0.9097 | 0.9488 | 0.8957 |
| 2 | rs1047889 | 0.422 | 172772034 | DLX1 | Promoter | 0.3109 | 0.4214 | 0.1845 |
| 2 | rs813720 | 0.350 | 172780248 | DLX1 | Downstream; intergenic | 0.6581 | 0.3154 | 0.4776 |
| 2 | rs10186317 | 0.280 | 172780518 | DLX1 | Downstream; intergenic | 0.2137 | 0.1403 | 0.2673 |
| 2 | rs13390848 | 0.195 | 172783150 | DLX1 | Downstream; intergenic | 0.9272 | 0.9877 | 0.6817 |
| 2 | rs2357322 | 0.339 | 172798335 | DLX2 | Promoter | 0.3293 | 0.9672 | 0.5198 |
| 2 | rs2016394 | 0.449 | 172798478 | DLX2 | Promoter | 0.1754 | 0.4183 | 0.3946 |
| 7 | rs886583 | 0.053 | 96269930 | DLX6 | Promoter | 0.0276 | 0.0172 | 0.0157 |
| 7 | rs17657924 | 0.428 | 96270240 | DLX6 | Promoter | 0.8178 | 0.7109 | 0.5555 |
| 7 | rs6957108 | 0.179 | 96270770 | DLX6 | Promoter | 0.6078 | 0.4701 | 0.6792 |
| 7 | rs11762736 | 0.344 | 96275288 | DLX6 | Promoter | 0.3065 | 0.1185 | 0.4159 |
| 7 | rs1005169 | 0.044 | 96287576 | DLX6 | Downstream; intergenic | 0.0736 | 0.0262 | 0.0494 |
| 7 | rs1207731 | 0.185 | 96294007 | DLX5 | Downstream; intergenic | 0.4725 | 0.1828 | 0.6017 |
| 7 | rs1207733 | 0.045 | 96296057 | DLX5 | Intron (boundary) | 0.0390 | 0.0076 | 0.0230 |
| 7 | rs9769385 | 0.056 | 96296636 | DLX5 | Intron 12 | 0.0892 | 0.2509 | 0.2885 |
| 7 | rs1207735 | 0.303 | 96300377 | DLX5 | Promoter | 0.9830 | 0.8499 | 0.9611 |
| 7 | rs6960249 | 0.419 | 96304783 | DLX5 | Promoter | 0.5898 | 0.2664 | 0.6090 |
| 7 | rs1207739 | 0.071 | 96304843 | DLX5 | Promoter | 0.2206 | 0.9539 | 0.1023 |
| 7 | rs17600042 | 0.069 | 96306041 | DLX5 | Promoter | 0.0295 | 0.1853 | 0.0447 |
| 17 | rs12453270 | 0.446 | 45391038 | DLX4 | Promoter | 0.2381 | 0.3679 | 0.2397 |
| 17 | rs985626 | 0.077 | 45395340 | DLX4 | Promoter | 0.1845 | 0.2225 | 0.2198 |
| 17 | rs919089 | 0.276 | 45402420 | DLX4 | Intron 1 | 0.4369 | 0.3612 | 0.5325 |
| 17 | rs1058564 | 0.206 | 45406771 | DLX4 | 3′ UTR | 0.8464 | 0.5213 | 0.8950 |
| 17 | rs11079881 | 0.124 | 45412872 | DLX4 | Downstream | 0.0894 | 0.0335 | 0.0798 |
| 17 | rs11654766 | 0.206 | 45413823 | DLX4 | Downstream | 0.1515 | 0.1089 | 0.1550 |
| 17 | rs16948563 | 0.067 | 45420140 | DLX3 | Downstream; intergenic | 0.7637 | 0.7582 | 0.8382 |
| 17 | rs11871663 | 0.482 | 45420289 | DLX3 | Downstream; intergenic | 0.7357 | 0.3123 | 0.8862 |
| 17 | rs3891034 | 0.189 | 45425224 | DLX3 | Intron 2 | 0.7421 | 0.1327 | 0.2091 |
| 17 | rs11079884 | 0.129 | 45431434 | DLX3 | Promoter | 0.7179 | 0.1845 | 0.4963 |
| 17 | rs1991293 | 0.068 | 45433948 | DLX3 | Promoter | 0.2727 | 0.6935 | 0.3877 |
ASD: autism spectrum disorder; DWORD: delayed acquisition of first words; DPHRASE: delayed phrase speech
The best haplotype results, using sliding windows of 2 to 4 neighboring markers in each gene, are shown in Table 2. A 2-marker haplotype (rs788160-rs1047889) in the promoter region of DLX1 revealed the strongest association with ASD, with a global p-value of 0.0059. However, none of the 3 common haplotypes (>5%) had an individual p-value < 0.05, suggesting that the effect was due to the remaining rare haplotype, rs788160-rs1047889 (T-C), which yielded an individual haplotype p-value = 0.00098. The minimal-p test that evaluates the maximally significant statistic of the four single haplotype tests also yielded a significant p-value of 0.000012. However, this result should be interpreted with caution due to the rare frequency of this haplotype, estimated at 0.5%. Although several other haplotypes also had nominal p-values < 0.05, none of them yield associations stronger than the single-marker effects. No significant MDR-PDT multi-locus interactions were identified after correction for multiple testing by permutation.
Table 2. Top Results of Haplotype Associations with ASD in 2-, 3-, and 4-Marker Sliding Windows.
| Gene | Markers | Allele(s)* | Freq* | PIndividual* | PGlobal |
|---|---|---|---|---|---|
| GAD1 | rs2241164-rs10191129 | G-G | 0.335 | 0.0086 | 0.0314 |
| rs769407-rs7561581-rs4668331 | C-A-A | 0.696 | 0.0125 | 0.0233 | |
| rs10191129-rs769407-rs7561581-rs4668331 | G-G-G-T | 0.251 | 0.0157 | 0.0520 | |
| DLX1/2 | rs788160-rs1047889 | T-T | 0.187 | 0.3084 | 0.0059 |
| rs788160-rs1047889-rs813720 | T-T-G | 0.181 | 0.1217 | 0.0250 | |
| rs788160-rs1047889-rs813720-rs10186317 | T-T-G-A | 0.182 | 0.1304 | 0.6619 | |
| DLX3/4 | rs11654766-rs16948563 | G-C | 0.140 | 0.0605 | 0.3005 |
| rs11654766-rs16948563-rs11871663 | G-C-T | 0.109 | 0.0201 | 0.1874 | |
| rs12453270-rs985626-rs919089-rs1058564 | A-C-A-C | 0.536 | 0.1187 | 0.1571 | |
| DLX5/6 | rs1207733-rs9769385 | C-C | 0.901 | 0.0088 | 0.0301 |
| rs1207731-rs1207733-rs9769385 | T-C-C | 0.716 | 0.0136 | 0.0360 | |
| rs1207735-rs6960249-rs1207739-rs17600042 | G-A-C-C | 0.216 | 0.0511 | 0.0304 |
The allele combination, frequency, and p-value of the individual haplotype that yields the smallest p-value in the specific n-marker sliding window of the gene.
To the best of our knowledge, this is the largest and most comprehensive investigation of common genetic variation in the entire DLX gene family and the GAD1 gene, and the first study to examine DLX3/4 effects in relation to ASD. Several earlier investigations examined one single SNP, rare nonsynonymous SNPs, and/or SNPs identified by mutation screening or in non-coding ultraconserved elements in one or some DLX genes (Bacchelli et al. 2003; Nabi et al. 2003; Rabionet et al. 2004; Hamilton et al. 2005; Richler et al. 2006), and two previous studies tested GAD1 as a candidate gene by one or a few single markers (Rabionet et al. 2004; Buttenschon et al. 2009), but most found no significant associations with ASD. Unlike these studies, we applied a comprehensive tagging approach to adequately cover all the common genetic variation in the genes, utilized haplotype analysis, which enables the testing of omitted variants indirectly, and examined potential cross-locus and cross-gene interactions. Nevertheless, consistent with these earlier studies, our findings do not support the involvement of common variation in the DLX and GAD1 genes in ASD susceptibility.
During preparation of this manuscript, a study examining the effect of DLX1/2 genes on ASD risk using 6 tag SNPs in multiplex families from both the United States (AGRE) and Canada (ASD-CARC) was published, and showed significant allelic and haplotypic associations of 4 common alleles (Liu et al. 2009). Although one of these markers, rs813720, was directly genotyped in our study, we observed no association with ASD at this locus (uncorrected p = 0.66). We did not directly genotype the other three SNPs, but the LD structure in the region showed that two markers genotyped in our study, rs813720 and rs10186317, were highly correlated with these three markers (r2=0.81-0.92) (Figure 1); however, neither were significantly associated with ASD in our sample (uncorrected p > 0.2). A closer look at the single SNP results reported by Liu et al. (2009) showed the significant allelic associations they found at rs788172, rs788173, and rs813720 were only observed in the ASD-CARC sample whereas significant association at rs4519482 was only seen in the AGRE sample (Table 1 in (Liu et al. 2009)). The authors speculated that these differences in association could be due to the different types of families included in the two samples since, in their study, AGRE families were chosen if they had few or no unaffected males, while that criterion was not applied to the selection of ASD-CARC families (personal communication). In contrast, our study included virtually all idiopathic ASD families that were in the AGRE repository at the time our study began. We did not identify any significant effects when restricting our analysis to either male-affected trios or male-affected-only families. Based on the available information, we were unable to replicate the positive findings by Liu and colleagues in our large sample of unselected AGRE families.
Figure 1.
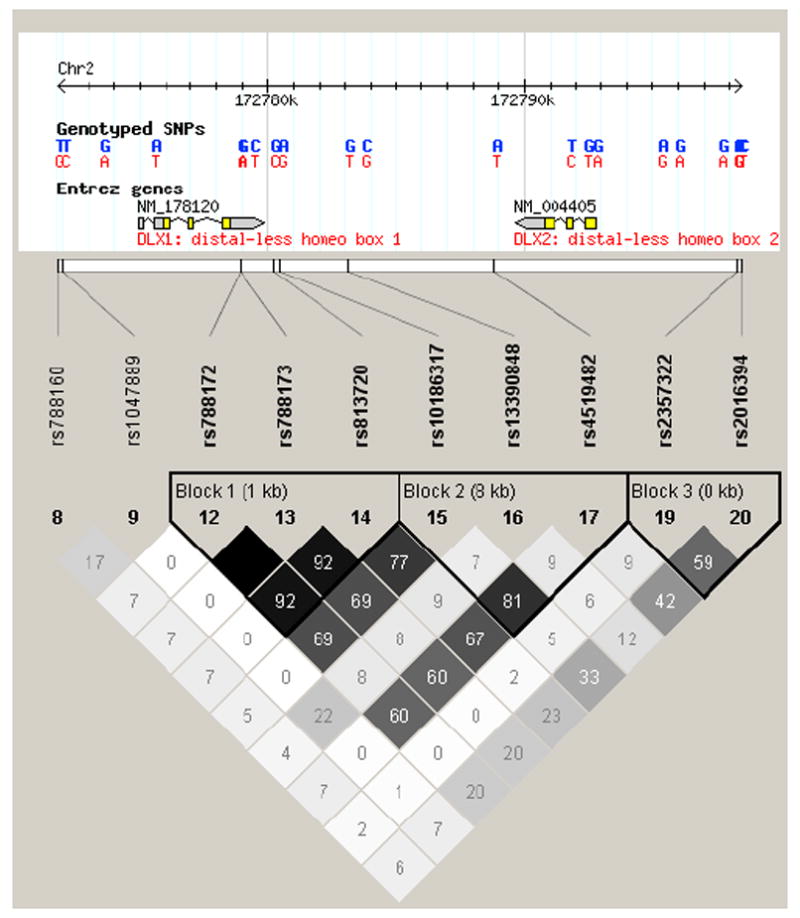
Linkage disequilibrium (LD) map of the DLX1/2 genes. Pairwise LD between SNPs was estimated using r2 (to measure correlation) with Haploview. SNP rs743605 is no longer in HapMap database.
Some limitations of our study should be considered. First, we used the tagging approach to select SNPs from the HapMap database, which is not optimal for identification of rare variants; we did not study nonsynonymous SNPs annotated in the dbSNP database. Hamilton and colleagues (Hamilton et al. 2005) carried out resequencing of the exons, exon/intron boundaries, and known enhancers of the DLX-1, -2, -5, and -6 genes. Although they identified several rare nonsynonymous variants, they were unable to provide statistically meaningful inferences, due to the small number of samples they examined.
Another potential limitation involves the phenotypes. The ASD qualitative outcomes are all derived from the ADI-R data which are available for virtually all affected individuals in the AGRE dataset. Only 77% of them also had ADOS data available, and discrepancies in diagnosis are often observed between the two assessments (for example, 6% of ASD defined by ADI-R did not meet either autism or spectrum using ADOS data). To mitigate this issue, two experienced reviewers (SLS and RS) carefully reviewed all relevant measures on a case by case basis to make the best consensus diagnosis for all probands with discrepant diagnoses. Individuals were excluded if their ADI-R and ADOS affection status classifications were widely discrepant and could not be further reconciled. The results were similar for the reviewed and original phenotype classifications.
The study has approximately 80% power to detect a difference corresponding to an odds ratio (OR) of 1.2 for α = 0.05, if the allele frequency in founders is between 0.2 and 0.65, assuming ASD prevalence of 0.6%, an additive genetic model, and complete LD between a genotyped and casual genetic variant (Lange et al. 2004). More specifically, the study has reasonable power to detect the effect of 8 SNPs that revealed nominal significance (power = 0.8 to detect the strongest association reported for rs2241164, for which OR = 1.2). However, the study is likely underpowered to detect smaller effects (OR < 1.2), despite the fact that it is by far the largest to examine the effects of GAD1 and DLX genetic variation on ASD susceptibility.
To summarize, this study does not support a major contribution of the DLX1/2, DLX3/4, DLX5/6 bigene clusters, or the GAD1 gene to autism susceptibility in a large family-based sample from the AGRE repository. However, we observed a rare two-marker haplotype in the promoter region of the DLX1 gene which was associated with ASD. Given the importance of rare variants in the etiology of autism in recent studies, the observed rare haplotype may be relevant to future investigations.
Acknowledgments
This work was supported by National Alliance for Autism Research (now merged with Autism Speaks) Pre-doctoral Mentor-based Training Fellowship.
We gratefully acknowledge support from Autism Speaks and the resources provided by the AGRE consortium and the participating AGRE families. The Autism Genetic Resource Exchange (AGRE) is a program of Autism Speaks and is supported, in part, by grant 1U24MH081810 from the National Institute of Mental Health to Clara M. Lajonchere (PI).
References
- Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Hum Mol Genet. 2001a;10:973–982. doi: 10.1093/hmg/10.9.973. [DOI] [PubMed] [Google Scholar]
- A genomewide screen for autism: strong evidence for linkage to chromosomes 2q, 7q, and 16p. Am J Hum Genet. 2001b;69:570–581. doi: 10.1086/323264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon M, Cantor RM, Liu J, Gilliam TC, Geschwind DH. Evidence for a language quantitative trait locus on chromosome 7q in multiplex autism families. Am J Hum Genet. 2002;70:60–71. doi: 10.1086/338241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon M, Yonan AL, Gilliam TC, Cantor RM, Geschwind DH. Quantitative genome scan and Ordered-Subsets Analysis of autism endophenotypes support language QTLs. Mol Psychiatry. 2005;10:747–757. doi: 10.1038/sj.mp.4001666. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997a;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Anderson SA, Qiu M, Bulfone A, Eisenstat DD, Meneses J, Pedersen R, Rubenstein JL. Mutations of the homeobox genes Dlx-1 and Dlx-2 disrupt the striatal subventricular zone and differentiation of late born striatal neurons. Neuron. 1997b;19:27–37. doi: 10.1016/s0896-6273(00)80345-1. [DOI] [PubMed] [Google Scholar]
- Bacchelli E, Blasi F, Biondolillo M, Lamb JA, Bonora E, Barnby G, Parr J, Beyer KS, Klauck SM, Poustka A, Bailey AJ, Monaco AP, Maestrini E. Screening of nine candidate genes for autism on chromosome 2q reveals rare nonsynonymous variants in the cAMP-GEFII gene. Mol Psychiatry. 2003;8:916–924. doi: 10.1038/sj.mp.4001340. [DOI] [PubMed] [Google Scholar]
- Bailey A, Le Couteur A, Gottesman I, Bolton P, Simonoff E, Yuzda E, Rutter M. Autism as a strongly genetic disorder: evidence from a British twin study. Psychol Med. 1995;25:63–77. doi: 10.1017/s0033291700028099. [DOI] [PubMed] [Google Scholar]
- Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord. 2001;31:537–543. doi: 10.1023/a:1013238809666. [DOI] [PubMed] [Google Scholar]
- Bradford Y, Haines J, Hutcheson H, Gardiner M, Braun T, Sheffield V, Cassavant T, Huang W, Wang K, Vieland V, Folstein S, Santangelo S, Piven J. Incorporating language phenotypes strengthens evidence of linkage to autism. Am J Med Genet. 2001;105:539–547. [PubMed] [Google Scholar]
- Buttenschon HN, Lauritsen MB, Daoud AE, Hollegaard M, Jorgensen M, Tvedegaard K, Hougaard D, Borglum A, Thorsen P, Mors O. A population-based association study of glutamate decarboxylase 1 as a candidate gene for autism. J Neural Transm. 2009;116:381–388. doi: 10.1007/s00702-008-0142-4. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Greenberg DA, Kilifarski M, Reichert J, Cook EH, Jr, Fang Y, Song CY, Vitale R. Association between a GABRB3 polymorphism and autism. Mol Psychiatry. 2002;7:311–316. doi: 10.1038/sj.mp.4001011. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Silverman JM, Smith CJ, Kilifarski M, Reichert J, Hollander E, Lawlor BA, Fitzgerald M, Greenberg DA, Davis KL. Evidence for a susceptibility gene for autism on chromosome 2 and for genetic heterogeneity. Am J Hum Genet. 2001;68:1514–1520. doi: 10.1086/320588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor RM, Kono N, Duvall JA, Alvarez-Retuerto A, Stone JL, Alarcon M, Nelson SF, Geschwind DH. Replication of autism linkage: fine-mapping peak at 17q21. Am J Hum Genet. 2005;76:1050–1056. doi: 10.1086/430278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Ma D, Whitehead PL, Martin ER, Wright HH, Abramson RK, Hussman JP, Haines JL, Cuccaro ML, Gilbert JR, Pericak-Vance MA. Investigation of autism and GABA receptor subunit genes in multiple ethnic groups. Neurogenetics. 2006;7:167–174. doi: 10.1007/s10048-006-0045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depew MJ, Simpson CA, Morasso M, Rubenstein JL. Reassessing the Dlx code: the genetic regulation of branchial arch skeletal pattern and development. J Anat. 2005;207:501–561. doi: 10.1111/j.1469-7580.2005.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVito TJ, Drost DJ, Neufeld RW, Rajakumar N, Pavlosky W, Williamson P, Nicolson R. Evidence for cortical dysfunction in autism: a proton magnetic resonance spectroscopic imaging study. Biol Psychiatry. 2007;61:465–473. doi: 10.1016/j.biopsych.2006.07.022. [DOI] [PubMed] [Google Scholar]
- Dhossche D, Applegate H, Abraham A, Maertens P, Bland L, Bencsath A, Martinez J. Elevated plasma gamma-aminobutyric acid (GABA) levels in autistic youngsters: stimulus for a GABA hypothesis of autism. Med Sci Monit. 2002;8:PR1–6. [PubMed] [Google Scholar]
- Fatemi SH, Halt AR, Stary JM, Kanodia R, Schulz SC, Realmuto GR. Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol Psychiatry. 2002;52:805–810. doi: 10.1016/s0006-3223(02)01430-0. [DOI] [PubMed] [Google Scholar]
- Feldblum S, Erlander MG, Tobin AJ. Different distributions of GAD65 and GAD67 mRNAs suggest that the two glutamate decarboxylases play distinctive functional roles. J Neurosci Res. 1993;34:689–706. doi: 10.1002/jnr.490340612. [DOI] [PubMed] [Google Scholar]
- Folstein S, Rutter M. Infantile autism: a genetic study of 21 twin pairs. J Child Psychol Psychiatry. 1977;18:297–321. doi: 10.1111/j.1469-7610.1977.tb00443.x. [DOI] [PubMed] [Google Scholar]
- Folstein SE, Rosen-Sheidley B. Genetics of autism: complex aetiology for a heterogeneous disorder. Nat Rev Genet. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- Hamilton SP, Woo JM, Carlson EJ, Ghanem N, Ekker M, Rubenstein JL. Analysis of four DLX homeobox genes in autistic probands. BMC Genet. 2005;6:52. doi: 10.1186/1471-2156-6-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussman JP. Suppressed GABAergic inhibition as a common factor in suspected etiologies of autism. J Autism Dev Disord. 2001;31:247–248. doi: 10.1023/a:1010715619091. [DOI] [PubMed] [Google Scholar]
- Jamain S, Betancur C, Quach H, Philippe A, Fellous M, Giros B, Gillberg C, Leboyer M, Bourgeron T. Linkage and association of the glutamate receptor 6 gene with autism. Mol Psychiatry. 2002;7:302–310. doi: 10.1038/sj.mp.4000979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SA, Kim JH, Park M, Cho IH, Yoo HJ. Association of GABRB3 Polymorphisms with Autism Spectrum Disorders in Korean Trios. Neuropsychobiology. 2006;54:160–165. doi: 10.1159/000098651. [DOI] [PubMed] [Google Scholar]
- Laird NM, Horvath S, Xu X. Implementing a unified approach to family-based tests of association. Genet Epidemiol. 2000;19 1:S36–42. doi: 10.1002/1098-2272(2000)19:1+<::AID-GEPI6>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM. PBAT: tools for family-based association studies. Am J Hum Genet. 2004;74:367–369. doi: 10.1086/381563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ji L. Adjusting multiple testing in multilocus analyses using the eigenvalues of a correlation matrix. Heredity. 2005;95:221–227. doi: 10.1038/sj.hdy.6800717. [DOI] [PubMed] [Google Scholar]
- Liu X, Novosedlik N, Wang A, Hudson ML, Cohen IL, Chudley AE, Forster-Gibson CJ, Lewis SM, Holden JJ. The DLX1and DLX2 genes and susceptibility to autism spectrum disorders. Eur J Hum Genet. 2009;17:228–235. doi: 10.1038/ejhg.2008.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH, Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord. 2000;30:205–223. [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord. 1994;24:659–685. doi: 10.1007/BF02172145. [DOI] [PubMed] [Google Scholar]
- Ma DQ, Whitehead PL, Menold MM, Martin ER, Ashley-Koch AE, Mei H, Ritchie MD, Delong GR, Abramson RK, Wright HH, Cuccaro ML, Hussman JP, Gilbert JR, Pericak-Vance MA. Identification of significant association and gene-gene interaction of GABA receptor subunit genes in autism. Am J Hum Genet. 2005;77:377–388. doi: 10.1086/433195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin ER, Ritchie MD, Hahn L, Kang S, Moore JH. A novel method to identify gene-gene effects in nuclear families: the MDR-PDT. Genet Epidemiol. 2006;30:111–123. doi: 10.1002/gepi.20128. [DOI] [PubMed] [Google Scholar]
- Menold MM, Shao Y, Wolpert CM, Donnelly SL, Raiford KL, Martin ER, Ravan SA, Abramson RK, Wright HH, Delong GR, Cuccaro ML, Pericak-Vance MA, Gilbert JR. Association analysis of chromosome 15 gabaa receptor subunit genes in autistic disorder. J Neurogenet. 2001;15:245–259. doi: 10.3109/01677060109167380. [DOI] [PubMed] [Google Scholar]
- Nabi R, Zhong H, Serajee FJ, Huq AH. No association between single nucleotide polymorphisms in DLX6 and Piccolo genes at 7q21-q22 and autism. Am J Med Genet B Neuropsychiatr Genet. 2003;119:98–101. doi: 10.1002/ajmg.b.10012. [DOI] [PubMed] [Google Scholar]
- Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004;74:765–769. doi: 10.1086/383251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panganiban G, Rubenstein JL. Developmental functions of the Distal-less/Dlx homeobox genes. Development. 2002;129:4371–4386. doi: 10.1242/dev.129.19.4371. [DOI] [PubMed] [Google Scholar]
- Pleasure SJ, Anderson S, Hevner R, Bagri A, Marin O, Lowenstein DH, Rubenstein JL. Cell migration from the ganglionic eminences is required for the development of hippocampal GABAergic interneurons. Neuron. 2000;28:727–740. doi: 10.1016/s0896-6273(00)00149-5. [DOI] [PubMed] [Google Scholar]
- Purcell AE, Jeon OH, Zimmerman AW, Blue ME, Pevsner J. Postmortem brain abnormalities of the glutamate neurotransmitter system in autism. Neurology. 2001;57:1618–1628. doi: 10.1212/wnl.57.9.1618. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabionet R, Jaworski JM, Ashley-Koch AE, Martin ER, Sutcliffe JS, Haines JL, Delong GR, Abramson RK, Wright HH, Cuccaro ML, Gilbert JR, Pericak-Vance MA. Analysis of the autism chromosome 2 linkage region: GAD1 and other candidate genes. Neurosci Lett. 2004;372:209–214. doi: 10.1016/j.neulet.2004.09.037. [DOI] [PubMed] [Google Scholar]
- Richler E, Reichert JG, Buxbaum JD, McInnes LA. Autism and ultraconserved non-coding sequence on chromosome 7q. Psychiatr Genet. 2006;16:19–23. doi: 10.1097/01.ypg.0000180683.18665.ef. [DOI] [PubMed] [Google Scholar]
- Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, et al. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–267. doi: 10.1034/j.1601-183x.2003.00037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo SL, Tsatsanis K. What is known about autism: genes, brain, and behavior. Am J Pharmacogenomics. 2005;5:71–92. doi: 10.2165/00129785-200505020-00001. [DOI] [PubMed] [Google Scholar]
- Shinohe A, Hashimoto K, Nakamura K, Tsujii M, Iwata Y, Tsuchiya KJ, Sekine Y, Suda S, Suzuki K, Sugihara G, Matsuzaki H, Minabe Y, Sugiyama T, Kawai M, Iyo M, Takei N, Mori N. Increased serum levels of glutamate in adult patients with autism. Prog Neuropsychopharmacol Biol Psychiatry. 2006;30:1472–1477. doi: 10.1016/j.pnpbp.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Shuang M, Liu J, Jia MX, Yang JZ, Wu SP, Gong XH, Ling YS, Ruan Y, Yang XL, Zhang D. Family-based association study between autism and glutamate receptor 6 gene in Chinese Han trios. Am J Med Genet B Neuropsychiatr Genet. 2004;131:48–50. doi: 10.1002/ajmg.b.30025. [DOI] [PubMed] [Google Scholar]
- Steffenburg S, Gillberg C, Hellgren L, Andersson L, Gillberg IC, Jakobsson G, Bohman M. A twin study of autism in Denmark, Finland, Iceland, Norway and Sweden. J Child Psychol Psychiatry. 1989;30:405–416. doi: 10.1111/j.1469-7610.1989.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Strutz-Seebohm N, Korniychuk G, Schwarz R, Baltaev R, Ureche ON, Mack AF, Ma ZL, Hollmann M, Lang F, Seebohm G. Functional significance of the kainate receptor GluR6(M836I) mutation that is linked to autism. Cell Physiol Biochem. 2006;18:287–294. doi: 10.1159/000097675. [DOI] [PubMed] [Google Scholar]
- Stuhmer T, Anderson SA, Ekker M, Rubenstein JL. Ectopic expression of the Dlx genes induces glutamic acid decarboxylase and Dlx expression. Development. 2002;129:245–252. doi: 10.1242/dev.129.1.245. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Arking DE, Daly MJ, Chakravarti A, Arking DE, Brune CW, West K, et al. A genome-wide linkage and association scan reveals novel loci for autism. Nature. 2009;461:802–808. doi: 10.1038/nature08490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip J, Soghomonian JJ, Blatt GJ. Decreased GAD67 mRNA levels in cerebellar Purkinje cells in autism: pathophysiological implications. Acta Neuropathol (Berl) 2007 doi: 10.1007/s00401-006-0176-3. [DOI] [PubMed] [Google Scholar]
- Zhu H, Bendall AJ. Dlx3 is expressed in the ventral forebrain of chicken embryos: implications for the evolution of the Dlx gene family. Int J Dev Biol. 2006;50:71–75. doi: 10.1387/ijdb.052087hz. [DOI] [PubMed] [Google Scholar]