

Abstract
Vibrational coherence spectroscopy (VCS) is used to investigate the low frequency dynamics of camphor-free and camphor-bound cytochrome P450cam (CYP 101) and its L358P mutant. The low frequency heme vibrations are found to be perturbed upon binding to the electron transfer partner putidaredoxin (Pdx). A strong correlation between the “detuned” vibrational coherence spectrum, which monitors frequencies between 100–400 cm−1, and the lower frequency part of the Raman spectrum is also demonstrated. The very low frequency region ≤ 200 cm−1, uniquely accessed by open-band VCS measurements, reveals a mode near 103 cm−1 in P450cam when camphor is not present in the distal pocket. This reflects the presence of a specific heme distortion, such as saddling or ruffling, in the substrate-free state where water is coordinated to the low-spin iron atom. Such distortions are likely to retard the rate of electron transfer to the substrate-free protein. The presence of strong mode near ~33 cm−1 in the camphor bound form suggests a significant heme doming distortion, which is supported by analysis using normal coordinate structural decomposition. Pdx also displays a strong coherent vibration near 30 cm−1 that could, in principle, be involved in vibrational resonance with its electron transfer target. A splitting of the 33 cm−1 feature and intensification of a mode near 78 cm−1 appear when the P450cam/Pdx complex is formed. These observations are consistent with vibrational mixing and heme geometric distortions upon Pdx binding that are coincident with the increased thiolate electron donation to the heme. The appearance of a mode near 65 cm−1 in the coherence spectra of the L358P mutant is comparable to the mode at 78 cm−1 seen in the P450cam/Pdx complex and is consistent with the view that the heme and its environment in the L358P mutant are similar to the Pdx-bound native protein. Resonance Raman spectra are presented for both P450cam and the L358P mutant and the changes are correlated with an increased amount of thiolate electron donation to the heme in the mutant sample.
Keywords: cytochrome P450, femtosecond vibrational coherence spectroscopy, CYP101 (L358P)mutant, putidaredoxin, low frequency modes, heme distort ion
Introduction
Cytochrome P450cam from the bacterium Pseudomonas putida (CYP101) is a heme protein monooxygenase that has been studied extensively to understand the catalytic mechanism of its superfamily, which is involved in drug metabolism, toxicity, xenobiotic degradation and biosynthesis1. A key distinctive feature of the structure of cytochrome P450cam is the coordination of a thiolate anion, from the proximal cysteine residue (Cys357) to the heme iron as a fifth ligand2;3. The active site of wild type P450cam, including heme and its axial ligand, Cys357, and the nearest proximal amino acids Leu358, Gly359 and Gln360 is shown along with d-camphor and Thr252 in Fig. 1. The major function of CYP101 is to catalyze the regio- and stereo-specific hydroxylation of its substrate, d-camphor, via oxygen atom insertion at the 5-exo position. The transfer of two electrons from NADH, which are needed for this process, is sequentially mediated by the flavin group of putidaredoxin reductase and the [2Fe-2S] centre of putidaredoxin (Pdx). The role of Pdx is unique because most low potential iron-sulfur proteins such as spinach ferredoxin and bovine adrenodoxin can donate the first electron but not the second electron. There have been a large number of kinetic and spectroscopic studies carried out to characterize the binding interaction between P450cam and Pdx in order to better understand the effector role of Pdx. It is thought that formation of the complex between P450cam and Pdx is mediated by electrostatic interactions 4–6, with a salt bridge between Asp38 of Pdx and Arg112 of P450camplaying a major role in the bin ding and electron transfer4;7.
Figure 1.
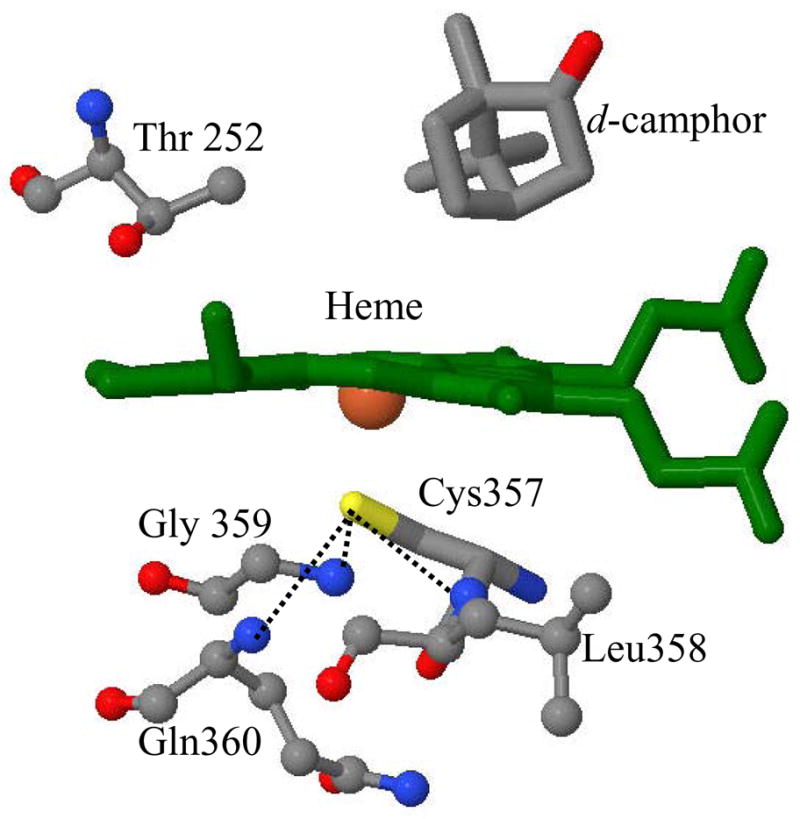
The crystal structure of the camphor bound cytochrome P450cam is shown for heme, its axial ligand Cys357 and nearby amino acids (coordinates taken from the protein data bank (PDB) 2CPP72). The position of substrate, d-camphor and Thr 252 are also shown. The dotted lines represent the three hydrogen bonds between the thiolate sulfur and amide nitrogens on the proximal side.
The spectral perturbations induced by formation of the P450cam/Pdx complex have been documented using different techniques, including EPR8;9, NMR10–12, resonance Raman13–15 and infrared16 spectroscopies. A perturbation of the heme iron-proximal Cys357 (Fe-S) bond upon P450cam/Pdx complex formation was observed by resonance Raman spectroscopy and revealed a ~3 cm−1 upshift of the Fe-S stretching mode, assigned to increased electron donation from the axial thiolate to the heme iron14. The increased electron donation was also supported by the observation that the Fe-CO and C-O stretching modes are shifted up and down respectively, upon binding of Pdx to P450cam-CO17. Unno et al.15 suggested that the electron donation of the proximal cysteine in the P450cam/Pdx complex is affected by changes in the P450cam protein conformation. Nagano et al.16 interpreted the enhanced infrared (IR) intensity of the CO stretching mode at 1932 cm−1 in the CO-P450cam/Pdx complex to increased electron back-donation from the proximal thiolate ligand, which could also facilitate the O2 scission reaction.
In order to investigate the Pdx-induced heme environmental changes in P450cam and the importance of hydrogen bonds that stabilize the axial thiolate (see Fig. 1), the L358P mutant of cytochrome P450cam was studied extensively18–21. In L358P, where leucine is replaced by proline, one of the amide hydrogen bonds with the thiolate ligand is removed. This reduces the stabilization of the negatively charged thiolate ligand and enhances electron donation to the heme iron. Yoshioka et al. 18;19 reported the lowering of the reduction potential by 36 mV, from −134 mV to −170 mV when L358P is compared to the wild type. This presumably reflects the same increased electron donation from the thiolate ligand in L358P that was inferred from the NMR spectra21. Evidently, a key function of the —NH---S− hydrogen bonding in P450cam (Fig. 1) is to regulate the thiolate electron donation to the heme iron, which must affect its reduction potential.
Crystal structures of the wild type and L358P mutant were compared for the ferrous and ferrous-CO bound forms20. In the L358P mutant, there were conformational rearrangements, such as the movement of Arg 112 that push the heme toward the substrate and ligand binding pocket, setting up a favorable heme-substrate position for regio-selective hydroxylation. Based on these observations, it was suggested that Pdx binding optimizes the distal pocket for monooxygenation of camphor. In the 1H NMR spectra of the L358P mutant10;21, the same trend of structural changes in heme environment was seen as in the Pdx bound wild type. This was based on chemical shift changes that reflect movements of the β-proton of the axial cysteine, the 9-methyl and 5-exo-protons of the camphor, and the γ-methyl group of Thr-252, relative to the heme. Although the magnitude of the structural changes was larger in wild type, the close pattern of chemical shift changes in the NMR spectra suggests that the heme environment of the L358P mutant mimics that of the Pdx-bound enzyme. It also suggests that Pdx binding acts as a structural trigger for thiolate electron donation to the oxygenated P450cam.
The aim of this study is to use vibrational coherence spectroscopy (VCS) to better understand how Pdx perturbs the active site of P450cam by inducing distortions of the heme geometry. The theoretical and experimental details of this nonlinear optical spectroscopy can be found elsewhere22–28. VCS is able to effectively monitor vibrational modes below ~ 200 cm−1 in the aqueous phase, which are very difficult to access using frequency domain vibrational spectroscopies such as infrared or Raman spectroscopy. At room temperature, kBT~200cm−1, these low frequency modes are thermally excited and are likely involved as reaction coordinates for protein function. For example, the doming mode near 40 cm−1 has been shown to play an important role in diatomic ligand binding to heme systems29;30. For electron transfer reactions, low frequency heme distortions, particularly along the ruffling and saddling modes, have been suggested to play a role in modulating both the heme reduction potential 31–36 and the reaction rates37. Such modes may turn out to be important thermally accessible reaction coordinates for electron transfer involving the heme. In this paper, we have used VCS to investigate substrate and Pdx binding to WT P450cam and to make comparisons to the L358P mutant.
Experimental
Sample preparation
Ferric cytochrome P450cam from Pseudomonas putida (CYP101) and its L358P mutant were prepared as discussed previously38. The mutant was generated using QuikChange XL site-directed mutagenesis of the His-tagged wild type CYP101 P450cam in a pET-28a vector. The mutated plasmid was transformed into the high competency XL-Gold E. coli, DNA was purified using the Qiagen Miniprep Kit and transformed into high expression BL-21 E. coli cells. All purified proteins had Rz (= A392/A280) values greater than 1.6. The ferric sample was generated by diluting the stock protein in potassium phosphate buffer (0.05 M, 0.15 M KCl, pH 7.4) to the required concentration. Pdx was expressed in E. coli strain RRI and was purified by previously described methods39 The purified Pdx had an absorbance at 325 nm relative to 280 nm that was at least 0.6. The buffer used for Pdx is 50 mM Tris/HCl, with 20 mM BME, at pH 7.4. For the vibrational coherence spectroscopy measurements, the concentration of the protein samples was adjusted so that the optical density of the sample at the pump wavelength was between 0.7 and 1.0 O.D. in a 1-mm path length spinning sample cell. The typical concentration was between 100 and 150 μM.
For obtaining the ferric complex of P450cam with Pdx, we used a molar concentration ratio of 1:1. Because KD~2.9 μM for Pdx dissociation40 and the concentrations of protein are on the order of 150 μM, the bound complex dominates. The ratio (r) of 1:1 bound protein complex to the free protein can be found using: r ≈ sqrt[C0/KD] − 1 ≈ 6 so that ~86% of the protein material should be bound in the protein-protein complex. However, there have been other reports of larger KD values41–44, which would suggest that a smaller amount of protein-protein complex might be present in the sample.
The absorption spectra of all samples were recorded after the preparation procedure as well as after the experiments to check for possible photochemical damage and none was observed.
Optical System
The experimental optical system has been described elsewhere27;45–47 and more details are given in the supporting information. The vibrational coherence data are recorded using two different detection geometries, each providing for optimal enhancement of a different range of frequencies. An open-band detection scheme, which integrates over the full probe pulse bandwidth, improves the fidelity of the low frequency response in the range 20–100 cm−1 23;27;48;49. The detuned detection scheme selectively enhances the higher frequency response in the coherent signal23;27;50–52 with improved reliability in the ~200–400 cm−1 region. The detuned detection is accomplished using a monochomator that detects 5 nm away from the carrier wavelength of the probe pulse with a bandwidth of 0.5 nm. The detuned results allow for direct comparison with frequency domain techniques like resonance Raman spectroscopy, which probe modes above 200 cm−1.
Data Analysis
The femtosecond time resolved pump -probe data contain a superposition of population transfer and vibrational coherence signals. To obtain the residual coherence signal, the monotonic population decay components must be removed. Digitization of the experimental signal is done using a lock-in amplifier (LIA) on a 24 bit scale, allowing for a sufficient dynamic range to detect the low amplitude coherence signals. The fractional change of transmittance, ΔT/T, for the open band oscillatory signals is on the order of 10−4–10−5. The analysis of data utilizes linear predictive singular value decomposition (LPSVD), which simultaneously fits the exponential decay for population dynamics and the damped cosine functions that describe the vibrational oscillations. We also employ a maximum entropy method (MEM) algorithm, which removes assumptions regarding the number of exponential decay processes, in order to retrieve the oscillatory signal with a second independent method. When the two methods agree, we are assured that the extracted low frequency oscillations are contained within the signal-to-noise of the experimental measurement.
Resonance Raman spectra
Resonance Raman spectra were obtained using a standard setup with a 90° light-collection geometry and a single grating monochromator (model No. 1870B; Spex Industries, Edison, NJ). An optical polarization scrambler was inserted in front of the monochromator to obtain the intensity of the scattered light without bias from the polarization-sensitive grating. The monochromator output was coupled to a thermoelectrically-cooled charge-coupled detector (PIXIS 400B, Princeton Instruments). In order to improve detection in the low-frequency domain of Raman shifts, an interferometric notch filter (Kaiser Optical Systems, Ann Arbor, MI) was used to extinguish the elastically and quasi-elastically scattered laser light. Samples were excited with a 413.1 nm laser line generated by a krypton laser (Innova 300, Coherent) with power of ~ 6 mW. A standard quartz cuvette (NSG Precision Cells, Farmingdale, NY) was used for the experimental Raman measurements. All Raman spectra were frequency calibrated using neat fenchone with ± 1 cm−1spectral resolution.
Results
Ferric cytochrome P450cam
The equilibrium absorption spectra of ferric cytochrome P450cam with and without camphor are shown in Fig. 2A. Cytochrome P450cam without camphor bound shows a strong Soret absorption band maximum at 417 nm that is typical for a 6-coordinate low-spin (6C/LS) ferric heme protein. When camphor is bound to P450cam, the Soret absorption maximum shifts from 417 nm to 391 nm. This reflects the formation of a five coordinate high-spin (5C/HS) species, due to expulsion of the heme -bound water from the distal pocket53.
Figure 2.

(A). The normalized equilibrium absorption spectra of ferric cytochrome P450cam in 50 mM KPi buffer with 150 mM KCl at pH 7.4. The absorption band peaking at 417 nm is the camphor-free form and the spectrum peaking at 391 nm is the camphor-bound form. (B). Normalized femtosecond time-resolved optical transmittance of cytochrome P450cam in the camphor-free (black) and camphor-bound (red) states. The excitation wavelength is 412 nm for camphor-bound (red) and 423 nm for camphor-free (black). The kinetic traces show a bleach followed by a recovery to equilibrium (ΔT > 0) and fitted with bi-exponential decay starting from 200 fs. The time constants (τ) and amplitudes (a) used as fitting parameters for the camphor free species are τ1= 56 fs (a1 = 0.79), τ2= 1.5 ps (a2 = 0.18) and offset = 0.02 and for camphor bound complex τ1= 140 fs (a1 = 0.36), τ2= 1.1 ps (a2 = 0.67) and offset = −0.03. The shortest time constant and its amplitude are distorted due to convolution with the coherence coupling signal.
The femtosecond time resolved optical transmittance (ΔT) of the camphor free and camphor bound ferric P450cam were obtained by exciting at 423 nm and 412 nm, respectively. Figure 2B shows the transient pump-probe population response for the above samples, which have a prompt bleach and coherence coupling signal, followed by a recovery to equilibrium. The major components of the signals have time constants of 1.5 ps for camphor free and 1.1 ps for camphor bound P450cam.
The coherence coupling signal appears near zero time delay and is due to the interleaved interaction of the electric fields of the degenerate pump and probe pulses as they interact with the electronic system of the material. The coherence coupling signal is relatively stronger in the six-coordinate low-spin species of the camphor free complex than in the five-coordinate high-spin species of camphor bound complex. This is probably related to the broader Soret band linewidth in the camphor bound species. We expect that the electronic dephasing time22;54–56 associated with the broader Soret band of the camphor bound complex might be shorter than the pulse width of the laser and this could result in a coherence coupling signal that is diminished compared to the camphor free form.
Figure 3 shows the open band coherence spectra of cytochrome P450cam, with and without camphor, obtained by exciting at 412 nm and 423 nm, respectively. The experimental data were analyzed using LPSVD. The left panels show the oscillatory data (small circles) and the LPSVD fits (solid red line). The LPSVD components corresponding to the dominant modes (~ 30–35 cm−1) and their phases are also shown as a solid blue line. The right panels show the corresponding power spectrum amplitudes. The coherence spectrum of camphor free ferric cytochrome P450cam is very complex, with the three strongest modes at 30, 103 and 194 cm−1 along with smaller peaks observed at 53, 80, 126 and 161 cm−1. The 30 cm−1 and 103 cm−1 modes are thought to contain large components of heme doming26–28;57–59 and saddling27, respectively. In the presence of camphor, the modes at 103 and 194 cm−1 are eliminated and the relative intensity of the mode at 33 cm−1 is dominant.
Figure 3.

The open-band coherence spectra of the ferric state of the cytochrome P450cam. The pump/probe excitation wavelengths are 423 and 412 nm for camphor-free and camphor-bound protein, respectively. The experimental data were analyzed by using LPSVD and the left panels shows the oscillatory components (circles) and the LPSVD fit (solid red lines). The LPSVD components corresponding to the dominant low frequency modes and their phases are also shown. The right panel shows the corresponding power spectrum amplitudes. The strongest modes in the camphor-free form are located at 30, 103 and 194 cm−1. In the camphor-bound form, the 103 and 194 cm−1 modes are diminished relative to the mode at 33 cm−1.
The more rapid damping of the coherent mode at 33 cm−1 in the high-spin camphor-bound form, compared to the camphor-free mode at 30 cm−1, may reflect either a more inhomogeneous heme environment or a genuine decrease in the coherent state lifetime. A similar trend in the Soret band line broadening is also observed in Fig. 2. We suggest that, like ferrous heme compounds60, the 5-coordinate ferric heme may be inherently more flexible and prone to out-of-plane distortions and therefore more inhomogeneous.
Figure 4 shows the correlation between the vibrational coherence and Raman spectra of cytochrome P450cam. The camphor-free and camphor-bound forms of cytochrome P450cam are shown in the top (4A) and bottom (4B) panels, respectively. The Raman spectrum (red), obtained by exciting at 413.1 nm, is presented at the top of the each figure. The Raman spectra can be compared with the coherence data, which were collected using both open-band and detuned detection using pulses with a center (i.e., carrier) wavelength of 423 nm (panel A) and 412 nm (panel B). The detuned detection selectively enhances the higher-frequency components of the third-order polarization and helps us to make comparisons with the resonance Raman data. The spontaneous resonance Raman spectra are unable to probe effectively below ~150 cm−1 because of cancellation of oppositely signed Raman Franck-Condon scattering amplitudes (when electronic resonance widths are of order 200 cm−1)61 and limitations brought on by Rayleigh and quasielastic light scattering. The insets show the detuned (blue) and open band (green) coherence data after removal of the monotonic background decay. The LPSVD fits, used to generate the time resolved frequency spectra, are also shown as solid lines through the open data points.
Figure 4.

The correlations between the resonance Raman and the vibrational coherence spectra for the camphor-free (A) and camphor-bound (B) ferric state of cytochrome P450cam are presented. The Raman spectrum (red, upper curve) was measured with excitation at 413.1 nm, whereas open band (green) and detuned (blue) coherence spectra were measured at a carrier wavelength of 423 nm (A) and 412 nm (B). The detuned coherence data were collected with a 0.5 nm spectral window, detuned 5 nm to the blue of the carrier wavelength. The time domain oscillation data are shown in the inset as small circles and the LPSVD fits are the solid lines through the data. There is good correlation between the Raman and coherence spectral frequencies, with estimated errors of roughly ± 5 cm−1.
As seen in Fig. 4A, there is a good correlation between the detuned coherence spectra and the Raman spectra in the camphor-free form. Importantly, vibrational modes (such as 234, 259 and 345 cm−1) in the Raman spectrum are correlated to modes obtained in the detuned detection (228, 259 and 351 cm−1) as well as the open-band detection (225, 260 and 341 cm−1). There is also strong correlation of modes when the open-band and detuned detection are compared at lower frequency (161, 194 cm−1). The mode at 80 is a small sharp peak in the open band measurement, which is correlated to the mode at 82 cm−1 respectively in the detuned detection. However, the small sharp peaks at 53 and 126 cm−1 in the open band experiment do not correlate well to the detuned spectrum. The mode at 120 cm−1 in the detuned spectrum might be split into the peaks at 103 and 126 cm−1 in the open band experiment, but small sharp peaks can sometimes arise as artifacts. This type of variation between the detuned and open band spectra is probably due to a combination of experimental error from noise in the time domain measurement and from the fact that the two measurements select for different frequency components in the coherent response.
In the camphor bound form (Fig. 4B), there is also a very strong correlation between detuned detection and the Raman spectra. The vibrational modes at 258, 344, and 380 cm−1 in the detuned coherence measurement match very well (± 1 cm−1) with the peaks at 258, 345 and 379 cm−1 in the Raman spectra. The mode at 235 cm−1 in the Raman spectrum is also well-aligned with the mode at 231 cm−1 in the detuned coherence spectrum. The modes at 226, 260 and 379 cm−1 in the open band measurement also align well with the Raman spectrum. The mode at 183 cm−1 in the open band measurement correlates to the mode at 176 cm−1 in the detuned spectrum and the 316 cm−1 mode in the open band measurement correlates to the 313 cm−1 mode in the Raman spectra, but this mode is not seen in the detuned spectrum. The absence of the mode at 313–316 cm−1 in the detuned spectrum is not understood, but it could be related to the broad (short-lived) feature near 281 cm−1, which appears down-shifted and broadened due to noise or coherence coupling contamination in the time domain data.
L358P mutant
The electronic absorption spectra of the ferric L358P mutant in the camphor-free and camphor-bound forms are shown in Fig 5A. The Soret maxima and the extinction coefficients18 of the L358P mutant are nearly same as those of wild type P450cam. This indicates that the L358P mutation does not induce major structural perturbations on the heme environment or its structure. Figure 5B shows the substrate dependence of the time resolved transmittance (ΔT) for camphor-free and camphor-bound L358P by exciting at 423 and 412 nm, respectively. The trend of the optical response is nearly same as found in the wild type protein. There is a bleaching signal, convolved with coherence coupling, followed by recovery to equilibrium with primary time constants of approximately 1.5 ps for the camphor-free, and 1.1 ps for the camphor-bound, L358P protein. The trend of the coherence coupling signal in the L358P mutant, with and without camphor, is similar to that of the wild type. The longer electronic dephasing time (narrower Soret bandwidth) of the low-spin ferric camphor free form appears to enhance the coherence coupling signal compared to the camphor bound form, which has broader Soret band and presumably undergoes faster electronic dephasing.
Figure 5.

(A) Normalized equilibrium electronic spectra of the camphor-free and camphor-bound ferric state of L358P. The Soret absorption maxima are 417 and 391 nm, respectively. (B) Normalized time resolved transmittance (ΔT) of the above complexes obtained with pump/probe excitation at 423 and 412 nm respectively. The kinetic traces show a bleaching signal (ΔT > 0) that recovers to equilibrium with kinetics that are similar to the wild type. The fitting parameters are for camphor free species τ1= 62 fs (a1 = 0.75), τ2= 1.5 ps (a2 = 0.21) and offset = 0.04 and for camphor bound complex τ1= 118 fs (a1 = 0.33), τ2= 1.1 ps (a2 = 0.74) and offset = − 0.07. The shortest time constant and amplitude are distorted due to the convolution with coherence coupling signal.
Figure 6 shows the vibrational coherence response of the L358P mutant without camphor (top panel) and with camphor bound (bottom panel) at 423 and 412 nm, respectively. The left panels show the oscillatory signals (circles) and the LPSVD fits (solid red line). The LPSVD components corresponding to the mode near ~40 cm−1 are also shown (blue solid lines). For the camphor-free L358P mutant, there are strong modes at 36 cm−1 and 147 cm−1. In the camphor-bound L358P, the mode at 147 cm−1 has disappeared and a mode in the 60–70 cm−1 region is strengthened relative to the mode at 36–39 cm−1.
Figure 6.

The open band coherence spectra of the ferric form of the L358P mutant. The pump/probe excitation wavelengths are 423 nm and 412 nm for camphor-free and camphor-bound proteins, respectively. The left panels show the oscillatory components (circles) and the LPSVD fits (solid red lines). The LPSVD components corresponding to the dominant modes ~39 cm−1 and their phases are also shown. The right panel shows the corresponding power spectrum amplitudes. The modes near 36 and 147 cm−1 dominate the camphor-free form, whereas in the camphor-bound form, the modes near 147 cm−1 have disappeared and the modes at 39 cm−1 and ~ 64 cm−1 dominate the spectrum.
The correlations between the Raman and coherence spectra of the camphor-free and camphor-bound states of the L358P mutant are shown in Fig. 7A and 7B, respectively. The detuned experiments utilize a 0.5 nm spectral window 5 nm to the blue of the carrier frequency maximum. Figure 7 shows good correlation between the open band and detuned coherence spectra and the Raman spectra. For camphor free L358P, the modes at 234, 259, 298, 350 and 379 cm−1 in the Raman spectra are especially well matched within the error of the detuned experiment (~ ±5 cm−1), which shows modes at 237, 254, 291, 347 and 382 cm−1. These modes are also reasonably well-correlated with the modes at 227, 256, 292 and 346 cm−1 observed in the open band measurements. The low frequency modes at 36 and 62 cm−1 in the open band measurement of the camphor-free sample are also well correlated to the detuned spectra.
Figure 7.

The correlation between the Raman and coherence spectra for the camphor-free (A) and camphor-bound (B) form of ferric L358P. The Raman spectrum (red, upper curve) was measured with excitation at 413.1 nm, whereas the open band (green) and detuned (blue) coherence spectra were measured at a carrier wavelength of 423 nm for the camphor-free and 412 nm for the camphor-bound form. The detuned coherence data were collected with a 0.5 nm spectral window, detuned 5 nm to the blue of the carrier wavelength. The time domain oscillation data are shown in the inset as small circles and the LPSVD fits as the solid lines through the data. There is good correlation between the Raman and coherence spectral frequencies, with estimated errors of roughly ± 5 cm−1.
When we examine the vibrational modes of the L358P mutant in the presence of camphor, the modes such as 235, 258 and 348 cm−1 in the Raman spectra are matched to the detuned coherence spectra at 234, 267 and 351 cm−1, respectively. There is also good correlation between the open band and the detuned coherence spectra in the lower frequency region.
Comparison of Raman spectra
Figure 8 compares the high and low frequency resonance Raman spectra of wild type and L358P mutant in the presence and absence of camphor. In the high frequency region of all four samples, the oxidation marker band, ν4, falls in the 1373–1371 cm−1 range, indicating the ferric oxidation state62;63. In camphor free P450cam, the ν3, ν2 and ν10 modes appear at 1502 cm−1, 1584 cm−1 and 1632 cm−1, respectively. This is consistent with a six-coordinate low spin (6C/LS) state in which both the thiolate sulfur of Cys357 and a water molecule are coordinated to the heme iron. Whereas, in the camphor free L358P sample, the ν2 (1582 cm−1) and ν10 (1638 cm−1) modes were down- and up-shifted, respectively with respect to the wild type protein. The low frequency modes of the camphor free L358P and WT are also very similar. However, a shift of the ν8 mode at 345 cm−1 to 350 cm −1 is noteworthy.
Figure 8.

Resonance Raman spectra of camphor-free and camphor-bound forms of ferric cytochrome P450cam and its L358P mutant. The spectra were obtained by exciting at 413.1 nm with a power of 6 mW. The left panel shows the low frequency and right panel shows the high frequency region. The descriptions of the modes are given in the text.
In the camphor-bound proteins, the ν3, ν2 and ν10 modes were enhanced and downshifted to 1487, 1570 and 1624 cm−1, respectively, indicating the formation of a five-coordinate high-spin (5C/HS) state in which the iron-coordinated water molecule has been expelled.
The mid- to low- frequency mode assignments for heme proteins have been discussed by Hu et al.64;65. The intense Raman bands at 677 and 345 cm−1 are assigned to ν7 and ν8, respectively. The ν8 mode is a combination of Fe-N(pyrrole) stretching and pyrrole substituent bending coordinates, while ν7 is due to symmetric porphyrin-stretching. The modes at 379 cm−1 and 425 cm−1 are assigned to the bending of the heme propionates, δ(CβCcCd), and bending of the heme vinyl group, δ(CβCaCb), respectively. The modes at 234 cm−1, 259 cm−1 and 522 cm−1 are tentatively assigned to γ24, ν9 and γ12.
The following general changes were noted in the low-frequency Raman spectra when camphor was added: The vinyl bending modes (at 425 cm−1 for WT P450cam and 426 cm−1 for L358P mutant) undergo a down-shift of 3 cm−1 and new modes appear at 366 cm−1 in WT and at 364 cm−1 in L358P. There is an upshift (~1 cm−1) of the 345 cm−1 mode in native P450cam and a downshift (~2 cm−1) of the 350 cm−1 mode in the mutant so that ν8 in the mutant and WT differ by ~5 cm−1 and ~2 cm−1 in the camphor free and bound state respectively. In WT, the mode at 308 cm−1 shifts to 313 cm−1 and intensifies, while the same mode at 305 cm−1 in L358P intensifies and shifts to 314 cm−1. Finally, the relative intensity of ν15 (754 cm−1) is increased in both WT and mutant upon camphor addition.
P450cam complexed with Pdx and L358P
In order to check the spectral similarities between the P450cam/Pdx complex and the L358P mutant, coherence spectra of ferric P450cam complexed with oxidized Pdx were measured by exciting at 412 nm. Figure 9 shows the oscillatory data and power spectra of the P450cam/Pdx complex. The coherence spectra of camphor-bound P450cam, L358P, and Pdx alone are also shown in the figure. When Pdx is added to P450cam, there is an extension of the coherence lifetimes and enhancement of the mode near 78 cm−1. This mode may be activated in response to a heme ruffling distortion, which has been suggested to affect the efficiency of electron transfer to the heme35;37;66. The appearance of the mode near 65–70 cm−1 in the L358P mutant resembles the 78 cm−1 mode in the P450cam/Pdx complex, but it is down-shifted by ~10 cm−1. This mode could still have significant ruffling content, but mode mixing due to structural perturbations can also lead to shifting force constants, which would account for the frequency disparity.
Figure 9.

The open band coherence spectra of the ferric form of the complex of Pdx with cytochrome P450cam is shown, along with Pdx, the camphor bound form of P450cam, and its L358P mutant. The pump/probe wavelength is 412 nm for all of the complexes. When compared to camphor bound P450cam, there is a mode at 78 cm−1 that is enhanced in the P450cam/Pdx complex. This mode probably corresponds to the mode at ~ 64 cm−1 seen in the camphor-bound form of L358P.
The power spectrum amplitudes of Pdx show a strong mode at ~ 31 cm−1 and weak activity at 53, 101, 121 and 291 cm−1. The weak mode ~ 291 cm−1 is likely associated with the labile sulfur-iron stretching vibration. The strong mode around 27 cm−1 in the P450cam/Pdx complex may arise from the 31 cm−1 mode of Pdx or the 33 cm−1 mode of the camphor bound P450cam. It is also conceivable that mode coupling between Pdx and P450cam leads to mixing and splitting of the low frequency modes, resulting in the appearance of the peaks at 27 cm−1 and 46 cm−1 upon formation of the P450 cam/Pdx complex.
NSD Analysis
The normal coordinate structural decomposition (NSD) analysis for ferric cytochrome P450cam in the camphor-free and camphor-bound states are compared in Fig. 10. NSD analysis quantifies the geometric distortion of the heme ring in different protein environments by extracting the displacements along low-frequency normal modes that are referenced to the planar protoporphyrin IX core (porphine). The details of the NSD analysis are given in the supporting information. Basically, the x-ray structure is mapped onto the low-frequency, out-of-plane (OOP), modes of different symmetry (such as propellering, ruffling, saddling, waving(x), waving(y), doming and inverse doming). There is a significant increase of heme doming and saddling distortions when camphor is bound to P450cam. The presence of an increased heme doming distortion in the camphor-bound sample is consistent with the increased relative intensity of the ~33 cm−1 doming mode observed in the coherence spectra of Fig. 3.
Figure 10.

Crystal structures and NSD analysis of the camphor-free and camphor-bound forms of the ferric heme of cytochrome P450cam. The displacement along each of the low frequency normal mode unit vectors of Fe porphine is given in mass weighted coordinates (amu1/2 Å). The color coding for the modes is pro: propellering (blue), ruf: ruffling (green), sad: saddling (red), wav(x): wavingx (light blue), wav(y): wavingy (yellow), dom: doming (purple), invdom: inverse doming (gray). The crystal structures are extracted from the protein data bank: 1PHC79 for the camphor-free and 2CPP72 for the camphor-bound form. One of the major differences is that the camphor-bound form has a strong heme doming distortion, which is consistent with the appearance of the ~33 cm−1 mode in the coherence spectra of camphor-bound P450cam (Fig. 3)
However, it is important to note that the quantitative correlation between the coherence intensities and the heme distortion 25 is based upon the assumption that the ratio of the ground and excited state force constants is independent of the protein induced distortions. Insofar as this assumption holds, the coherence intensities arising from distortion-induced Raman scattering will scale as the square of the protein induced distortions along the various low-frequency normal modes as determined by NSD analysis25.
Discussion
In this paper we have presented an investigation of the low frequency dynamics of cytochrome P450cam, its L358P mutant, and the P450cam/Pdx complex by using resonance Raman and vibrational coherence spectroscopy. At 300 K, heme vibrational modes below 200 cm−1 are thermally excited. The observation of vibrational coherence demonstrates that these modes are not over-damped and diffusive due to rapid dissipation of energy to the environment. As a result of their recurrent thermally driven oscillatory behavior, such modes are important candidates for reaction coordinates that are associated with the broad class of biochemical reactions mediated by heme proteins, as demonstrated for the CO ligand binding reaction29;30.
Cytochrome P450cam
The vibrational coherence spectrum of camphor-free cytochrome P450cam shown in Fig. 3 reveals strong modes at 30, 103, 161 and 194 cm−1 along with small sharp peaks observed at 53, 80, 126 cm−1. The 103 cm−1 mode is upshifted by 7 cm−1 compared to the 96 cm−1 mode of HRP-CN, another low-spin ferric heme system. The 96 cm−1 mode was suggested to involve heme saddling67 and, by analogy, we suggest that the 103 cm−1 mode is activated by a similar heme distortion, probably involving saddling and/or ruffling. Distortions along the heme ruffling coordinate have been suggested to affect both the reduction potential and the electron transfer rate31;35;36. As an example, mutation studies of a heme nitric oxide/oxygen binding protein showed the increase of the reduction potential as the saddling distortion of the heme cofactor decreases32. Analogous behavior has also been observed for non-planar model compounds33;34 which are harder to reduce than planar porphyrins. Recent NMR studies of spin density delocalization on the β-pyrrole carbon atoms of the heme clearly show more potential for overlap with an electron donor when the heme is in a more planar configuration37. The strong activity of the 103 cm−1 mode in the absence of camphor is a suggestive of a heme distortion that helps to stabilize the ferric state. When camphor is bound to cytochrome P450cam, the higher frequency modes are diminished and the relative intensity of the mode at 33 cm−1, assigned to heme doming, is dramatically increased. It is also known that, when camphor binds, water is lost as an axial ligand and the heme becomes 5-coordinate high-spin and distorts along the doming coordinate. The increased doming distortion upon camphor addition (strong 33 cm−1 mode) is consistent with the NSD analysis of the x-ray structure. This demonstrates the connection between heme distortions and the intensities of the low frequency modes observed in the coherence spectra.25
A reduction of the heme ruffling/saddling distortion that activates the 103 cm−1 mode (Fig. 3) is also indicated by its decreased intensity in the camphor bound state. This is consistent with an increased reduction potential upon camphor binding. The reduction potential increases from −340 mV to −170 mV upon camphor binding, which suggests that the ruffling/saddling distortion stabilizes the ferric state. However, it must be acknowledged that a much larger fraction of the change in reduction potential is probably due to the change in ligation and spin-state of the heme that takes place when camphor binds and displaces the water ligand.
L358P mutant
Based on electronic absorption, resonance Raman and NMR spectroscopy, it is clear that the L358P mutation does not lead to major electronic or geometric structural perturbations of the heme environment18. Figure 6 compares the coherence spectra of L358P in the camphor-bound and camphor-free forms. When camphor is bound, the doming mode locates at ~39 cm and a mode near 64 cm−1 appears. This mode probably contains a significant ruffling contribution, which may be involved in stabilizing the ferric heme system35;66. It is possible that the ruffling mode could also be involved at some level as an electron transfer reaction coordinate. When the heme ruffles, the mixing of the d-orbitals of the iron atom and the p-orbitals of the porphyrin nitrogen atoms is perturbed. This can modulate the electron density in the iron d-orbitals and potentially the amount of thiolate electron donation.
Since there are only two thiolate hydrogen bonds in L358P compared to three in the wild type, the reduction potential of the heme iron is lowered18 by about 36 mV compared to the wild type, and Cys357 moves closer to the heme iron 20;21. This presumably arises due to increased electron donation from the axial cysteine when only two H-bonds are present and is consistent with previous report14 of an increase in anionic character of the thiolate ligand in the P450cam/Pdxcomplex.
The heme ruffling distortion in the camphor bound state of L358P, as signaled by the increased intensity of the mode at 64 cm−1, should stabilize its ferric form relative to the wild type. Insofar as L358P is a mimic of the Pdx bound complex, this would indicate that (at this stage of the catalytic cycle) a heme structural perturbation takes place to help limit the amount of electron donation from the thiolate to the heme iron. One possibility is that heme ruffling may help to control the amount of thiolate electron donation into the iron d-orbitals that is allowed when only two (rather than three) thiolate H-bonds are present in the ferric state. It is also likely that heme distortion alters both the localization of the electron hole molecular orbitals on the iron37 and the nuclear reorganization energy, which are involved in regulating the electron transfer process. The localization of the vacant iron-porphyrin molecular orbital acceptor state on the iron will tend to reduce donor-acceptor overlap at the heme periphery and, thus, the associated electron transfer rate. Such effects potentially offer a finely tuned mechanism for controlling the rate of heme reduction, even when the distortion does not greatly affect the reduction potential. For complex interacting systems, where the electron delivery is coupled to the binding and release of the electron transfer partner, control of the relative reaction rates can be as important as the driving force in regulating the overall dynamics of the system.
In contrast to the native protein, the heme in the Pdx complex and the L358P mutant is more distorted in the camphor bound state68. This is probably related to the fact that the camphor bound ferric state is 5-coordinate and more easily distorted by the forces that arise from Pdx binding or L358P mutation. Important catalytic consequences of the Pdx-induced heme distortion involve Asp251 rearrangement and water reordering in the proximal pocket68. As mentioned above, an additional role of the heme distortion may be to control the amount of electron donation to the ferric iron from the more basic thiolate when it is stabilized by only two H-bonds in the L358P mutant or the Pdx bound form. We expect that the reduction potential of the heme should reflect the loss of the H-bond to the thiolate. However, by limiting the amount of thiolate electron donation, the distortion might minimize the changes in the reduction potential. The question of precisely how much the reduction potential of the heme changes upon binding of Pdx remains open as there are several differing reports for the redox potential of camphor bound P450cam 4;18;69, which make the comparison (−36 mV) with the reduction potential of the L358P mutant18 somewhat ambiguous.
P450cam/Pdx complex
Figure 9 shows the vibrational coherence spectra of the P450cam/Pdx complex. When Pdx is added to the cytochrome P450cam, there is an enhancement of the 78 cm−1 mode, which is believed to involve the heme ruffling distortion. When the P450/Pdx complex is formed, Arg 112 of P450 shifts and aids in the formation of a salt bridge with the Asp38 of Pdx4;7;70. Arg 112 is also connected to the heme via a hydrogen bond with one of the heme propionates71;72. This interaction creates a positional change of the propionate and evidently leads to changes in the protein-induced heme distortions that activate the 78 cm−1 mode. This occurs because Pdx binding also disrupts the H-bonding, shown in Fig. 1, which stabilizes the thiolate ligand. Electron density transfer upon Pdx binding is consistent with the report of movement of Cys357 toward the heme iron in the P450cam/Pdx complex11, and its lowered reduction potential18. This is also consistent with the earlier observation of a 3 cm−1 up-shift of the Fe-S mode in the P450cam/Pdx complex14.
It is unclear at this time what changes take place in the heme reduction potential when either oxidized or reduced Pdx binds to ferric P450cam. Early reports suggested that the reduction potential of the heme was not changed during complexation based on the assumption that the binding of the two proteins is unaffected by the redox state of cytochrome P450cam73. On the other hand, some reports have suggested that the heme reduction potential is reduced by about 90 mV when Pdx binds11.
The spectral perturbations observed in the coherence measurements are consistent with earlier comparisons between the P450/Pdx complex and the L358P mutant21, which show that the heme and its environment are similar in these two situations. Thus, we suggest that the appearance of the modes at 64 cm−1 and 78 cm−1, in the coherence spectra of the camphor bound L358P mutant and the P450cam/Pdx complex, respectively, reflects a similar heme structural perturbation, probably involving a heme ruffling and/or saddling distortion25. The ruffling and/or saddling distortions are a signature of the forces on the heme that arise from either Pdx binding or L358P substitution. In addition to triggering the proximal pocket water rearrangements68, these low frequency out-of-plane distortions may turn out to be important reaction coordinates that help to control electron transfer to the heme. For CO binding to heme, it has been shown that the heme doming distortion (a different low frequency heme mode near 40 cm−1) is a key reaction coordinate that plays a significant role in determining the barrier to ligand binding29.
For electron transfer to heme, one must consider both electronic effects, such as distortion-induced mixing of the iron-porphyrin orbitals, as well as structural changes that alter the nuclear reorganization energy. Insofar as the distortions of the heme induced by Pdx binding move the system toward the configuration of the reduced state (at fixed reduction potential) this would reduce the barrier for electron transport, thus helping to ensure that the electron transfer reaction takes place prior to dissociation of the protein-protein complex.
Finally, we note that the low frequency coherence spectra of Pdx and P450cam each have a strong mode near 31–33 cm−1. This leaves open the possibility that vibrational resonance can occur so that mixing and splitting of these modes accounts for the observed spectrum in the protein-protein complex. It is conceivable that such vibrational coupling could be related to the electron transfer process. Since the modes near 30 cm−1 are highly excited by thermal fluctuations at 300 K (200 cm−1), and they involve heme out-of-plane distortions that can modulate the relative energies of the iron orbitals and lead to localization or delocalization of the iron hole wavefunctions, we must consider the possibility that we are observing an important electron transfer reaction coordinate in the P450cam/Pdxsystem.
Raman Spectra
The binding of the camphor substrate can also be observed by changes in the resonance Raman spectra of the heme. In the low frequency region, we observe the bending motions of the vinyl and/or propionate substituents and other out-of-plane modes activated by perturbations induced on the heme geometry. It was found previously that the Raman excitation profile of the 351 cm−1 Fe-S axial ligand mode of the substrate bound cytochrome P450cam has a doublet structure that is shifted by about 2000 cm−1 to the blue of the 391 nm Soret band maximum74. Hence, the Fe-S mode does not appear with 413.1 nm excitation in Fig. 8 because there is no significant resonance enhancement. Thus, the mode near 345–350 cm−1 is assigned to ν8, based on the similarity of its frequency to the ν8 mode seen in the resonance Raman spectra of ferric samples with visible excitation. This mode, which involves Fe-N(pyrrole) stretching and pyrrole substituent bending, is shifted from 345 cm−1 to 350 cm−1 in the camphor-free WT to L358P comparison. The same mode at 346 cm−1 in the camphor bound P450cam is shifted to 348 cm−1 in the camphor bound L358P. The systematic upshift of this mode in the L358P mutant may be due to the thiolate ligand being pushed towards the heme in the mutant sample resulting in electronic donation from the thiolate to the heme. This would lead to bond strengthening that is reflected as an up-shift of 2–5 cm−1 of the ν8 mode.
The bending modes of the heme propionates, δ(CβCcCd) for all the complexes studied appeared at the same position (379 cm−1) indicating insensitivity to the presence of substrate and mutation. The frequency of this mode is sensitive to the hydrogen bonding of the propionate groups and suggests H-bonds to well-ordered amino acid residues, rather than to more disordered bulk solvent molecules75;76. The addition of camphor to cytochrome P450cam induces a new vibration at 366 cm−1. This is analogous to cytochrome P450arom, where the Raman lines at 377 and 371 cm−1 were assigned to the bending modes of heme propionate with and without hydrogen-bonding interaction with the protein environment, respectively77. The bending mode of the heme propionate at 371 cm−1 in substrate-free P450arom is absent upon the addition of the substrate, whereas the peak at 377 cm−1 is observed both with and without substrate. Hence, in cytochrome P450cam, the new mode 366 cm−1 has been correlated to partial population of a bending mode of propionate without a protein hydrogen-bonding interaction. There is a similar effect observed in the L358P mutant, but with a slightly lower propionic bending mode at 364 cm−1 for the species without H-bonding.
The mode at 425 cm−1 assigned to vinyl bending modes in the camphor free cytochrome P450cam is slightly down-shifted to 422 cm−1 when camphor binds. A similar down-shift was observed in the mutant. This may be due to a hydrophobic interaction between the substrate and the vinyl group. The appearance of the ν7 modes at the same position (~676 cm−1) for all four complexes represents that the symmetric porphyrin-stretching modes do not change with addition of substrate or with mutation. The increase in the intensity of ν15 at ~755 cm−1 in the camphor bound forms of both proteins indicates that the porphyrin symmetric breathing mode is coupled more strongly to the Soret excitation when the water ligand is lost and the high-spin heme is in a more domed 5-coordinate high-spin configuration.
Summary
In summary, we have investigated the low frequency dynamics of cytochrome P450cam, its mutant L358P, and its electron-transfer complex (P450cam/Pdx) using both resonance Raman and vibrational coherence spectroscopy. Vibrational modes in the low-frequency region (< 200 cm−1) are “soft” (weak force constants) and they are easily excited by thermal fluctuations. Because the force constants are weak, protein-induced distortions along these low-frequency normal coordinates also occur and offer a variety of control mechanisms for both ligand and electron transfer to and from the heme group. Generally, the low-frequency heme modes involve out-of-plane motions that are activated in the Raman spectrum by symmetry breaking distortions of the normally planar heme25. Because of this, the low frequency heme modes are sensitive reporters of distortions in the heme geometry. The vibrational coherence spectra and steady state Raman spectra show a strong correlation to each other (± 5 cm−1) in the range above 200 cm−1 where both spectra can be obtained. This helps to validate the low frequency modes that are extracted in the region below 200 cm−1, which are not accessible to Raman spectroscopy.
It has been postulated27;35;66;78 that heme out-of-plane distortions along the ruffling and/or saddling coordinates can help to stabilize the ferric state. The appearance of the strong ruffling or saddling mode at 103 cm−1 in the camphor free cytochrome P450cam sample is consistent with such a description, since it is well known that the camphor free state has a reduction potential that is reduced by ~150 mV compared to the camphor bound form. Clearly the ligation and spin state of the central iron atom are major contributors to the heme reduction potential, but the d-π orbital interactions that are modulated by non-planar heme distortions are also expected to play a role, particularly in the determination of the rates of electron transfer37. After the addition of the camphor, the increased relative intensity of the doming mode at ~33 cm−1 is consistent with larger heme doming distortions, as revealed by the NSD analysis.
The enhancement of the mode near 78 cm−1 in the P450cam/Pdx complex probably reflects a heme distortion with ruffling components. Such a heme distortion may offer a method to control and/or redirect the increased thiolate electron donation in the ferric complex when the H-bonding from surrounding amino acids is decreased upon Pdx binding. The increased heme ruffling will localize the vacant iron-porphyrin molecular orbital on the iron, reducing the electron transfer spatial overlap with the heme periphery. Such distortions may serve to both control the rate of electron transfer to the heme as well as to help focus the available electron density during an iron-localized catalytic event (such as dioxygen scission). Thus, the soft, thermally accessible, heme out-of-plane distortions (or vibrational excitations of such modes) can serve as a mechanism to control the dynamics of the various processes involved in heme protein reactions.
The addition of Pdx evidently induces conformational changes of the heme environment and the H-bonding arrangement of the thiolate sulfur that are similar to those found in the L358P mutant. The resonance Raman spectra of P450cam and its L358P mutant were compared and the observed up-shift of the ν8 mode (~345 cm−1) in L358P is consistent with the thiolate ligand forming a stronger bond and moving closer to the heme in the mutant sample. The effect of camphor on the coherence spectra of the L358P mutant revealed the appearance of a mode at ~65 cm−1, which also reinforces the similarities between the L358P mutant and the P450cam/Pdx complex and is thus consistent with earlier studies21.
Finally, the first vibrational coherence spectrum of Pdx is presented in this work (Fig. 9) and a strong mode at ~31 cm−1 is observed. Given the presence of a similar mode frequency in the spectrum of its heme chromophore redox partner, mode mixing and splitting to generate the modes 27 and 46 cm−1 observed in the P450cam/Pdx complex is a possibility that remains under consideration.
Supplementary Material
Acknowledgments
This work is supported by grants from the NIH (DK35090 to PMC and GM31756 to SGS) and NSF(MCB-0744738 to PMC). The authors thank Mr. Alexander Demidov for the NSD calculation, Dr. Abdelkrim Benabbas and Mr. Yuhan Sun for useful discussion and Jason Bugno, Yelena V. Grinkova and Dr. Aditi Das for expression and purification of native and mutant proteins.
Footnotes
Supporting information available
The experimental setup and the procedure for the NSD analysis are presented in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
Reference List
- 1.Denisov IG, Makris TM, Sligar SG, Schlichting I. Chem Rev. 2005;105:2253–2277. doi: 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- 2.Champion PM, Stallard BR, Wagner GC, Gunsalus IC. J Am Chem Soc. 1982;104:5469–5472. [Google Scholar]
- 3.Sono M, Andersson LA, Dawson JH. J Biol Chem. 1982;257:8308–8320. [PubMed] [Google Scholar]
- 4.Unno M, Shimada H, Toba Y, Makino R, Ishimura Y. J Biol Chem. 1996;271:17869–17874. doi: 10.1074/jbc.271.30.17869. [DOI] [PubMed] [Google Scholar]
- 5.Pochapsky TC, Lyons TA, Kazanis S, Arakaki T, Ratnaswamy G. Biochimie. 1996;78:723–733. doi: 10.1016/s0300-9084(97)82530-8. [DOI] [PubMed] [Google Scholar]
- 6.Furukawa Y, Ishimori K, Morishima I. Biochemistry. 2000;39:10996–11004. doi: 10.1021/bi000874y. [DOI] [PubMed] [Google Scholar]
- 7.Roitberg AE, Holden MJ, Mayhew MP, Kurnikov IV, Beratan DN, Vilker VL. J Am Chem Soc. 1998;120:8927–8932. [Google Scholar]
- 8.Shimada H, Nagano S, Ariga Y, Unno M, Egawa T, Hishiki T, Ishimura Y. J Biol Chem. 1999;274:9363–9369. doi: 10.1074/jbc.274.14.9363. [DOI] [PubMed] [Google Scholar]
- 9.Lipscomb JD. Biochemistry. 1980;19:3590–3599. doi: 10.1021/bi00556a027. [DOI] [PubMed] [Google Scholar]
- 10.Pochapsky SS, Pochapsky TC, Wei JW. Biochemistry. 2003;42:5649–5656. doi: 10.1021/bi034263s. [DOI] [PubMed] [Google Scholar]
- 11.Tosha T, Yoshioka S, Takahashi S, Ishimori K, Shimada H, Morishima I. J Biol Chem. 2003;278:39809–39821. doi: 10.1074/jbc.M304265200. [DOI] [PubMed] [Google Scholar]
- 12.Zhang W, Pochapsky SS, Pochapsky TC, Jain NU. J Mol Biol. 2008;384:349–363. doi: 10.1016/j.jmb.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sjodin T, Christian JF, Macdonald IDG, Davydov R, Unno M, Sligar SC, Hoffman BM, Champion PM. Biochemistry. 2001;40:6852–6859. doi: 10.1021/bi002510b. [DOI] [PubMed] [Google Scholar]
- 14.Unno M, Christian JF, Benson DE, Gerber NC, Sligar SG, Champion PM. J Am Chem Soc. 1997;119:6614–6620. [Google Scholar]
- 15.Unno M, Christian JF, Sjodin T, Benson DE, Macdonald IDG, Sligar SG, Champion PM. J Biol Chem. 2002;277:2547–2553. doi: 10.1074/jbc.M108917200. [DOI] [PubMed] [Google Scholar]
- 16.Nagano S, Shimada H, Tarumi A, Hishiki T, Kimata-Ariga Y, Egawa T, Suematsu M, Park SY, Adachi S, Shiro Y, Ishimura Y. Biochemistry. 2003;42:14507–14514. doi: 10.1021/bi035410p. [DOI] [PubMed] [Google Scholar]
- 17.Makino R, Iizuka S, Sakaguchi K, Ishimura Y. Oxygenases and Oxygen Metabolism. In: Nozaki M, Yamamoto S, Ishimura Y, Coon MJ, Ernster L, Estabrook RW, editors. Oxygenases and Oxygen Metabolism. Academic Press; New York: 1982. pp. 467–477. [Google Scholar]
- 18.Yoshioka S, Takahashi S, Ishimori K, Morishima I. J Inorg Biochem. 2000;81:141–151. doi: 10.1016/s0162-0134(00)00097-0. [DOI] [PubMed] [Google Scholar]
- 19.Yoshioka S, Tosha T, Takahashi S, Ishimori K, Hori H, Morishima I. J Am Chem Soc. 2002;124:14571–14579. doi: 10.1021/ja0265409. [DOI] [PubMed] [Google Scholar]
- 20.Nagano S, Tosha T, Ishimori K, Morishima I, Poulos TL. J Biol Chem. 2004;279:42844–42849. doi: 10.1074/jbc.M404217200. [DOI] [PubMed] [Google Scholar]
- 21.Tosha T, Yoshioka S, Ishimori K, Morishima I. J Biol Chem. 2004;279:42836–42843. doi: 10.1074/jbc.M404216200. [DOI] [PubMed] [Google Scholar]
- 22.Mukamel S. Principles of Nonlinear Optical Spectroscopy. Oxford University Press; New York: 1995. [Google Scholar]
- 23.Kumar ATN, Rosca F, Widom A, Champion PM. J Chem Phys. 2001;114:701–724. [Google Scholar]
- 24.Kumar ATN, Rosca F, Widom A, Champion PM. J Chem Phys. 2001;114:6795–6815. [Google Scholar]
- 25.Kubo M, Gruia F, Benabbas A, Barabanschikov A, Montfort WR, Maes EM, Champion PM. J Am Chem Soc. 2008;130:9800–9811. doi: 10.1021/ja800916d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gruia F, Kubo M, Ye X, Champion PM. Biophys J. 2008;94:2252–2268. doi: 10.1529/biophysj.107.122119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gruia F, Kubo M, Ye X, Ionascu D, Lu C, Poole RK, Yeh SR, Champion PM. J Am Chem Soc. 2008;130:5231–5244. doi: 10.1021/ja7104027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gruia F, Ionascu D, Kubo M, Ye X, Dawson J, Osborne RL, Sligar SG, Denisov I, Das A, Poulos TL, Terner J, Champion PM. Biochemistry. 2008;47:5156–5167. doi: 10.1021/bi7025485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye X, Ionascu D, Gruia F, Yu A, Benabbas A, Champion PM. Proc Natl Acad Sci US A. 2007;104:14682–14687. doi: 10.1073/pnas.0702622104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srajer V, Reinisch L, Champion PM. J Am Chem Soc. 1988;110:6656–6670. [Google Scholar]
- 31.Ma JG, Zhang J, Franco R, Jia SL, Moura I, Moura JJ, Kroneck PM, Shelnutt JA. Biochemistry. 1998;37:12431–12442. doi: 10.1021/bi981189i. [DOI] [PubMed] [Google Scholar]
- 32.Olea C, Jr, Kuriyan J, Marletta MA. J Am Chem Soc. 2010;132:12794–12795. doi: 10.1021/ja106252b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Barkigia KM, Chantranupong L, Smith KM, Fajer J. J Am Chem Soc. 1988;110:7566–7567. [Google Scholar]
- 34.Ravikanth M, Chandrashekar TK. Struct Bonding. 1995:105–188. [Google Scholar]
- 35.Walker FA. Coord Chem Rev. 1999;186:471–534. [Google Scholar]
- 36.Shelnutt JA, Song XZ, Ma JG, Jia SL, Jentzen W, Medforth CJ. Chem Soc Rev. 1998;27:31–41. [Google Scholar]
- 37.Liptak MD, Wen X, Bren KL. J Am Chem Soc. 2010;132:9753–9763. doi: 10.1021/ja102098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Makris TM, von Koenig K, Schlichting I, Sligar SG. Biochemistry. 2007;46:14129–14140. doi: 10.1021/bi7013695. [DOI] [PubMed] [Google Scholar]
- 39.Gerber NC, Sligar SG. J Biol Chem. 1994;269:4260–4266. [PubMed] [Google Scholar]
- 40.Sligar SG, Debrunner PG, Lipscomb JD, Namtvedt MJ, Gunsalus IC. Proc Natl Acad Sci US A. 1974;71:3906–3910. doi: 10.1073/pnas.71.10.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Furukawa Y, Morishima I. J Biol Chem. 2001;276:12983–12990. doi: 10.1074/jbc.M010217200. [DOI] [PubMed] [Google Scholar]
- 42.Koga H, Sagara Y, Yaoi T, Tsujimura M, Nakamura K, Sekimizu K, Makino R, Shimada H, Ishimura Y, Yura K, Go M, Ikeguchi M, Horiuchi T. FEBS Letters. 1993;331:109–113. doi: 10.1016/0014-5793(93)80307-g. [DOI] [PubMed] [Google Scholar]
- 43.Hintz MJ, Mock DM, Peterson LL, Tuttle K, Peterson JA. J Biol Chem. 1982;257:14324–14332. [PubMed] [Google Scholar]
- 44.Aoki M, Ishimori K, Morishima I, Wada Y. Inorg Chim Acta. 1998;272:80–88. [Google Scholar]
- 45.Rosca F, Kumar ATN, Ye X, Sjodin T, Demidov AA, Champion PM. J Phys Chem A. 2000;104:4280–4290. [Google Scholar]
- 46.Gruia F, Ye X, Ionascu D, Kubo M, Champion PM. Biophys J. 2007;93:4404–4413. doi: 10.1529/biophysj.107.114736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang W, Ye X, Demidov AA, Rosca F, Sjodin T, Cao WX, Sheeran M, Champion PM. J Phys Chem B. 2000;104:10789–10801. [Google Scholar]
- 48.Rosca F, Kumar ATN, Ye X, Sjodin T, Demidov AA, Champion PM. J Phys Chem A. 2000;104:4280–4290. [Google Scholar]
- 49.Gruia F, Ye X, Ionascu D, Kubo M, Champion PM. Biophys J. 2007;93:4404–4413. doi: 10.1529/biophysj.107.114736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosca F, Kumar ATN, Ye X, Sjodin T, Demidov AA, Champion PM. J Phys Chem A. 2000;104:4280–4290. [Google Scholar]
- 51.Wang W, Ye X, Demidov AA, Rosca F, Sjodin T, Cao WX, Sheeran M, Champion PM. J Phys Chem B. 2000;104:10789–10801. [Google Scholar]
- 52.Constantine S, Zhou Y, Morais J, Ziegler LD. J Phys Chem A. 1997;101:5456–5462. [Google Scholar]
- 53.Wells AV, Li P, Champion PM, Martinis SA, Sligar SG. Biochemistry. 1992;31:4384–4393. doi: 10.1021/bi00133a002. [DOI] [PubMed] [Google Scholar]
- 54.Chachisvilis M, Fidder H, Sundström V. Chem Phys Lett. 1995;234:141–150. [Google Scholar]
- 55.Balk MW, Fleming GR. J Chem Phys. 1985;83:4300–4307. [Google Scholar]
- 56.Gu YG, Widom A, Champion PM. J Chem Phys. 1994;100:2547–2560. [Google Scholar]
- 57.Zhu L, Li P, Huang M, Sage JT, Champion PM. Phys Rev Lett. 1994;72:301–304. doi: 10.1103/PhysRevLett.72.301. [DOI] [PubMed] [Google Scholar]
- 58.Liebl U, Lipowski G, Negrerie M, Lambry JC, Martin JL, Vos MH. Nature. 1999;401:181–184. doi: 10.1038/43699. [DOI] [PubMed] [Google Scholar]
- 59.Rosca F, Kumar ATN, Ionascu D, Ye X, Demidov AA, Sjodin T, Wharton D, Barrick D, Sligar SG, Yonetani T, Champion PM. J Phys Chem A. 2002;106:3540–3552. [Google Scholar]
- 60.Srajer V, Schomacker KT, Champion PM. Phys Rev Lett. 1986;57:1267–1270. doi: 10.1103/PhysRevLett.57.1267. [DOI] [PubMed] [Google Scholar]
- 61.Schomacker KT, Bangcharoenpaurpong O, Champion PM. J Chem Phys. 1984;80:4701–4717. [Google Scholar]
- 62.Spaulding LD, Chang CC, Yu NT, Felton RH. J Am Chem Soc. 2002;97:2517–2525. [Google Scholar]
- 63.Spiro TG. Iron Porphyrins. Addison-Wesley; New York: 1983. p. 91. [Google Scholar]
- 64.Hu S, Smith KM, Spiro TG. J Am Chem Soc. 1996;118:12638–12646. [Google Scholar]
- 65.Hu S, Morris IK, Singh JP, Smith KM, Spiro TG. J Am Chem Soc. 2002;115:12446–12458. [Google Scholar]
- 66.Roberts SA, Weichsel A, Qiu Y, Shelnutt JA, Walker FA, Montfort WR. Biochemistry. 2001;40:11327–11337. doi: 10.1021/bi0109257. [DOI] [PubMed] [Google Scholar]
- 67.Gruia F, Kubo M, Ye X, Ionascu D, Lu C, Poole RK, Yeh SR, Champion PM. J Am Chem Soc. 2008;130:5231–5244. doi: 10.1021/ja7104027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sevrioukova IF, Poulos TL. Arch Biochem Biophys. 2010 In Press, Corrected Proof. [Google Scholar]
- 69.Fisher MT, Sligar SG. J Am Chem Soc. 1985;107:5018–5019. [Google Scholar]
- 70.Pochapsky TC, Lyons TA, Kazanis S, Arakaki T, Ratnaswamy G. Biochimie. 1996;78:723–733. doi: 10.1016/s0300-9084(97)82530-8. [DOI] [PubMed] [Google Scholar]
- 71.Poulos TL, Finzel BC, Gunsalus IC, Wagner GC, Kraut J. J Biol Chem. 1985;260:16122–16130. [PubMed] [Google Scholar]
- 72.Poulos TL, Finzel BC, Howard AJ. J Mol Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. [DOI] [PubMed] [Google Scholar]
- 73.Sligar SG, Gunsalus IC. Proc Natl Acad Sci USA. 1976;73:1078–1082. doi: 10.1073/pnas.73.4.1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bangcharoenpaurpong O, Champion PM, Martinis SA, Sligar SG. J Chem Phys. 1987;87:4273–4284. [Google Scholar]
- 75.Chen Z, Ost TWB, Schelvis JPM. Biochemistry. 2004;43:1798–1808. doi: 10.1021/bi034920g. [DOI] [PubMed] [Google Scholar]
- 76.Peterson ES, Friedman JM, Chien EYT, Sligar SG. Biochemistry. 1998;37:12301–12319. doi: 10.1021/bi980752u. [DOI] [PubMed] [Google Scholar]
- 77.Tosha T, Kagawa N, Ohta T, Yoshioka S, Waterman MR, Kitagawa T. Biochemistry. 2006;45:5631–5640. doi: 10.1021/bi060094a. [DOI] [PubMed] [Google Scholar]
- 78.Karunakaran V, Benabbas A, Sun Y, Zhang Z, Singh S, Banerjee R, Champion PM. J Phys Chem B. 2010;114:3294–3306. doi: 10.1021/jp909700r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poulos TL, Finzel BC, Howard AJ. Biochemistry. 2002;25:5314–5322. doi: 10.1021/bi00366a049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.